
Co-Encapsulation of Methylene Blue and PARP-Inhibitor into Poly(Lactic-Co-Glycolic Acid) Nanoparticles for Enhanced PDT of Cancer



Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of PLGA NPs
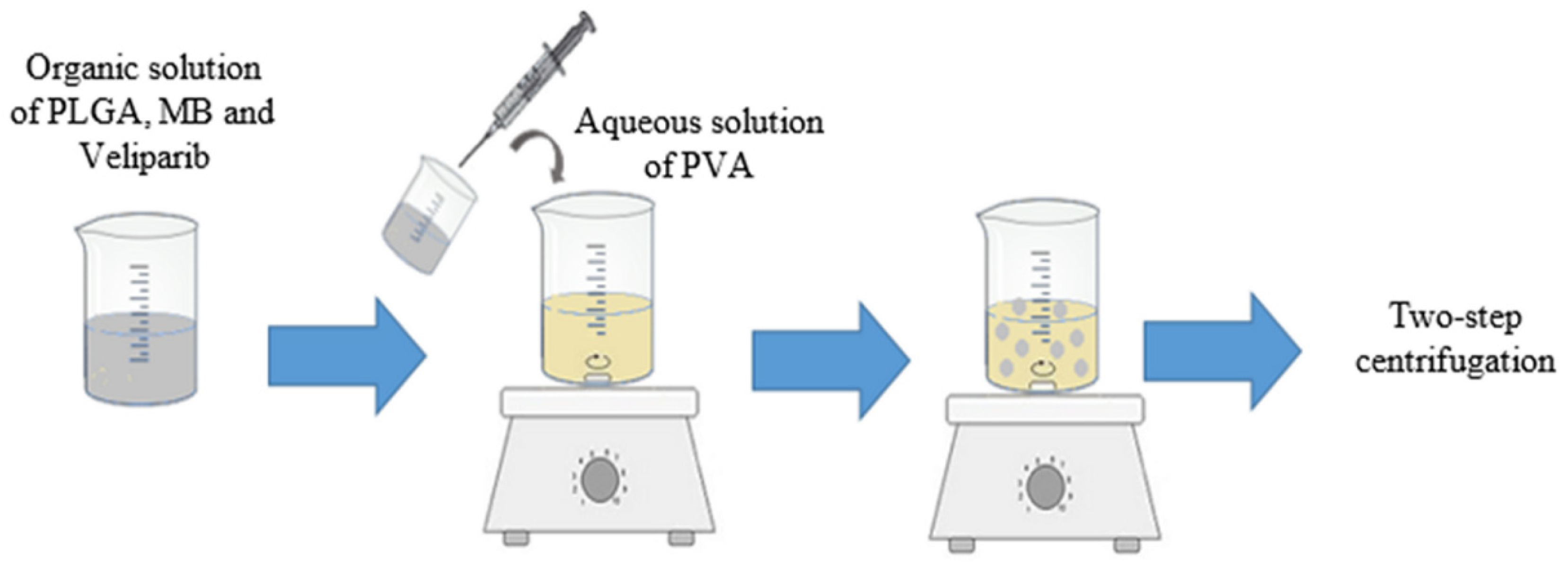
2.3. Synthesis of PLGA NPs with MB and Veliparib Co-Loaded (VMB-NPs)
2.4. Evaluation of NPs Physicochemical Properties
2.5. Determination of Encapsulation Efficiency (%EE)
2.6. In Vitro Drug Release
2.7. Cell Viability Assays
3. Results and Discussion
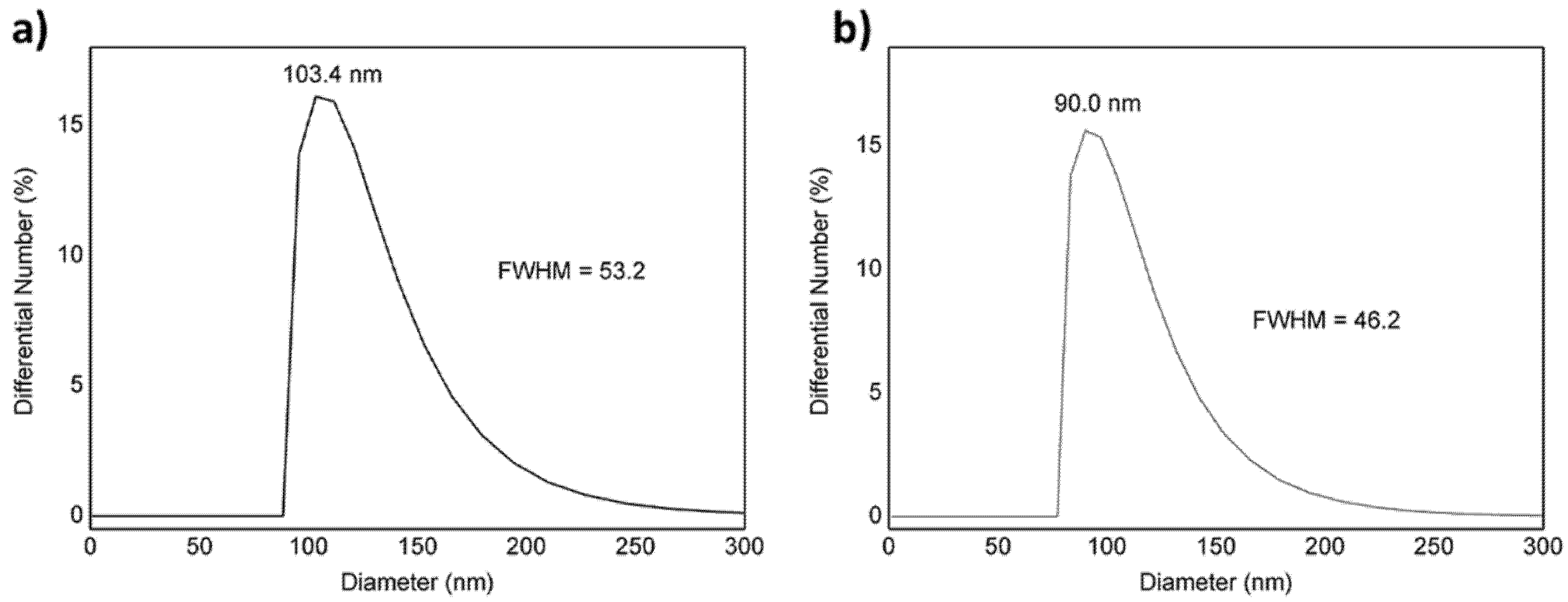
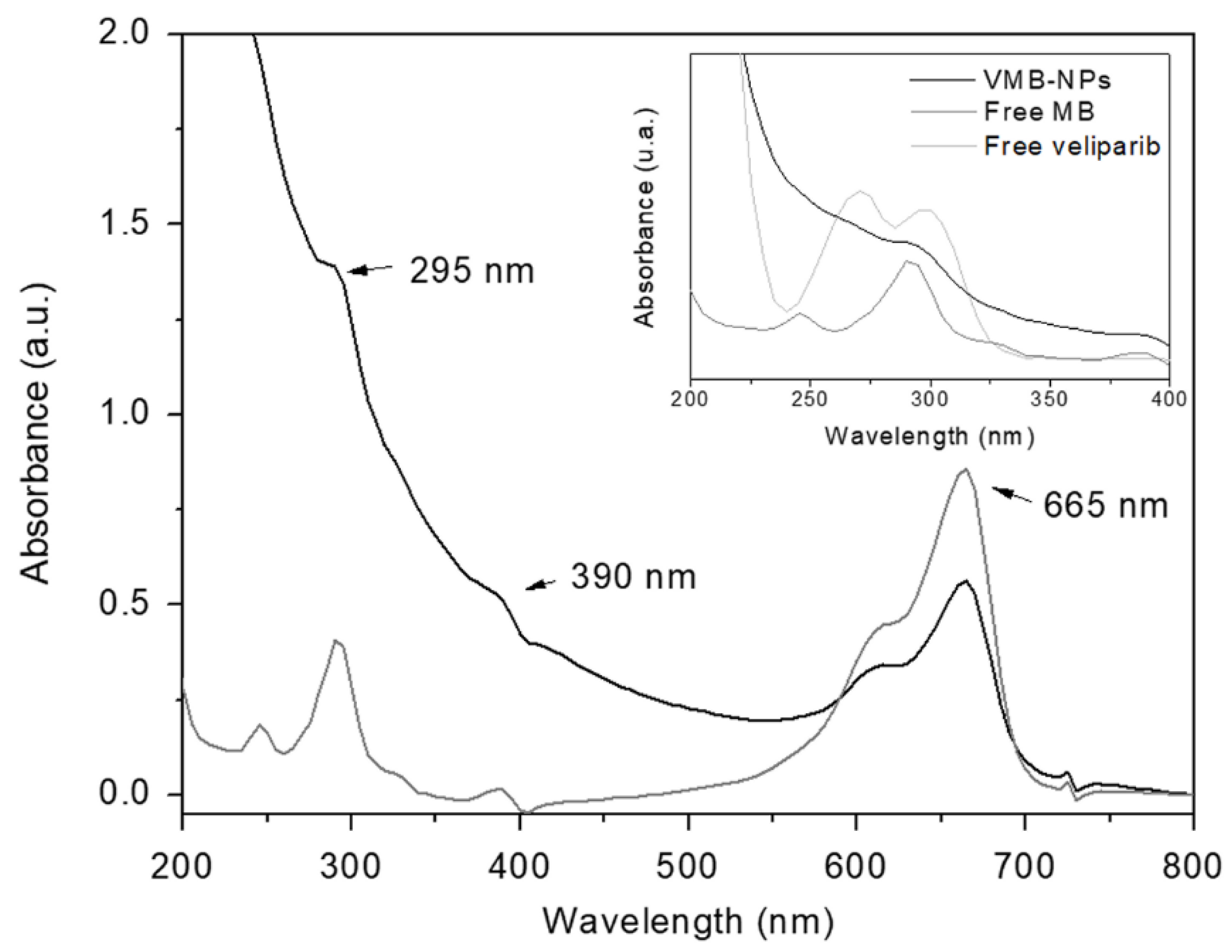
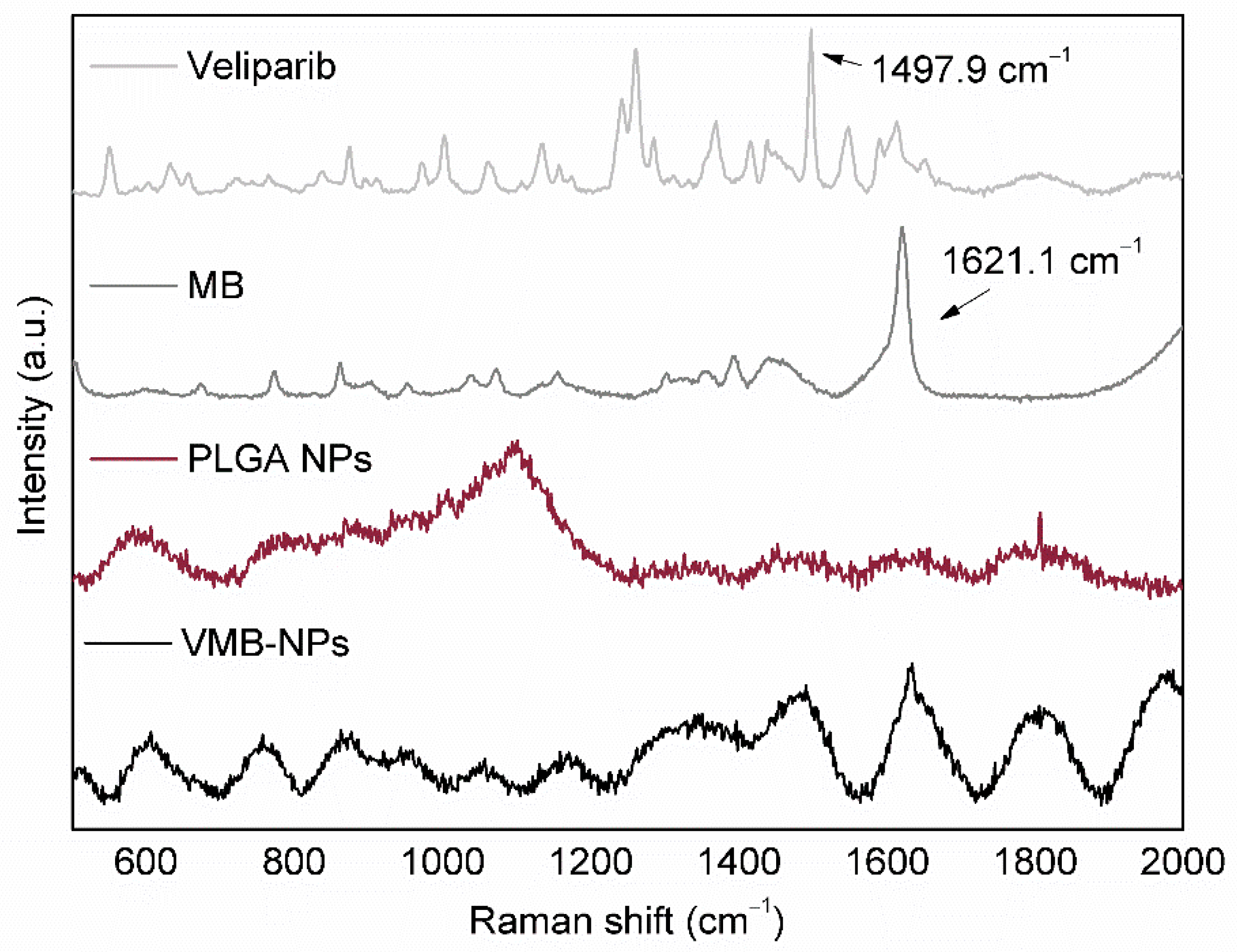
3.1. Physicochemical Properties
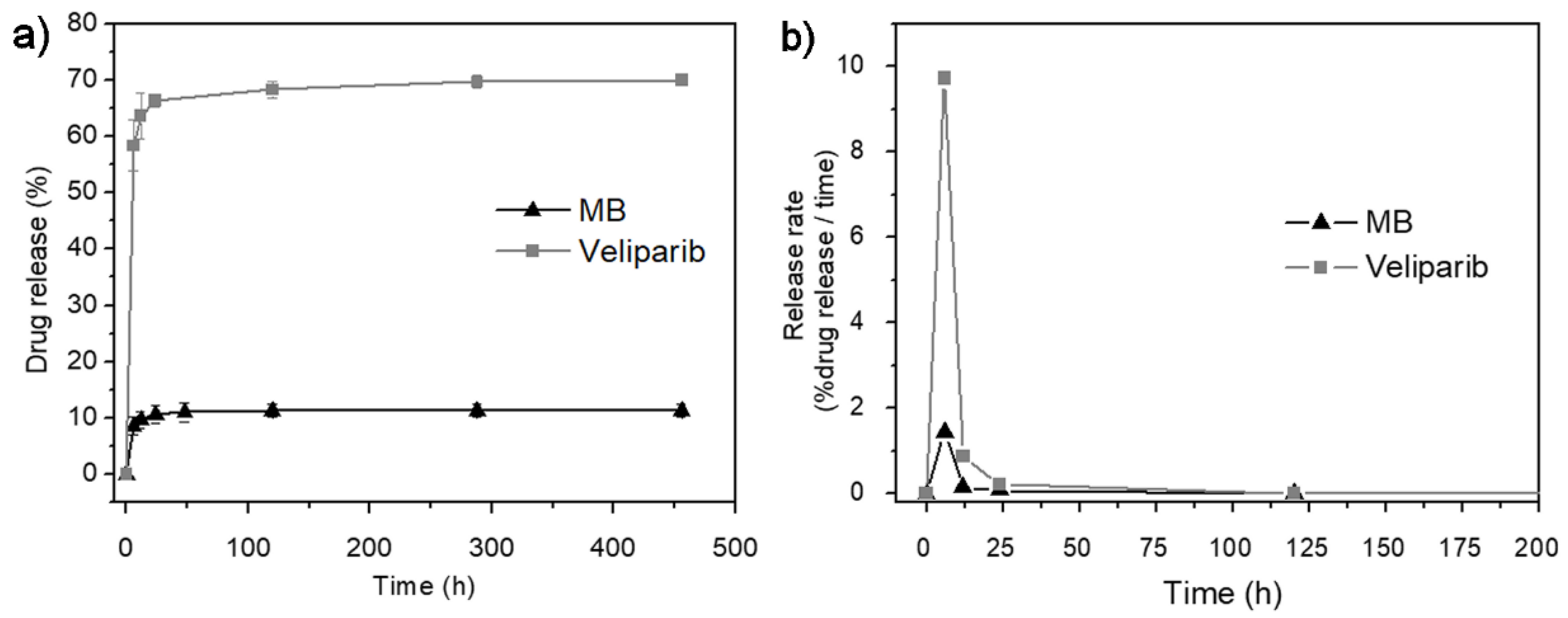
3.2. In Vitro Drug-Release
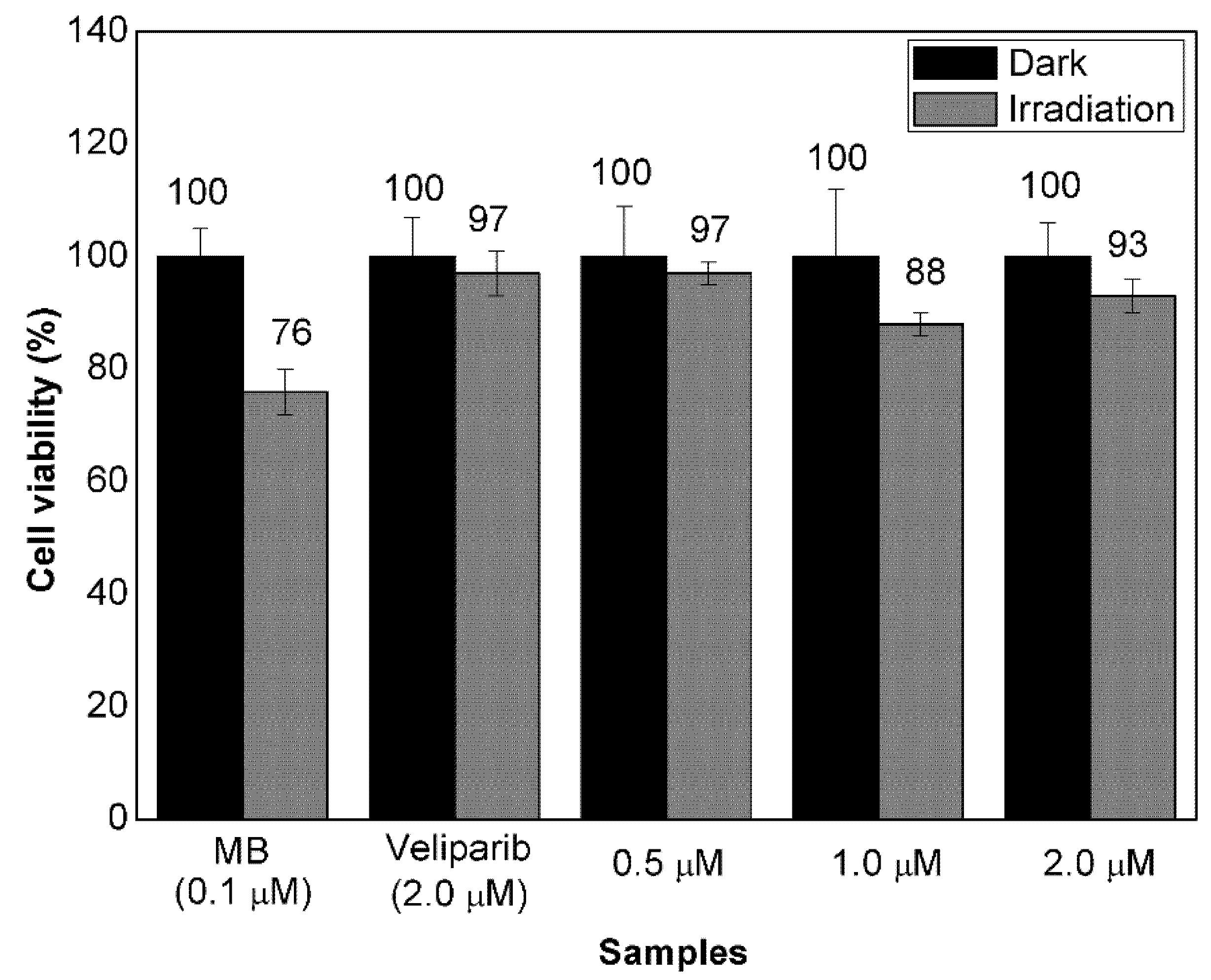
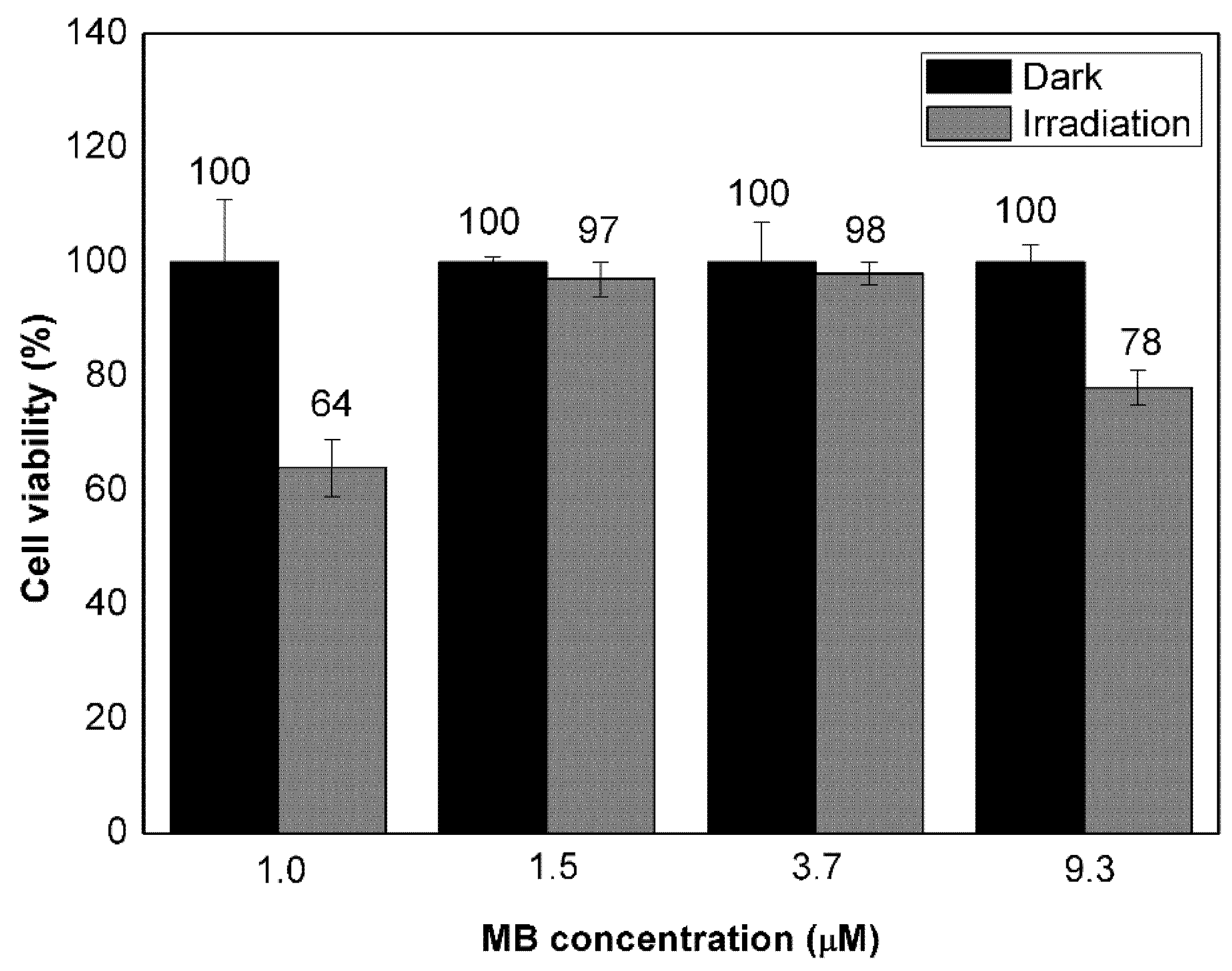
3.3. Evaluation of Cell Response to Non-Encapsulated Molecules and VMB-NPs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McFarland, S.A.; Mandel, A.; Dumoulin-White, R.; Gasser, G. Metal-based photosensitizers for photodynamic therapy: The future of multimodal oncology? Curr. Opin. Chem. Biol. 2020, 56, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Qidwai, A.; Nabi, B.; Kotta, S.; Narang, J.K.; Baboota, S.; Ali, J. Role of nanocarriers in photodynamic therapy. Photodiagn. Photodyn. Ther. 2020, 30, 101782. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Gao, Y.; Liu, N.; Suo, Y. Nanoparticles Loading Porphyrin Sensitizers in Improvement of Photodynamic Therapy for Ovarian Cancer. Photodiagn. Photodyn. Ther. 2020, 33, 102156. [Google Scholar] [CrossRef]
- Najlah, M.; Ahmed, Z.; Iqbal, M.; Wang, Z.; Tawari, P.; Wang, W.; McConville, C. Development and characterisation of disulfiram-loaded PLGA nanoparticles for the treatment of non-small cell lung cancer. Eur. J. Pharm. Biopharm. 2017, 112, 224–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalyane, D.; Raval, N.; Maheshwari, R.; Tambe, V.; Kalia, K.; Tekade, R.K. Employment of enhanced permeability and retention effect (EPR): Nanoparticle-based precision tools for targeting of therapeutic and diagnostic agent in cancer. Mater. Sci. Eng. C 2019, 98, 1252–1276. [Google Scholar] [CrossRef]
- Acharya, S.; Sahoo, S.K. PLGA nanoparticles containing various anticancer agents and tumour delivery by EPR effect. Adv. Drug Deliv. Rev. 2011, 63, 170–183. [Google Scholar] [CrossRef] [PubMed]
- Fukumori, Y.; Ichikawa, H. Nanoparticles for cancer therapy and diagnosis. Adv. Powder Technol. 2006, 17, 1–28. [Google Scholar] [CrossRef]
- Sanna, V.; Pala, N.; Sechi, M. Targeted therapy using nanotechnology: Focus on cancer. Int. J. Nanomed. 2014, 9, 467–483. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, Y.; Mochida, A.; Choyke, P.L.; Kobayashi, H. Nanodrug delivery: Is the enhanced permeability and retention effect sufficient for curing cancer? Bioconjug. Chem. 2016, 27, 2225–2238. [Google Scholar] [CrossRef] [PubMed]
- Han, R.; Tang, K.; Hou, Y.; Yu, J.; Wang, C.; Wang, Y. Fabrication of core/shell/shell structure nanoparticle with anticancer drug and dual-photosensitizer co-loading for synergistic chemotherapy and photodynamic therapy. Microporous Mesoporous Mater. 2020, 297, 110049. [Google Scholar] [CrossRef]
- Doustvandi, M.A.; Mohammadnejad, F.; Mansoori, B.; Tajalli, H.; Mohammadi, A.; Mokhtarzadeh, A.; Baghbani, E.; Khaze, V.; Hajiasgharzadeh, K.; Moghaddam, M.M.; et al. Photodynamic therapy using zinc phthalocyanine with low dose of diode laser combined with doxorubicin is a synergistic combination therapy for human SK-MEL-3 melanoma cells. Photodiagn. Photodyn.Ther. 2019, 28, 88–97. [Google Scholar] [CrossRef]
- Soriano, J.; Mora-Espí, I.; Alea-Reyes, M.E.; Pérez-García, L.; Barrios, L.; Ibáñez, E.; Nogués, C. Cell Death Mechanisms in Tumoral and Non-Tumoral Human Cell Lines Triggered by Photodynamic Treatments: Apoptosis, Necrosis and Parthanatos. Sci. Rep. 2017, 7, 41340. [Google Scholar] [CrossRef] [Green Version]
- Kessel, D.; Oleinick, N.L. Cell Death Pathways Associated with Photodynamic Therapy: An Update. Photochem. Photobiol. 2018, 94, 213–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins, W.K.; Belotto, R.; Silva, M.N.; Grasso, D.; Suriani, M.D.; Lavor, T.S.; Itri, R.; Baptista, M.S.; Tsubone, T.M. Autophagy Regulation and Photodynamic Therapy: Insights to Improve Outcomes of Cancer Treatment. Front. Oncol. 2021, 10, 3121. [Google Scholar] [CrossRef]
- Castano, A.P.; Demidova, T.N.; Hamblin, M.R. Mechanisms in photodynamic therapy: Part two—Cellular signaling, cell metabolism and modes of cell death. Photodiagn. Photodyn. Ther. 2005, 2, 1–23. [Google Scholar] [CrossRef] [Green Version]
- Martins, W.K.; Santos, N.F.; Rocha, C.S.; Bacellar, I.O.L.; Tsubone, T.M.; Viotto, A.C.; Matsukuma, A.Y.; Abrantes, A.B.P.; Siani, P.; Dias, L.G.; et al. Parallel damage in mitochondria and lysosomes is an efficient way to photoinduce cell death. Autophagy 2019, 15, 259–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kessel, D.; Evans, C.L. Promotion of Pro-Apoptotic Signals by Lysosomal Photodamage: Mechanistic Aspects and Influence of Autophagy. Photochem. Photobiol. 2016, 92, 620–623. [Google Scholar] [CrossRef] [Green Version]
- Kessel, D. Sub-cellular Targeting as a Determinant of the Efficacy of Photodynamic Therapy. Photochem. Photobiol. 2017, 93, 609–612. [Google Scholar] [CrossRef] [Green Version]
- Kessel, D.; Reiners, J.J., Jr. Enhanced Efficacy of Photodynamic Therapy via a Sequential Targeting Protocol. Photochem. Photobiol. 2014, 90, 889–895. [Google Scholar] [CrossRef] [Green Version]
- Casas, A.; Di Venosa, G.; Hasan, T.; Batlle, A. Mechanisms of resistance to photodynamic therapy. Curr. Med. Chem. 2011, 18, 2486–2515. [Google Scholar] [CrossRef] [Green Version]
- Shanmugapriya, K.; Kim, H.; Kang, H.W. Nanoengineered chlorin e6 conjugated with hydrogel for photodynamic therapy on cancer. Colloids Surf. B Biointerfaces 2019, 181, 778–788. [Google Scholar] [CrossRef]
- Scalfi-Happ, C.; Zhu, Z.; Graefe, S.; Wiehe, A.; Ryabova, A.; Loschenov, V.; Wittig, R.; Steiner, R.W. Chlorin nanoparticles for tissue diagnostics and photodynamic therapy. Photodiagn. Photodyn. 2018, 22, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Korbelik, M.; Sun, J.; Payne, P.W. Activation of poly (adenosine di-phosphate-ribose) polymerase in mouse tumors treated by photodynamic therapy. Photochem. Photobiol. 2003, 78, 400–406. [Google Scholar] [CrossRef]
- Kim, J.; Lim, W.; Kim, S.; Jeon, S.; Hui, Z.; Ni, K.; Kim, C.; Im, Y.; Choi, H.; Kim, O. Photodynamic therapy (PDT) resistance by PARP1 regulation on PDT-induced apoptosis with autophagy in head and neck cancer cells. J. Oral Pathol. Med. 2014, 43, 675–684. [Google Scholar] [CrossRef]
- Kästle, M.; Grimm, S.; Nagel, R.; Breusing, N.; Grune, T. Combination of PDT and inhibitor treatment affects melanoma cells and spares keratinocytes. Free Radic. Biol. Med. 2011, 50, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.J.; Oak, C.H.; Heo, J.; Kim, Y.H. Methylene blue-mediated photodynamic therapy enhances apoptosis in lung cancer cells. Oncol. Rep. 2013, 30, 856–862. [Google Scholar] [CrossRef]
- Arruda, D.C.; de Oliveira, T.D.; Cursino, P.H.F.; Maia, V.S.C.; Berzaghi, R.; Travassos, L.R.; Tada, D.B. Inhibition of melanoma metastasis by dual-peptide PLGA NPs. Biopolymers 2017, 108, e23029. [Google Scholar] [CrossRef]
- Danhier, F.; Ansorena, E.; Silva, J.M.; Coco, R.; LeBreton, A.; Préat, V. PLGA-based nanoparticles: An overview of biomedical applications. J. Control. Release 2012, 161, 505–522. [Google Scholar] [CrossRef]
- Almoustafa, H.A.; Alshawsh, M.A.; Chik, Z. Technical aspects of preparing PEG-PLGA nanoparticles as carrier for chemotherapeutic agents by nanoprecipitation method. Int. J. Pharm. 2017, 533, 275–284. [Google Scholar] [CrossRef]
- Jinwal, U.K.; Groshev, A.; Zhang, J.; Grover, A.; Sutariya, V.B. Preparation and characterization of methylene blue nanoparticles for Alzheimer’s disease and other tauopathies. Curr. Drug Deliv. 2014, 11, 541–550. [Google Scholar] [CrossRef]
- Bergmann, K.; O’Konski, C.T. A spectroscopy study of methylene blue monomer, dimer, and complexes with montmorillonite. J. Phys. Chem. 1963, 67, 2169–2177. [Google Scholar] [CrossRef]
- Hong, E.J.; Choi, D.G.; Shim, M.S. Targeted and effective photodynamic therapy for cancer using functionalized nanomaterials. Acta Pharm. Sin. B 2016, 6, 297–307. [Google Scholar] [CrossRef] [Green Version]
- Rui, L.L.; Cao, H.L.; Xue, Y.D.; Liu, L.C.; Xu, L.; Gao, Y.; Zhang, W.A. Functional organic nanoparticles for photodynamic therapy. Chin. Chem. Lett. 2016, 27, 1412–1420. [Google Scholar] [CrossRef]
- Huang, W.; Zhang, C. Tuning the size of poly (lactic-co-glycolic acid) (PLGA) nanoparticles fabricated by nanoprecipitation. Biotechnol. J. 2018, 13, 1700203. [Google Scholar] [CrossRef]
- Pérez-Herrero, E.; Fernández-Medarde, A. Advanced targeted therapies in cancer: Drug nanocarriers, the future of chemotherapy. Eur. J. Pharm. Biopharm. 2015, 93, 52–79. [Google Scholar] [CrossRef] [Green Version]
- Sadat, S.M.A.; Jahan, S.; Haddadi, A. Effects of size and surface charge of polymeric nanoparticles on in vitro and in vivo applications. J. Biomater. Nanobiotechnol. 2016, 7, 91–108. [Google Scholar] [CrossRef] [Green Version]
- Klepac-Ceraj, V.; Patel, N.; Song, X.; Holewa, C.; Patel, C.; Kent, R.; Amiji, M.M.; Soukos, N.S. Photodynamic effects of methylene blue-loaded polymeric nanoparticles on dental plaque bacteria. Lasers Surg. Med. 2011, 43, 600–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutiérrez-Valenzuela, C.A.; Esquivel, R.; Guerrero-Germán, P.; Zavala-Rivera, P.; Rodríguez-Figueroa, J.C.; Guzmán-Z, R.; Lucero-Acuña, A. Evaluation of a combined emulsion process to encapsulate methylene blue into PLGA nanoparticles. RSC Adv. 2018, 8, 414–422. [Google Scholar] [CrossRef] [Green Version]
- Castañeda-Gill, J.M.; Ranjan, A.P.; Vishwanatha, J.K. Development and Characterization of Methylene Blue Oleate Salt-Loaded Polymeric Nanoparticles and their Potential Application as a Treatment for Glioblastoma. J. Nanomed. Nanotechnol. 2017, 8, 449. [Google Scholar] [CrossRef] [Green Version]
- Muñoz-Gamez, J.A.; Viota, J.L.; Barrientos, A.; Carazo, Á.; Sanjuán-Nuñez, L.; Quiles-Perez, R.; Muñoz-de-Rueda, P.; Delgado, Á.; Ruiz-Extremera, A.; Salmerón, J. Synergistic cytotoxicity of the poly (ADP-ribose) polymerase inhibitor ABT-888 and temozolomide in dual-drug targeted magnetic nanoparticles. Liver Int. 2015, 35, 1430–1441. [Google Scholar] [CrossRef]
- Hines, D.J.; Kaplan, D.L. Poly (lactic-co-glycolic acid) controlled release systems: Experimental and modeling insights. Crit. Rev. Ther. Drug Carr. Syst. 2013, 30, 257–276. [Google Scholar] [CrossRef] [PubMed]
- Bruschi, M.L. Mathematical models of drug release. In Strategies to Modify the Drug Release from Pharmaceutical Systems; Woodhead Publishing: Cambridge, UK, 2015; pp. 63–86. [Google Scholar]
- Chiarelli-Neto, O.; Pavani, C.; Ferreira, A.S.; Uchoa, A.F.; Severino, D.; Baptista, M.S. Generation and suppression of singlet oxygen in hair by photosensitization of melanin. Free Radic. Biol. Med. 2011, 51, 1195–1202. [Google Scholar] [CrossRef] [PubMed]
- Wagner, L.M. Profile of veliparib and its potential in the treatment of solid tumors. Onco Targets Ther. 2015, 8, 1931–1939. [Google Scholar] [CrossRef] [Green Version]
- Stewart, E.; Goshorn, R.; Bradley, C.; Griffiths, L.M.; Benavente, C.; Twarog, N.R.; Miller, G.M.; Caufield, W.; Freeman, B.B.; Bahrami, A.; et al. Targeting the DNA repair pathway in Ewing sarcoma. Cell Rep. 2014, 9, 829–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, Z.; Chang, H.; Li, H.; Wang, S. Induction of reactive oxygen species: An emerging approach for cancer therapy. Apoptosis 2017, 22, 1321–1335. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Hydrodynamic Diameter (nm) | PDI | Zeta Potential (mV) | Encapsulation Efficiency (%) | |
|---|---|---|---|---|---|
| PLGA NPs | 103.4 | 0.07 ± 0.03 | −6.8 ± 0.6 | ||
| VMB-NPs | 90.0 | 0.08 ± 0.03 | −3.7 ± 0.2 | MB 23 | Veliparib 58 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Magalhães, J.A.; Arruda, D.C.; Baptista, M.S.; Tada, D.B. Co-Encapsulation of Methylene Blue and PARP-Inhibitor into Poly(Lactic-Co-Glycolic Acid) Nanoparticles for Enhanced PDT of Cancer. Nanomaterials 2021, 11, 1514. https://doi.org/10.3390/nano11061514
Magalhães JA, Arruda DC, Baptista MS, Tada DB. Co-Encapsulation of Methylene Blue and PARP-Inhibitor into Poly(Lactic-Co-Glycolic Acid) Nanoparticles for Enhanced PDT of Cancer. Nanomaterials. 2021; 11(6):1514. https://doi.org/10.3390/nano11061514
Chicago/Turabian StyleMagalhães, Jéssica A., Denise C. Arruda, Maurício S. Baptista, and Dayane B. Tada. 2021. "Co-Encapsulation of Methylene Blue and PARP-Inhibitor into Poly(Lactic-Co-Glycolic Acid) Nanoparticles for Enhanced PDT of Cancer" Nanomaterials 11, no. 6: 1514. https://doi.org/10.3390/nano11061514
APA StyleMagalhães, J. A., Arruda, D. C., Baptista, M. S., & Tada, D. B. (2021). Co-Encapsulation of Methylene Blue and PARP-Inhibitor into Poly(Lactic-Co-Glycolic Acid) Nanoparticles for Enhanced PDT of Cancer. Nanomaterials, 11(6), 1514. https://doi.org/10.3390/nano11061514