
NOx Storage on BaTi0.8Cu0.2O3 Perovskite Catalysts: Addressing a Feasible Mechanism

 , ,
, ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Synthesis and Characterization of Catalysts
2.2. NOx Storage Tests
2.3. In Situ Diffuse Reflectance Infrared Fourier Transform Spectroscopy (DRIFTS) Experiments
3. Results
3.1. Catalyst Characterization
3.2. NOx Storage under Temperature-Programmed Reaction Conditions
3.3. NOx Storage under Isothermal Reaction Conditions
- At 350 and 400 °C, the lifetime of the nitrite species, formed at the beginning of the NOx storage cycle, becomes shorter. Thus, the intensity of the nitrite band increases until 30 s of NO/O2 exposition and, afterwards, the nitrite band intensity drastically drops. This trend suggests that the oxidation of nitrites to nitrates becomes faster at these temperatures than at 300 °C. In fact, at 450 °C, nitrites are not detected even at the very beginning of the NOx storage step.
3.4. Identification of Active Phase for NOx Storage
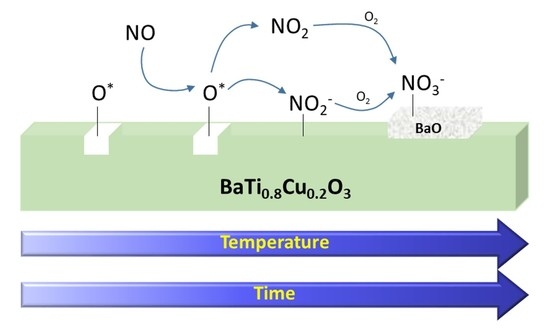
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Epling, W.S.; Campbell, L.E.; Yezerets, A.; Currier, N.W.; Parks, J.E. Overview of the Fundamental Reactions and Degradation Mechanisms of NOx Storage/Reduction Catalysts. Catal. Rev. 2004, 46, 163–245. [Google Scholar] [CrossRef]
- Roy, S.; Baiker, A. NOx Storage−Reduction Catalysis: From Mechanism and Materials Properties to Storage−Reduction Performance. Chem. Rev. 2009, 109, 4054–4091. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Gao, P.-X. A review of NOx storage/reduction catalysts: Mechanism, materials and degradation studies. Catal. Sci. Technol. 2011, 1, 552–568. [Google Scholar] [CrossRef]
- Pereda-Ayo, B.; González-Velasco, J.R. NOx Storage and Reduction for Diesel Engine Exhaust Aftertreatment. In Diesel Engine-Combustion, Emissions and Condition Monitoring; InTechOpen: London, UK, 2013; p. 266. [Google Scholar] [CrossRef] [Green Version]
- Fridell, E.; Skoglundh, M.; Björnwesterbergab, B.; Johanssonac, S.; Smedlera, G. NOxStorage in Barium-Containing Catalysts. J. Catal. 1999, 183, 196–209. [Google Scholar] [CrossRef]
- Broqvist, P.; Panas, I.; Fridell, E.; Persson, H. NOx Storage on BaO(100) Surface from First Principles: A Two Channel Scenario. J. Phys. Chem. B 2001, 106, 137–145. [Google Scholar] [CrossRef]
- Anderson, J.A.; Bachiller-Baeza, B.; Fernández-García, M. Role of Pt in Pt/Ba/Al2O3 NOx storage and reduction traps. Phys. Chem. Chem. Phys. 2003, 5, 4418–4427. [Google Scholar] [CrossRef]
- Liu, Z.; Anderson, J.A. Influence of reductant on the thermal stability of stored NOx in Pt/Ba/Al2O3 NOx storage and reduction traps. J. Catal. 2004, 224, 18–27. [Google Scholar] [CrossRef]
- Ayo, B.P.; De La Torre, U.; Marcos, M.P.G.; González-Velasco, J.R. Influence of ceria loading on the NOx storage and reduction performance of model Pt–Ba/Al2O3 NSR catalyst. Catal. Today 2015, 241, 133–142. [Google Scholar] [CrossRef]
- Ji, Y.; Toops, T.J.; Pihl, J.A.; Crocker, M. NOx storage and reduction in model lean NOx trap catalysts studied by in situ DRIFTS. Appl. Catal. B Environ. 2009, 91, 329–338. [Google Scholar] [CrossRef]
- Lietti, L.; Daturi, M.; Blasin-Aubé, V.; Ghiotti, G.; Prinetto, F.; Forzatti, P. Relevance of the Nitrite Route in the NOx Adsorption Mechanism over Pt-Ba/Al2O3 NOx Storage Reduction Catalysts Investigated by using Operando FTIR Spectroscopy. ChemCatChem 2011, 4, 55–58. [Google Scholar] [CrossRef]
- Fridell, E.; Persson, H.; Westerberg, B.; Olsson, L.; Skoglundh, M. The mechanism for NOx storage. Catal. Lett. 2000, 66, 71–74. [Google Scholar] [CrossRef]
- Castoldi, L.; Lietti, L.; Righini, L.; Forzatti, P.; Morandi, S.; Ghiotti, G. FTIR and Transient Reactivity Experiments of the Reduction by H2, CO and HCs of NO x Stored Over Pt–Ba/Al2O3 LNTs. Top. Catal. 2013, 56, 193–200. [Google Scholar] [CrossRef]
- Lietti, L.; Righini, L.; Castoldi, L.; Artioli, N.; Forzatti, P. Labeled 15NO Study on N2 and N2O Formation Over Pt–Ba/Al2O3 NSR Catalysts. Top. Catal. 2013, 56, 7–13. [Google Scholar] [CrossRef]
- Castoldi, L.; Nova, I.; Lietti, L.; Forzatti, P. Study of the effect of Ba loading for catalytic activity of Pt–Ba/Al2O3 model catalysts. Catal. Today 2004, 96, 43–52. [Google Scholar] [CrossRef]
- Shi, C.; Ji, Y.; Graham, U.M.; Jacobs, G.; Crocker, M.; Zhang, Z.; Wang, Y.; Toops, T.J. NOx storage and reduction properties of model ceria-based lean NOx trap catalysts. Appl. Catal. B Environ. 2012, 119–120, 183–196. [Google Scholar] [CrossRef]
- Su, Y.; Amiridis, M.D. In situ FTIR studies of the mechanism of NOx storage and reduction on Pt/Ba/Al2O3 catalysts. Catal. Today 2004, 96, 31–41. [Google Scholar] [CrossRef]
- Sedlmair, C.; Seshan, K.; Jentys, A.; Lercher, J.A. Elementary steps of NOx adsorption and surface reaction on a commercial storage–reduction catalyst. J. Catal. 2003, 214, 308–316. [Google Scholar] [CrossRef]
- Hodjati, S.; Petit, C.; Pitchon, V.; Kiennemann, A. Absorption/desorption of NOx process on perovskites: Nature and stability of the species formed on BaSnO3. Appl. Catal. B Environ. 2000, 27, 117–126. [Google Scholar] [CrossRef]
- Milt, V.; Ulla, M.; Miró, E. NOx trapping and soot combustion on BaCoO3−y perovskite: LRS and FTIR characterization. Appl. Catal. B Environ. 2005, 57, 13–21. [Google Scholar] [CrossRef]
- Abrahamsson, B.; Grönbeck, H. NOx Adsorption on ATiO3(001) Perovskite Surfaces. J. Phys. Chem. C 2015, 119, 18495–18503. [Google Scholar] [CrossRef]
- López-Suárez, F.; Illán-Gómez, M.; Bueno-López, A.; Anderson, J.A. NOx storage and reduction on a SrTiCuO3 perovskite catalyst studied by operando DRIFTS. Appl. Catal. B Environ. 2011, 104, 261–267. [Google Scholar] [CrossRef]
- Albaladejo-Fuentes, V.; E López-Suárez, F.; Sánchez-Adsuar, M.S.; Illán-Gómez, M.J. BaTi1−xCuxO3 perovskites: The effect of copper content in the properties and in the NOx storage capacity. Appl. Catal. A Gen. 2014, 488, 189–199. [Google Scholar] [CrossRef] [Green Version]
- Pechini, M.P. Method of Preparing Lead and Alkaline Earth Titanates and Niobates and Coating Method Using the Same to form a Capacitor. U.S. Patent No. 3330697, 11 July 1967. [Google Scholar]
- Kareiva, A.; Tautkus, S.; Rapalaviciute, R.; Jørgensen, J.-E.; Lundtoft, B. Sol-gel synthesis and characterization of barium titanate powders. J. Mater. Sci. 1999, 34, 4853–4857. [Google Scholar] [CrossRef]
- Narendar, Y.; Messing, G.L. Mechanisms of phase separation in gel-based synthesis of multicomponent metal oxides. Catal. Today 1997, 35, 247–268. [Google Scholar] [CrossRef]
- Hadjiivanov, K.I. Identification of Neutral and Charged NxOySurface Species by IR Spectroscopy. Catal. Rev. 2000, 42, 71–144. [Google Scholar] [CrossRef]
- Roedel, E.; Urakawa, A.; Kureti, S.; Baiker, A. On the local sensitivity of different IR techniques: Ba species relevant in NOx storage-reduction. Phys. Chem. Chem. Phys. 2008, 10, 6190–6198. [Google Scholar] [CrossRef] [PubMed]
- Frola, F.; Manzoli, M.; Prinetto, F.; Ghiotti, G.; Castoldi, L.; Lietti, L. Pt−Ba/Al2O3 NSR Catalysts at Different Ba Loading: Characterization of Morphological, Structural, and Surface Properties. J. Phys. Chem. C 2008, 112, 12869–12878. [Google Scholar] [CrossRef]
- Prinetto, F.; Ghiotti, G.; Nova, I.; Lietti, L.; Tronconi, E.; Forzatti, P. FT-IR and TPD Investigation of the NOx Storage Properties of BaO/Al2O3 and Pt−BaO/Al2O3 Catalysts. J. Phys. Chem. B 2001, 105, 12732–12745. [Google Scholar] [CrossRef]
- Wu, N.L.; Tschamber, V.; Limousy, L.; Michelin, L.; Westermann, A.; Azambre, B.; Fechete, I.; Garin, F. Combined Fixed-Bed Reactor and In Situ DRIFTS Tests of NO Adsorption on a NOx Storage-Reduction System Catalyst. Chem. Eng. Technol. 2013, 37, 204–212. [Google Scholar] [CrossRef]
- Kim, D.H.; Kwak, J.H.; Szanyi, J.; Burton, S.D.; Peden, C.H. Water-induced bulk Ba(NO3)2 formation from NO2 exposed thermally aged BaO/Al2O3. Appl. Catal. B Environ. 2007, 72, 233–239. [Google Scholar] [CrossRef]
- Briggs, D. Handbook of X-ray Photoelectron Spectroscopy C. D. Wanger, W. M. Riggs, L. E. Davis, J. F. Moulder and G. E.Muilenberg Perkin-Elmer Corp., Physical Electronics Division, Eden Prairie, Minnesota, USA, 1979. 190 pp. $195. Surf. Interface Anal. 1981, 3. [Google Scholar] [CrossRef]
- Merino, N.A.; Barbero, B.P.; Eloy, P.; Cadús, L.E. La1−xCaxCoO3 perovskite-type oxides: Identification of the surface oxygen species by XPS. Appl. Surf. Sci. 2006, 253, 1489–1493. [Google Scholar] [CrossRef]
- Lotnyk, A.; Senz, S.; Hesse, D. Formation of BaTiO3 thin films from (110) TiO2 rutile single crystals and BaCO3 by solid state reactions. Solid State Ionics 2006, 177, 429–436. [Google Scholar] [CrossRef]
- Leipner, H.S.; Abicht, H.-P.; Hollricher, O.; Gablenz, S. Raman microscopic investigations of BaTiO3 precursors with core?shell structure. Anal. Bioanal. Chem. 2004, 380, 157–162. [Google Scholar] [CrossRef]
- Templeton, L.K.; Pask, J.A. Formation of BaTiO3 from BaCO3 and TiO2 in Air and in CO2. J. Am. Ceram. Soc. 1959, 42, 212–216. [Google Scholar] [CrossRef]
- Niepce, J.C.; Thomas, G. About the mechanism of the solid-way synthesis of barium metatitanate. industrial consequences. Solid State Ion. 1990, 43, 69–76. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.M.; Chen, T.C. Surface chemical states of barium titanate: Influence of sample processing. J. Mater. Res. 1995, 10, 1502–1507. [Google Scholar] [CrossRef]
- Szanyi, J.; Kwak, J.H.; Hanson, J.; Wang, C.; Szailer, T.; Peden, C.H.F. Changing Morphology of BaO/Al2O3 during NO2Uptake and Release. J. Phys. Chem. B 2005, 109, 7339–7344. [Google Scholar] [CrossRef] [PubMed]
- Beauger, A.; Mutin, J.C.; Niepce, J.C. Role and behaviour of orthotitanate Ba2TiO4 during the processing of BaTiO3 based ferroelectric ceramics. J. Mater. Sci. 1984, 19, 195–201. [Google Scholar] [CrossRef]
- Schneider, W.F. Qualitative Differences in the Adsorption Chemistry of Acidic (CO2, SOx) and Amphiphilic (NOx) Species on the Alkaline Earth Oxides. J. Phys. Chem. B 2004, 108, 273–282. [Google Scholar] [CrossRef]
- Vovk, E.I.; Turksoy, A.; Bukhtiyarov, V.I.; Ozensoy, E. Interactive Surface Chemistry of CO2 and NO2 on Metal Oxide Surfaces: Competition for Catalytic Adsorption Sites and Reactivity. J. Phys. Chem. C 2013, 117, 7713–7720. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.H.; Chin, Y.-H.; Kwak, J.H.; Szanyi, J.; Peden, C.H. Changes in Ba Phases in BaO/Al2O3 upon Thermal Aging and H2O Treatment. Catal. Lett. 2005, 105, 259–268. [Google Scholar] [CrossRef]
- López, M.D.C.B.; Fourlaris, G.; Rand, B.; Riley, F.L. Characterization of Barium Titanate Powders: Barium Carbonate Identification. J. Am. Ceram. Soc. 1999, 82, 1777–1786. [Google Scholar] [CrossRef]
- Xian, H.; Zhang, X.; Li, X.; Zou, H.; Meng, M.; Zou, Z.; Guo, L.; Tsubaki, N. Raman microscopic investigations of BaTiO3 precursors with core-shell structure. Catal. Today 2010, 158, 215–219. [Google Scholar] [CrossRef]
- Emmez, E.; Vovk, E.I.; Bukhtiyarov, V.I.; Ozensoy, E. Direct Evidence for the Instability and Deactivation of Mixed-Oxide Systems: Influence of Surface Segregation and Subsurface Diffusion. J. Phys. Chem. C 2011, 115, 22438–22443. [Google Scholar] [CrossRef] [Green Version]
- Piacentini, M.; Maciejewski, M.; Baiker, A. Pt-Ba/alumina NOx storage-reduction catalysts: Effect of Ba-loading on build-up, stability and reactivity of Ba-containing phases. Appl. Catal. B Environ. 2005, 59, 187–195. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Temperature (°C) | NSC (µmol/g.cat.) | NO2/NOx (%) |
|---|---|---|
| 300 | 211 | 5 |
| 350 | 231 | 11 |
| 400 | 262 | 16 |
| 450 | 340 | 15 |
| Catalyst | ATR (w/w %) | TGA (w/w %) | XPS (w/w % of C) |
|---|---|---|---|
| BTCuO_2_NSR | 7.7 | 5.6 | 3.5 |
| BTCuO_2_sat | 4.9 | 4.9 | 1.9 |
| BTCuO_2_red | 6.5 | 5.8 | 2.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Albaladejo-Fuentes, V.; Sánchez-Adsuar, M.-S.; Anderson, J.A.; Illán-Gómez, M.-J. NOx Storage on BaTi0.8Cu0.2O3 Perovskite Catalysts: Addressing a Feasible Mechanism. Nanomaterials 2021, 11, 2133. https://doi.org/10.3390/nano11082133
Albaladejo-Fuentes V, Sánchez-Adsuar M-S, Anderson JA, Illán-Gómez M-J. NOx Storage on BaTi0.8Cu0.2O3 Perovskite Catalysts: Addressing a Feasible Mechanism. Nanomaterials. 2021; 11(8):2133. https://doi.org/10.3390/nano11082133
Chicago/Turabian StyleAlbaladejo-Fuentes, Vicente, María-Salvadora Sánchez-Adsuar, James A. Anderson, and María-José Illán-Gómez. 2021. "NOx Storage on BaTi0.8Cu0.2O3 Perovskite Catalysts: Addressing a Feasible Mechanism" Nanomaterials 11, no. 8: 2133. https://doi.org/10.3390/nano11082133
APA StyleAlbaladejo-Fuentes, V., Sánchez-Adsuar, M. -S., Anderson, J. A., & Illán-Gómez, M. -J. (2021). NOx Storage on BaTi0.8Cu0.2O3 Perovskite Catalysts: Addressing a Feasible Mechanism. Nanomaterials, 11(8), 2133. https://doi.org/10.3390/nano11082133