
NaCl-Templated Ultrathin 2D-Yttria Nanosheets Supported Pt Nanoparticles for Enhancing CO Oxidation Reaction


Abstract
:
1. Introduction
2. Materials and Methods
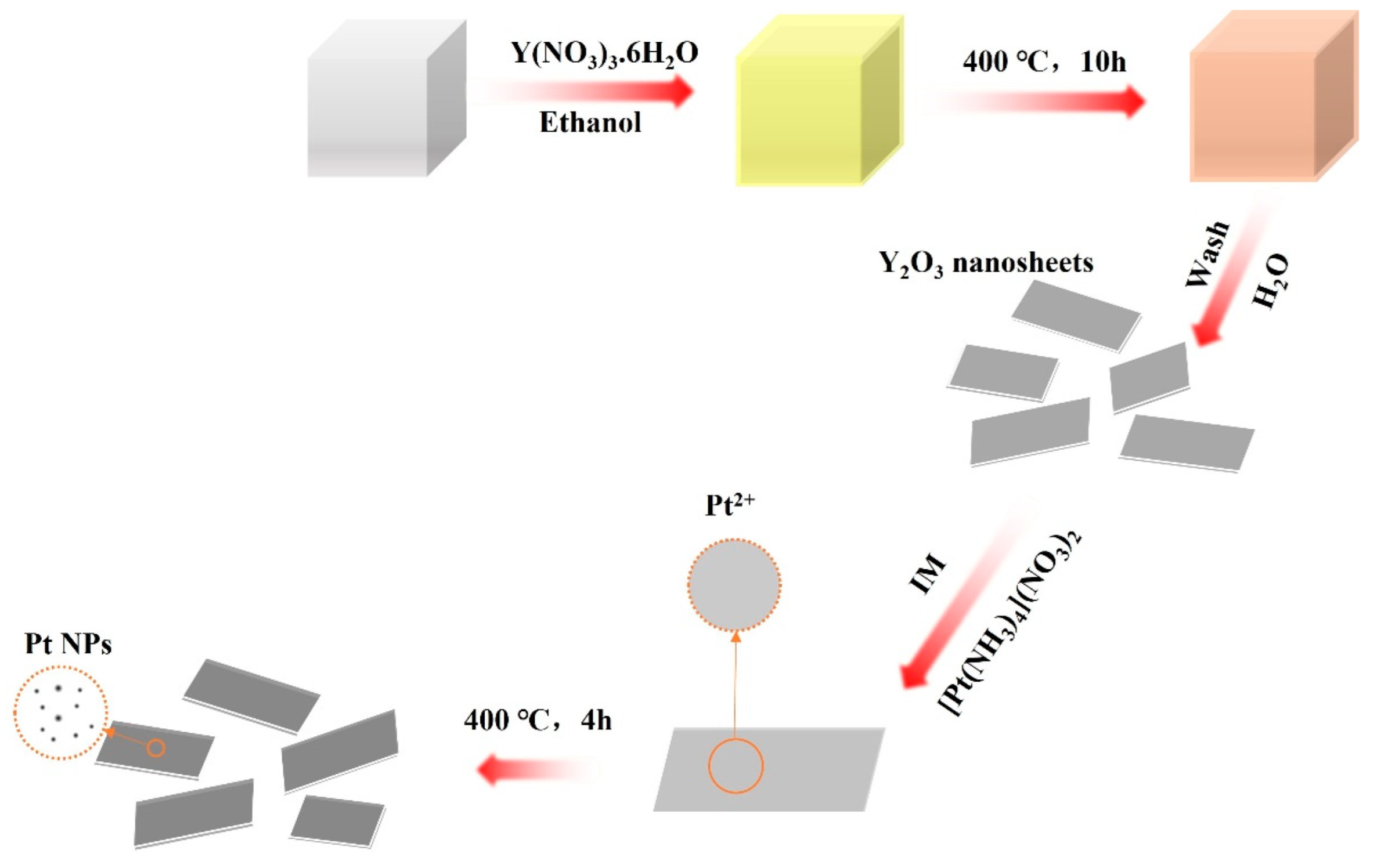
2.1. Catalyst Preparation
2.2. Characterization Methods
2.3. Catalytic Tests
3. Results and Discussion
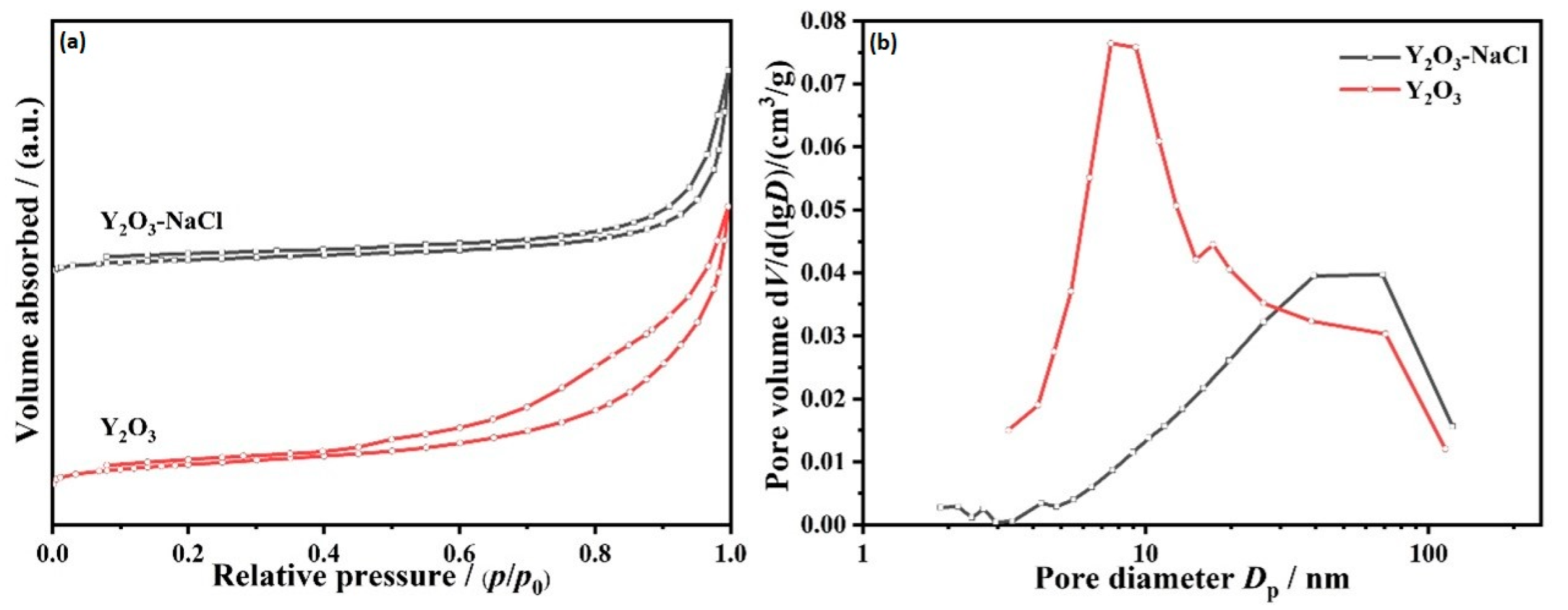
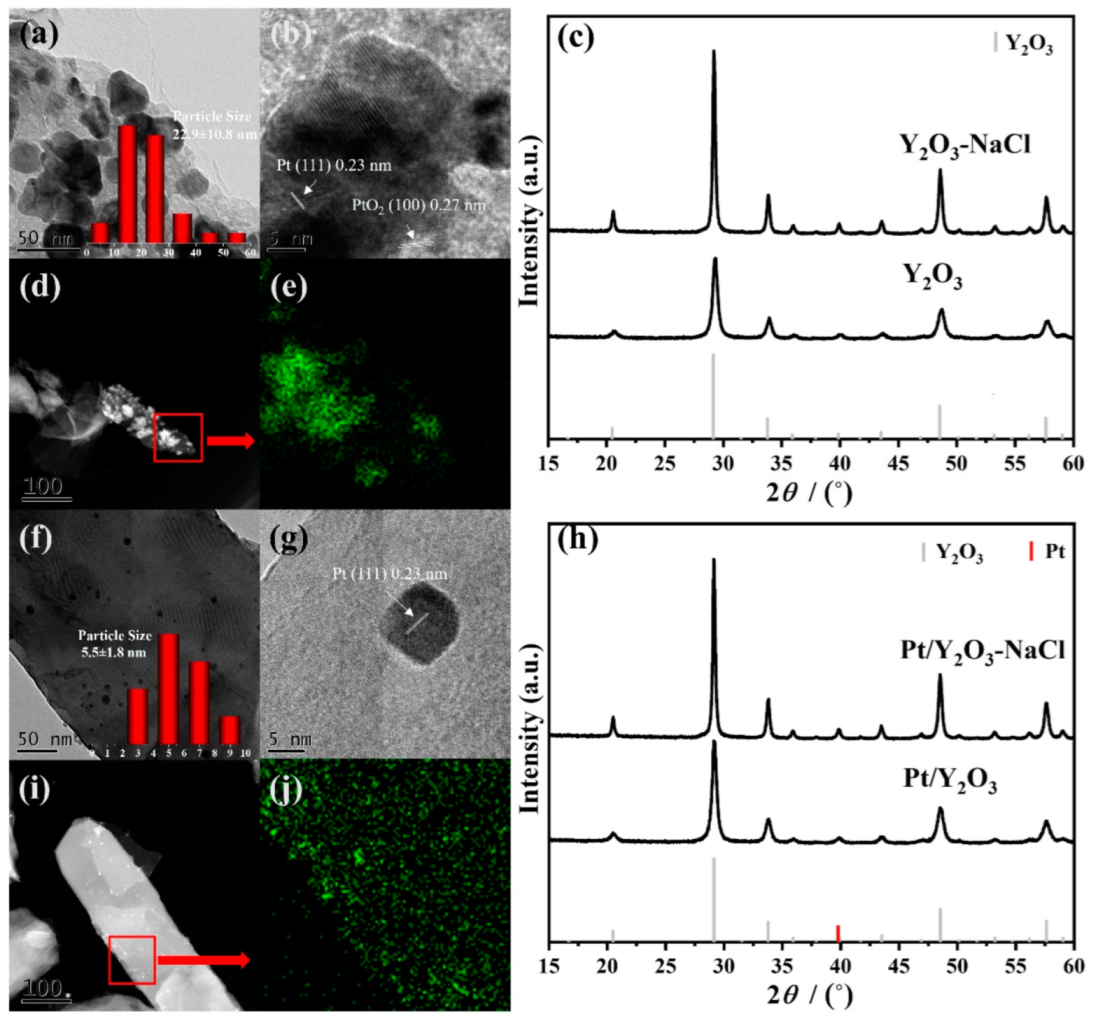
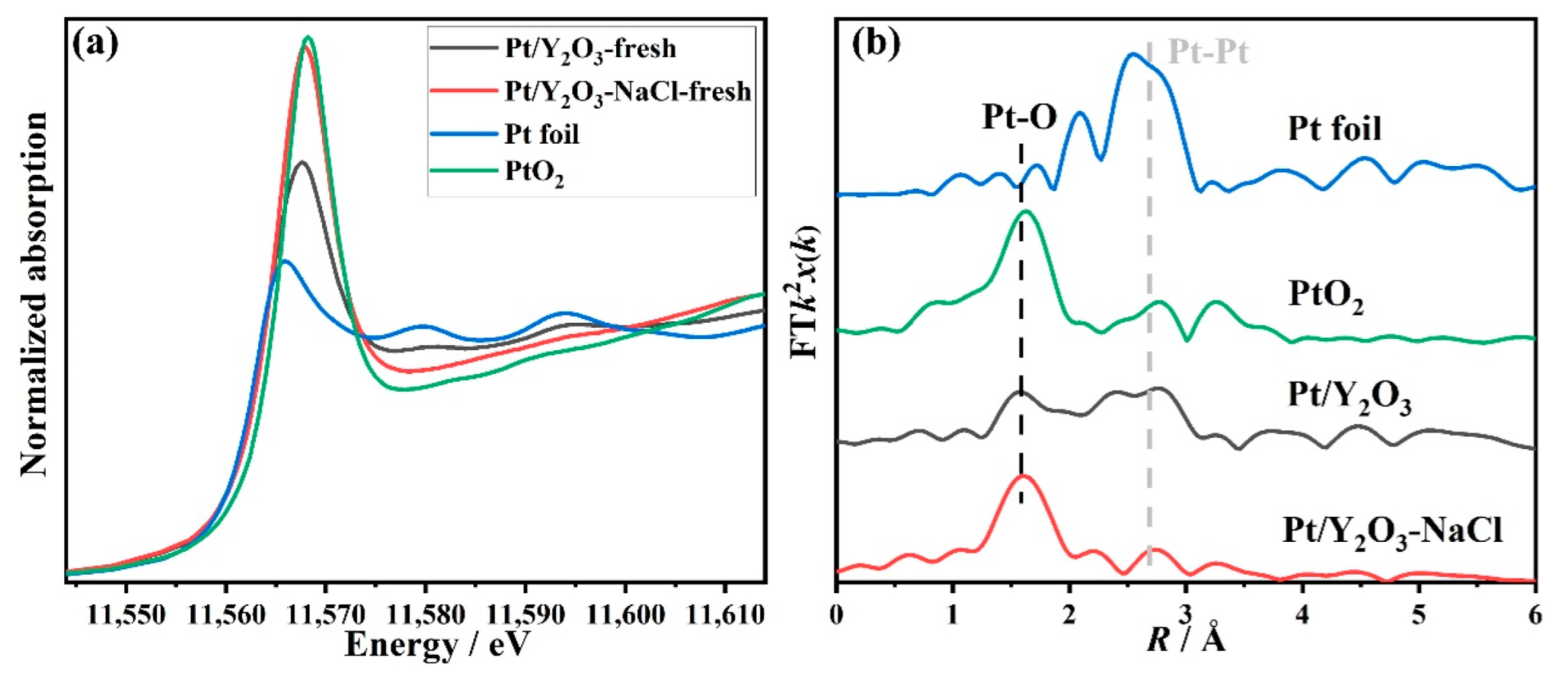
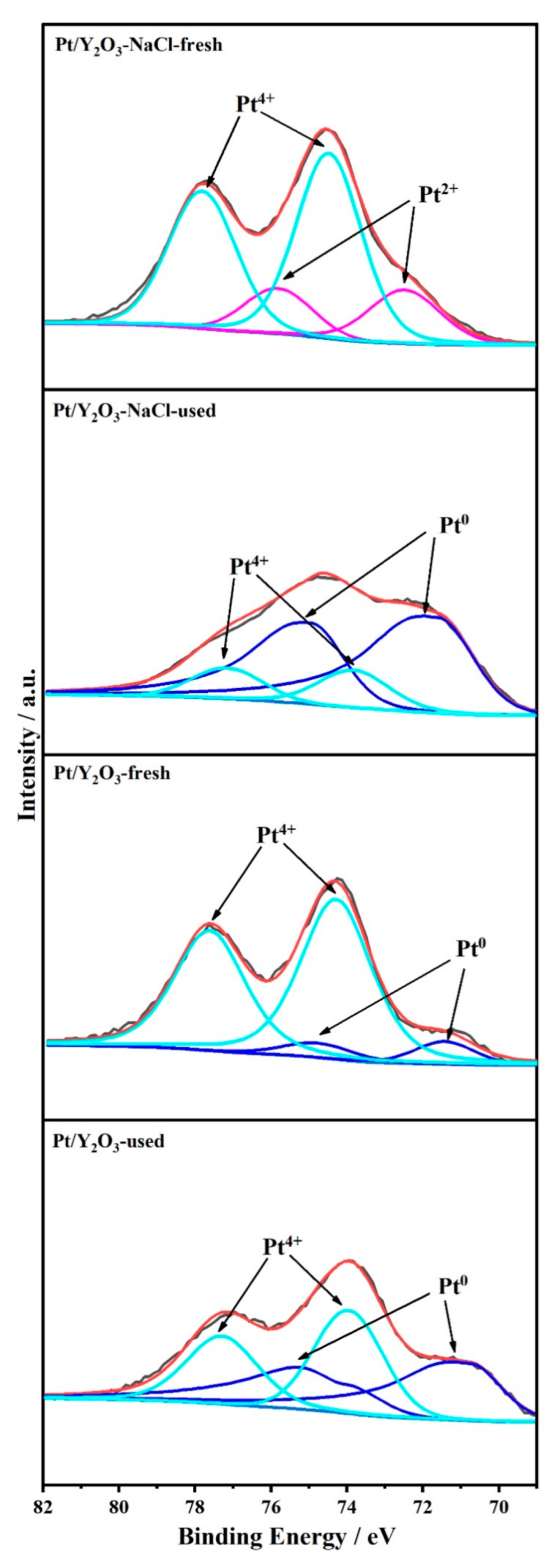
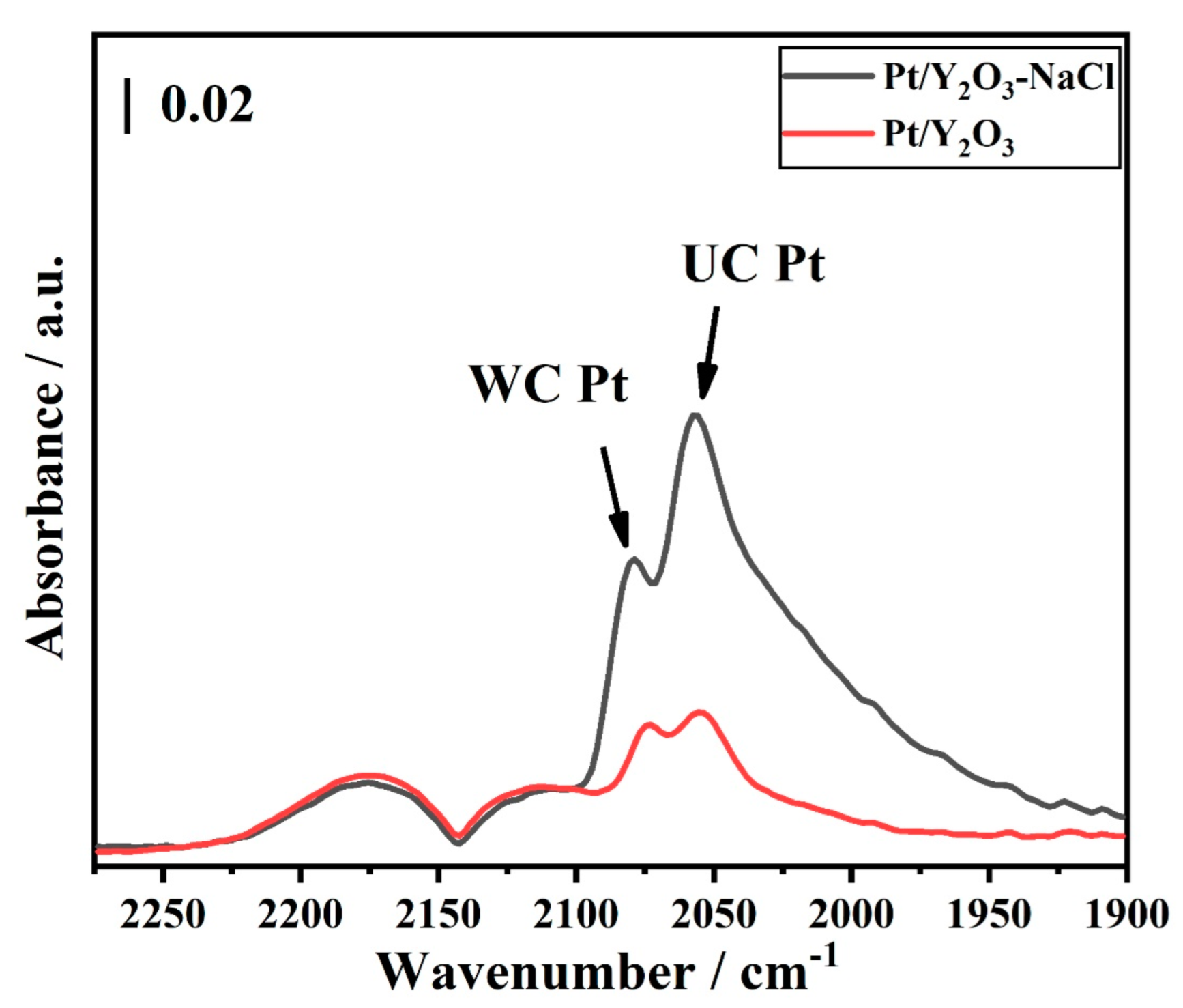
3.1. Structural Characterization of Fresh Platinum-Yttrium Oxide Catalysts
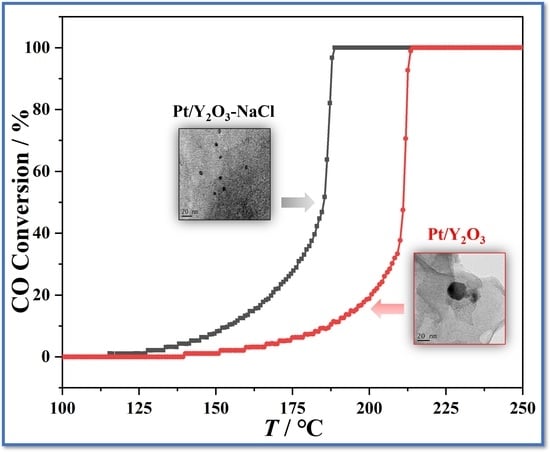
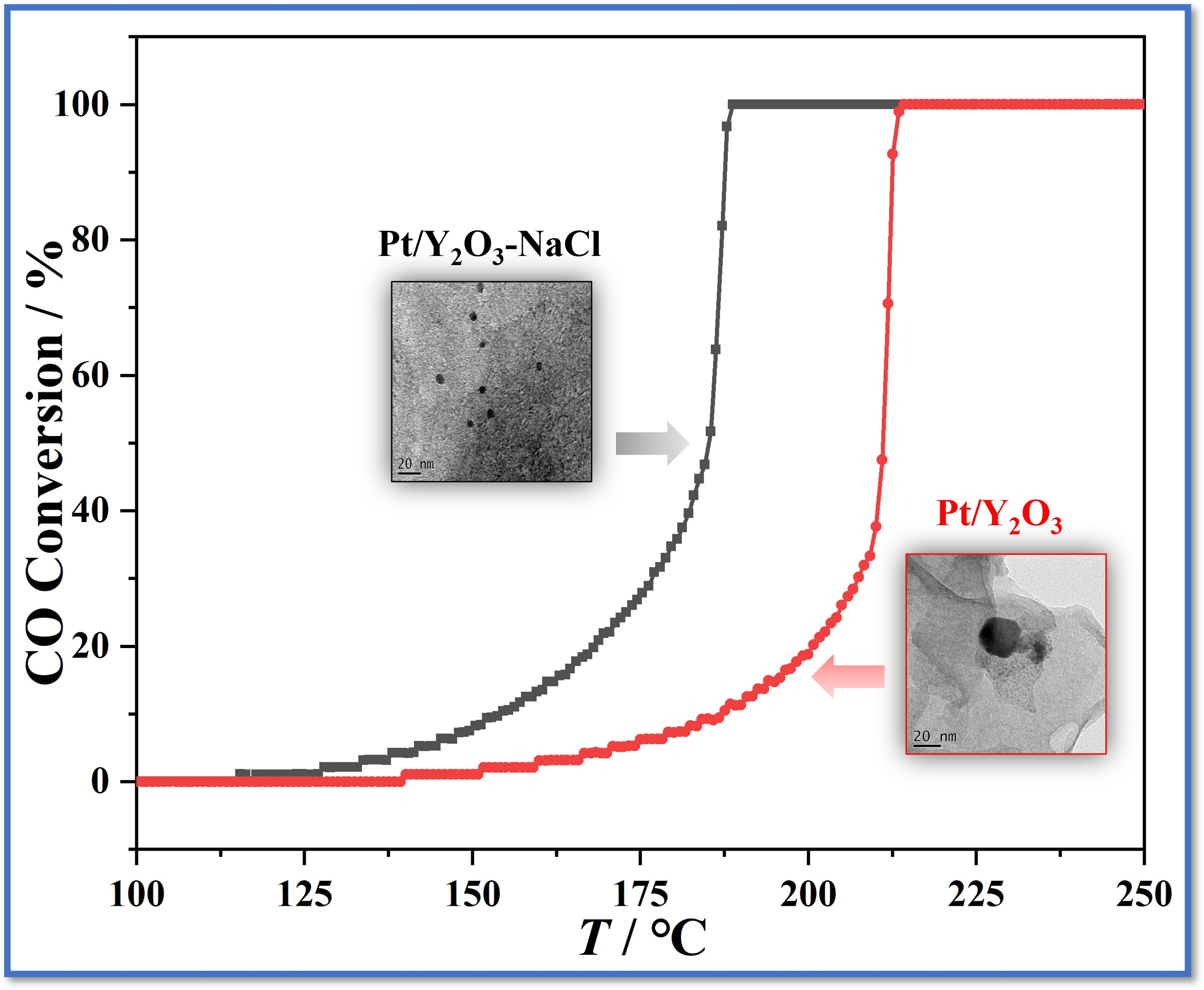
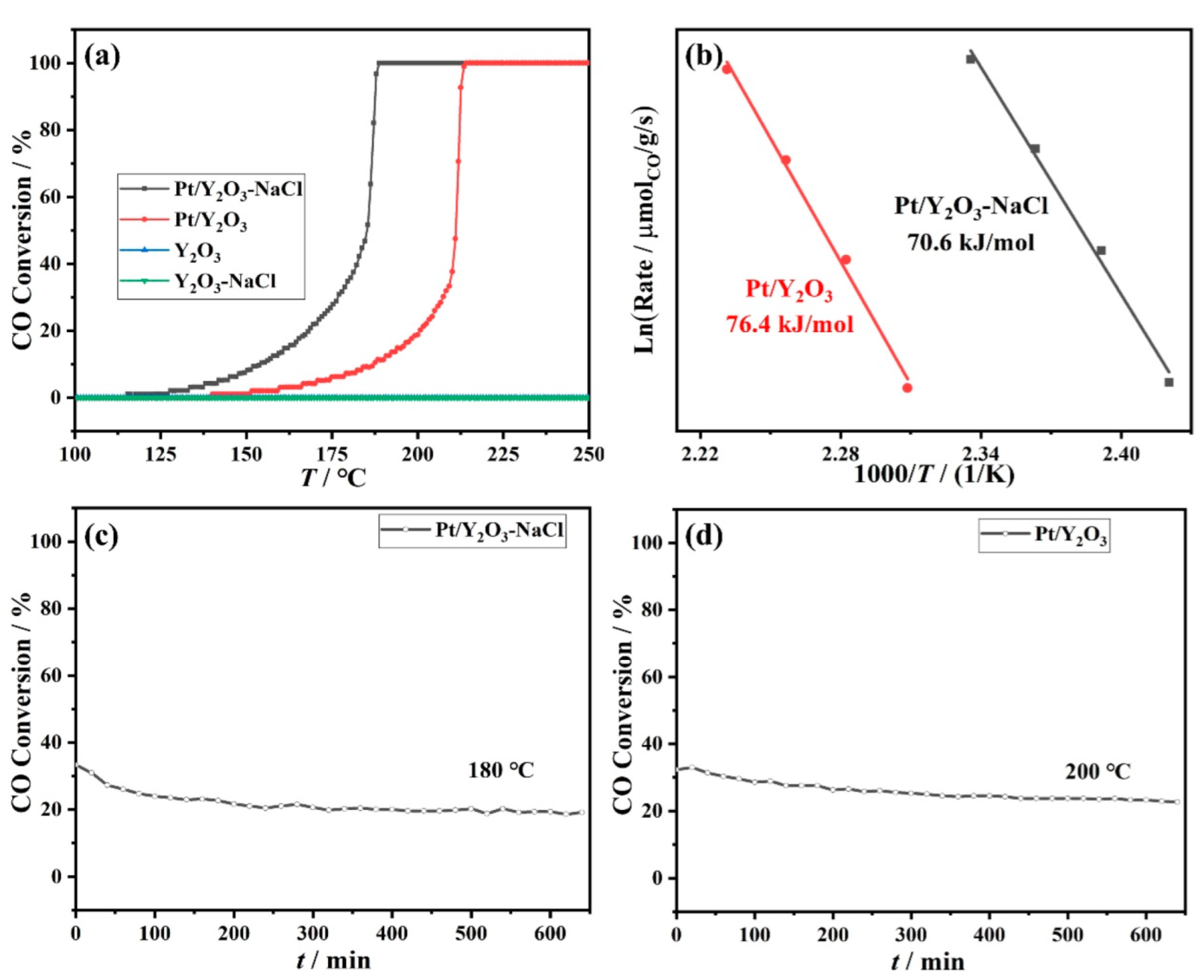
3.2. Catalytic Performance
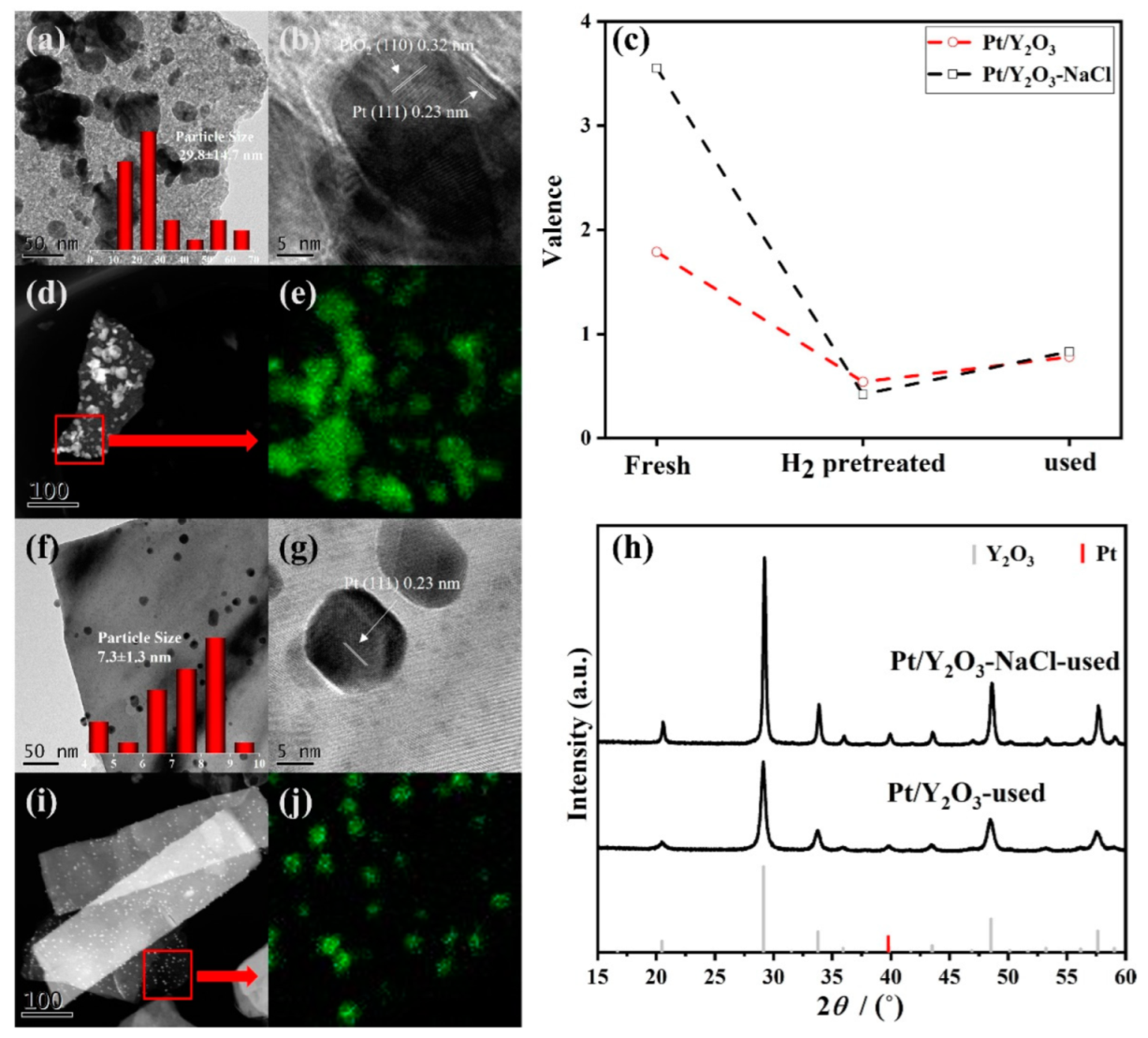
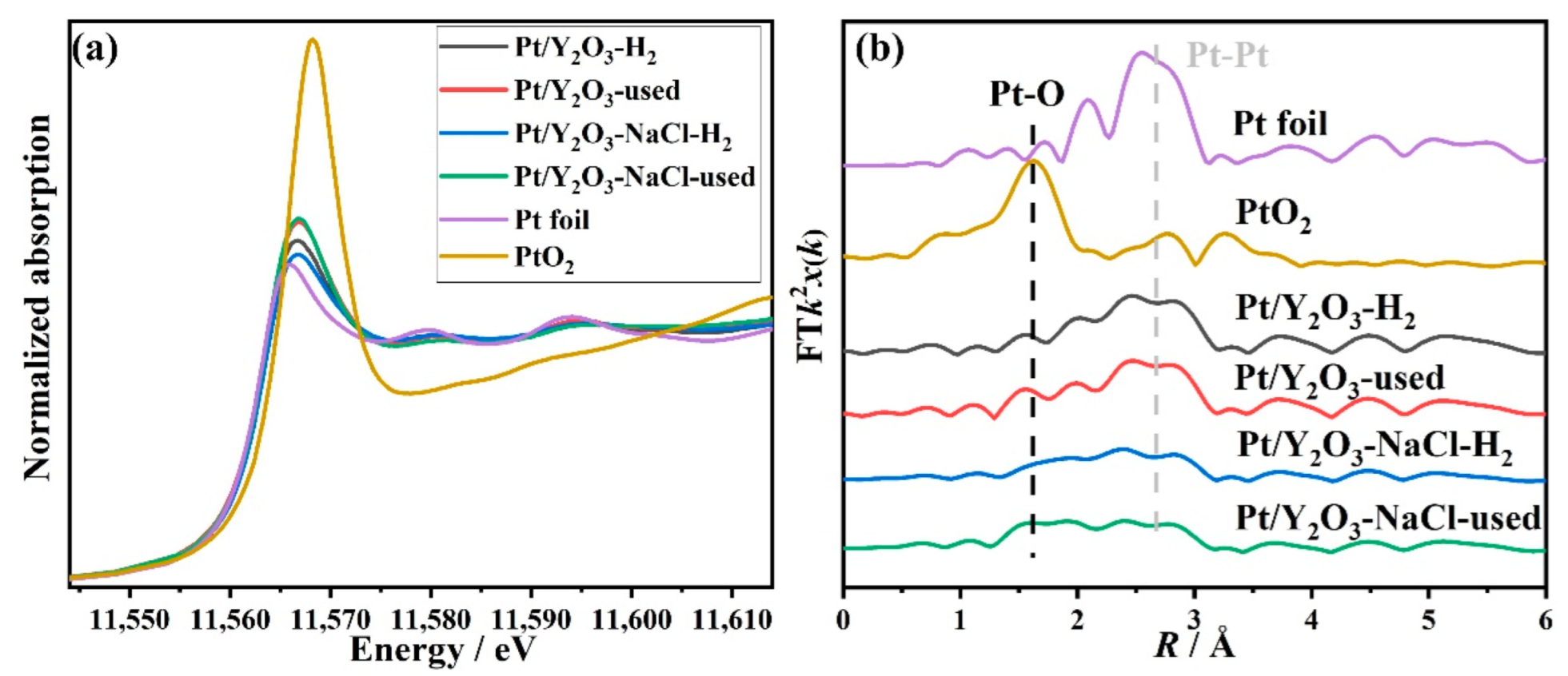
3.3. Structural Characterization of Used Platinum-Yttrium Oxide Catalysts
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Min, B.K.; Friend, C.M. Heterogeneous Gold-Based Catalysis for Green Chemistry: Low-Temperature CO Oxidation and Propene Oxidation. Chem. Rev. 2007, 107, 2709. [Google Scholar]
- Guo, Z.; Liu, B.; Zhang, Q.; Deng, W.; Wang, Y.; Yang, Y. Recent Advances in Heterogeneous Selective Oxidation Catalysis for Sustainable Chemistry. Chem. Soc. Rev. 2014, 43, 3480. [Google Scholar]
- Rodriguez, J.A.; Grinter, D.C.; Liu, Z.; Palomino, R.M.; Senanayake, S.D. Ceria-based Model Catalysts: Fundamental Studies on the Importance of the Metal-Ceria Interface in CO Oxidation, the Water-Gas Shift, CO2 hydrogenation, and methane and alcohol reforming. Chem. Soc. Rev. 2017, 46, 1824. [Google Scholar]
- Tan, P.; Hu, Z.; Lou, D.; Li, Z. Exhaust Emissions from a Light-Duty Diesel Engine with Jatropha Biodiesel Fuel. Energy 2012, 39, 356. [Google Scholar]
- Pan, T.; Wang, Y.; Xue, X.; Zhang, C. Rational Design of Allosteric Switchable Catalysts. Exploration 2022, 2, 20210095. [Google Scholar]
- Zheng, B.; Wu, S.; Yang, X.; Jia, M.; Zhang, W.; Liu, G. Room Temperature CO Oxidation over Pt/MgFe2O4: A Stable Inverse Spinel Oxide Support for Preparing Highly Efficient Pt Catalyst. ACS Appl. Mater. Interfaces 2016, 8, 26683. [Google Scholar]
- Boubnov, A.; Dahl, S.; Johnson, E.; Molina, A.P.; Simonsen, S.B.; Cano, F.M.; Helveg, S.; Lemus-Yegres, L.J.; Grunwaldt, J.-D. Structure-Activity Relationships of Pt/Al2O3 Catalysts for CO and NO Oxidation at Diesel Exhaust Conditions. Appl. Catal. B-Environ. 2012, 126, 315. [Google Scholar]
- Siwon, L.; Chao, L.; Seunghyun, K.; Xinyu, M.; Taeho, K.; Sang, J.K.; Raymond, J.G.; Woo, C.J. Manganese Oxide Overlayers Promote CO Oxidation on Pt. ACS Catal. 2021, 11, 13935. [Google Scholar]
- Almana, N.; Phivilay, S.P.; Laveille, P.; Hedhili, M.N.; Fornasiero, P.; Takanabe, K.; Basset, J.-M. Design of a Core-Shell Pt-SiO2 Catalyst in a Reverse Microemulsion System: Distinctive Kinetics on CO Oxidation at Low Temperature. J. Catal. 2016, 340, 368. [Google Scholar]
- Zheng, T.; Dong, Y.-R.; Nishiyama, N.; Egashira, Y.; Ueyama, K. Selective CO Oxidation Over Carbon-Coated Pt/SiO2-TiO2 Particles. Appl. Catal. A-Gen. 2006, 308, 210. [Google Scholar]
- Minemura, Y.; Ito, S.; Miyao, T.; Naito, S.; Tomishige, K.; Kunimori, K. Preferential CO Oxidation Promoted by the Presence of H2 over K-Pt/Al2O3. Chem. Commun. 2005, 11, 1429. [Google Scholar]
- Haneda, M.; Watanabe, T.; Kamiuchi, N.; Ozawa, M. Effect of Platinum Dispersion on the Catalytic Activity of Pt/Al2O3 for the Oxidation of Carbon Monoxide and Propene. Appl. Catal. B-Environ. 2013, 142, 8. [Google Scholar]
- Hong, X.; Sun, Y. Effect of Preparation Methods on the Performance of Pt/CeO2 Catalysts for the Catalytic Oxidation of Carbon Monoxide. Catal. Lett. 2016, 146, 2001. [Google Scholar]
- Lee, J.; Ryou, Y.; Kim, J.; Chan, X.; Kim, T.J.; Kim, D.H. Influence of the Defect Concentration of Ceria on the Pt Dispersion and the CO Oxidation Activity of Pt/CeO2. J. Phys. Chem. C 2018, 122, 4972. [Google Scholar]
- Zheng, B.; Liu, G.; Geng, L.; Cui, J.; Wu, S.; Wu, P.; Jia, M.; Yan, W.; Zhang, W. Role of the FeOx Support in Constructing High-Performance Pt/FeOx Catalysts for Low-Temperature CO Oxidation. Catal. Sci. Technol. 2016, 6, 1546. [Google Scholar]
- Liu, H.; Zakhtser, A.; Naitabdi, A.; Rochet, F.; Bournel, F.; Salzemann, C.; Petit, C.; Gallet, J.-J.; Jie, W. Operando Near-Ambient Pressure X-ray Photoelectron Spectroscopy Study of the CO Oxidation Reaction on the Oxide/Metal Model Catalyst ZnO/Pt(111). ACS Catal. 2019, 9, 10212. [Google Scholar]
- Guo, L.; Liu, B.H.; McBriarty, M.E.; Martynova, Y.; Groot, I.M.N.; Wang, S.; Bedzyk, M.J.; Shaikhutdinov, S.; Freund, H.J. Reactivity of Ultra-Thin ZnO Films Supported by Ag(111) and Cu(111): A Comparison to ZnO/Pt(111). Nanomaterials. 2022, 12, 1881. [Google Scholar]
- Xu, X.; Wang, Y.; Zeng, H.; Feng, Y.; Yang, X.; Zhang, S.; Xu, Y.; Wang, G.; Wang, Y.; Zhang, Z. Rational Design of SnO2 Hollow Microspheres Functionalized with Derivatives of Pt Loaded MOFs for Superior Formaldehyde Detection. Catal. Sci. Technol. 2014, 4, 3151. [Google Scholar]
- Xu, X.; Fu, Q.; Wei, M.; Wu, X.; Bao, X. Comparative Studies of Redox Behaviors of Pt-Co/SiO2 and Au-Co/SiO2 Catalysts and Their Activities in CO Oxidation. Catal. Sci. Technol. 2014, 4, 3151. [Google Scholar]
- Zhu Chen, J.; Talpade, A.; Canning, G.A.; Probus, P.R.; Ribeiro, F.H.; Datye, A.K.; Miller, J.T. Strong Metal-Support Interaction (SMSI) of Pt/CeO2 and Its Effect on Propane Dehydrogenation. Catal. Today 2021, 371, 4. [Google Scholar]
- Jiang, L.Z.; Chen, J.X.; Si, R. LaOx(OH)y Supported Platinum Catalysts for CO Oxidation: Deactivation by Formation of Lanthanum Carbonate. J. Rare Earths 2021, 39, 297. [Google Scholar]
- Liu, H.; Wu, H.; He, D. Methane Conversion to Syngas Over Ni/Y2O3 Catalysts—Effects of Calcination Temperatures of Y2O3 on Physicochemical Properties and Catalytic Performance. Fuel Process. Technol. 2014, 119, 81. [Google Scholar]
- Yan, Y.; Dai, Y.; Yang, Y.; Lapkin, A.A. Improved Stability of Y2O3 Supported Ni Catalysts for CO2 Methanation by Precursor-Determined Metal-Support Interaction. Appl. Catal. B-Environ. 2018, 237, 504. [Google Scholar]
- Li, J.; Wang, S.; Zhang, B.; Wang, W.; Feng, L. Highly Efficient Methanol Electrooxidation Catalyzed by Co-Action of Pd/Y2O3 in Alkaline Solution for Fuel Cells. Int. J. Hydrog. Energy 2017, 42, 12236. [Google Scholar]
- Ryoo, R.; Kim, J.; Jo, C.; Han, S.W.; Kim, J.C.; Park, H.; Han, J.; Shin, H.S.; Shin, J.W. Rare-Earth-Platinum Alloy Nanoparticles in Mesoporous Zeolite for Catalysis. Nature 2020, 585, 221. [Google Scholar]
- Guzman, J.; Corma, A. Nanocrystalline and Mesostructured Y2O3 as Supports for Gold Catalysts. Chem. Commun. 2005, 6, 743. [Google Scholar]
- Sreethawong, T.; Sitthiwechvijit, N.; Rattanachatchai, A.; Ouraipryvan, P.; Schwank, J.W.; Chavadej, S. Preparation of Au/Y2O3 and Au/NiO Catalysts by Co-Precipitation and Their Oxidation Activities. Mater. Chem. Phys. 2011, 126, 212. [Google Scholar]
- Huang, H.; Li, F.; Xue, Q.; Zhang, Y.; Yin, S.; Chen, Y. Salt-Templated Construction of Ultrathin Cobalt Doped Iron Thiophosphite Nanosheets toward Electrochemical Ammonia Synthesis. Small 2019, 15, 1903500. [Google Scholar]
- Ren, X.; Tong, L.; Chen, X.; Ding, H.; Yang, X.; Yang, H. Deposition of luminescent Y2O3: Eu3+ on Ferromagnetic Mesoporous CoFe2O4@mSiO2 Nanocomposites. Phys. Chem. Chem. Phys. 2014, 16, 10539. [Google Scholar]
- Nan, B.; Hu, X.C.; Wang, X.; Jia, C.J.; Ma, C.; Li, M.X.; Si, R. Effects of Multiple Platinum Species on Catalytic Reactivity Distinguished by Electron Microscopy and X-ray Absorption Spectroscopy Techniques. J. Phys. Chem. C 2017, 121, 25805. [Google Scholar]
- Ono, L.K.; Croy, J.R.; Heinrich, H.; Roldan Cuenya, B. Oxygen Chemisorption, Formation, and Thermal Stability of Pt Oxides on Pt Nanoparticles Supported on SiO2/Si(001): Size Effects. J. Phys. Chem. C 2011, 115, 16856. [Google Scholar]
- Ono, L.K.; Yuan, B.; Heinrich, H.; Cuenya, B.R. Formation and Thermal Stability of Platinum Oxides on Size-Selected Platinum Nanoparticles: Support Effects. J. Phys. Chem. C 2010, 114, 22119. [Google Scholar]
- Zhao, F.; Ikushima, Y.; Shirai, M.; Ebina, T.; Arai, M. Influence of Electronic State and Dispersion of Platinum Particles on the Conversion and Selectivity of Hydrogenation of an α, β-Unsaturated Aldehyde in Supercritical Carbon Dioxide. J. Mol. Catal. A-Chem. 2002, 180, 259. [Google Scholar]
- Brieger, C.; Melke, J.; Kaghazchi, P.; Roth, C. CO Adsorption on Platinum Nanoparticles—The Importance of Size Distribution Studied with in-Situ DRIFTS and DFT Calculations. ECS Trans. 2015, 69, 249. [Google Scholar]
- Garnier, A.; Sall, S.; Garin, F.; Chetcuti, M.J.; Petit, C. Site Effects in the Adsorption of Carbon Monoxide on Real 1.8 nm Pt Nanoparticles: An Infrared Investigation in Time and Temperature. J. Mol. Catal. A-Chem. 2013, 373, 127. [Google Scholar]
- Gracia, F.J.; Bollmann, L.; Wolf, E.E.; Miller, J.T.; Kropf, A.J. In situ FTIR, EXAFS, and Activity Studies of the Effect of Crystallite Size on Silica-Supported Pt Oxidation Catalysts. J. Catal. 2003, 220, 382. [Google Scholar]
- Calle-Vallejo, F.; Tymoczko, J.; Colic, V.; Vu, Q.H.; Pohl, M.D.; Morgenstern, K.; Loffreda, D.; Sautet, P.; Schuhmann, W.; Bandarenka, A.S. Finding Optimal Surface Sites on Heterogeneous Catalysts by Counting Nearest Neighbors. Science 2015, 350, 185. [Google Scholar]
- Panagiotopoulou, P.; Kondarides, D.I. Effects of Alkali Additives on the Physicochemical Characteristics and Chemisorptive Properties of Pt/TiO2 Catalysts. J. Catal. 2008, 260, 141. [Google Scholar]
- Avanesian, T.; Dai, S.; Kale, M.J.; Graham, G.W.; Pan, X.; Christopher, P. Quantitative and Atomic-Scale View of CO-Induced Pt Nanoparticle Surface Reconstruction at Saturation Coverage via DFT Calculations Coupled with in Situ TEM and IR. J. Am. Chem. Soc. 2017, 139, 4551. [Google Scholar]
- Liu, A.; Liu, X.; Liu, L.; Pu, Y.; Guo, K.; Tan, W.; Gao, S.; Luo, Y.; Yu, S.; Si, R.; et al. Getting Insights into the Temperature-Specific Active Sites on Platinum Nanoparticles for CO Oxidation: A Combined in Situ Spectroscopic and ab Initio Density Functional Theory Study. ACS Catal. 2019, 9, 7759. [Google Scholar]
- Kale, M.J.; Christopher, P. Utilizing Quantitative in Situ FTIR Spectroscopy to Identify Well-Coordinated Pt Atoms as the Active Site for CO Oxidation on Al2O3-Supported Pt Catalysts. ACS Catal. 2016, 6, 5599. [Google Scholar]
- Yoshinobu, J.; Tsukahara, N.; Yasui, F.; Mukai, K.; Yamashita, Y. Lateral Displacement by Transient Mobility in Chemisorption of CO on Pt(997). Phys. Rev. Lett. 2003, 90, 248301. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | CPt/Y | SBET | VP | a | DXRD |
|---|---|---|---|---|---|
| (wt %) a | (m2g−1) b | (cm3g−1) b | (Å) c | (nm) c | |
| Y2O3 | - | 19 | 0.07 | 10.5659(6) | 17 |
| Y2O3-NaCl | - | 10 | 0.05 | 10.5915(6) | 30 |
| Pt/Y2O3 | 0.62 | - | - | 10.6049(6) | 18 |
| Pt/Y2O3-NaCl | 0.68 | - | - | 10.6038(6) | 31 |
| Pt/Y2O3-used | - | - | - | 10.6069(6) | 19 |
| Pt/Y2O3-NaCl-used | - | - | - | 10.5900(6) | 30 |
| Sample | Shell | CN | R (Å) | σ2 (Å2) | ΔE0 |
|---|---|---|---|---|---|
| Pt/Y2O3-fresh | Pt-O | 2.0 ± 0.2 | 2.00 ± 0.01 | 0.0024 | 11.5 ± 1.4 |
| Pt-Pt | 7.0 ± 0.6 | 2.77 ± 0.01 | 0.0040 | ||
| Pt-O-O | 3.7 ± 1.1 | 2.81 ± 0.03 | 0.0076 | ||
| Pt-O-Pt | 5.4 ± 1.8 | 3.24 ± 0.03 | 0.0094 | ||
| Pt/Y2O3-NaCl-fresh | Pt-O | 4.9 ± 0.2 | 2.01 ± 0.01 | 0.0039 | 11.6 ± 1.0 |
| Pt-Pt | 3.2 ± 0.7 | 2.80 ± 0.01 | 0.0060 | ||
| Pt-O-O | 3.3 ± 1.0 | 2.99 ± 0.03 | 0.0094 |
| Sample | Shell | CN | R (Å) | σ2 (Å2) | ΔE0 |
|---|---|---|---|---|---|
| Pt/Y2O3-H2 | Pt-O | 1.0 ± 0.2 | 1.98 ± 0.01 | 0.0086 | 8.6 ± 0.9 |
| Pt-Pt | 9.3 ± 0.3 | 2.76 ± 0.01 | 0.0053 | ||
| Pt-O-Pt | 2.2 ± 1.4 | 3.09 ± 0.06 | 0.0157 | ||
| Pt/Y2O3-used | Pt-O | 1.1 ± 0.2 | 1.99 ± 0.01 | 0.0026 | 8.1 ± 1.0 |
| Pt-Pt | 7.8 ± 0.3 | 2.76 ± 0.01 | 0.0044 | ||
| Pt/Y2O3-NaCl-H2 | Pt-O | 0.6 ± 0.2 | 2.01 ± 0.02 | 0.0016 | 5.7 ± 1.9 |
| Pt-O1 | 0.5 ± 0.4 | 2.46 ± 0.05 | 0.0034 | ||
| Pt-Pt | 7.1 ± 0.4 | 2.74 ± 0.01 | 0.0077 | ||
| Pt/Y2O3-NaCl-used | Pt-O | 1.4 ± 0.2 | 1.98 ± 0.01 | 0.0038 | 3.1 ± 1.7 |
| Pt-O1 | 1.7 ± 0.5 | 2.47 ± 0.02 | 0.0047 | ||
| Pt-Pt | 6.0 ± 0.3 | 2.73 ± 0.01 | 0.0065 |
| Sample | CPt | CPt surface | Pt0 | Pt2+ | Pt4+ |
|---|---|---|---|---|---|
| (at. %) a | (at. %) b | (%) b | (%) b | (%) b | |
| Pt/Y2O3-fresh | 0.62 | 0.36 | 9.9 | - | 90.1 |
| Pt/Y2O3-NaCl-fresh | 0.68 | 1.22 | - | 23.9 | 76.1 |
| Pt/Y2O3-used | - | 0.36 | 50.0 | - | 50.0 |
| Pt/Y2O3-NaCl-used | - | 0.94 | 80.2 | - | 19.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, L.; Tian, C.; Li, Y.; Si, R.; Du, M.; Li, X.; Guo, L.; Li, L. NaCl-Templated Ultrathin 2D-Yttria Nanosheets Supported Pt Nanoparticles for Enhancing CO Oxidation Reaction. Nanomaterials 2022, 12, 2306. https://doi.org/10.3390/nano12132306
Jiang L, Tian C, Li Y, Si R, Du M, Li X, Guo L, Li L. NaCl-Templated Ultrathin 2D-Yttria Nanosheets Supported Pt Nanoparticles for Enhancing CO Oxidation Reaction. Nanomaterials. 2022; 12(13):2306. https://doi.org/10.3390/nano12132306
Chicago/Turabian StyleJiang, Luozhen, Chen Tian, Yunan Li, Rui Si, Meng Du, Xiuhong Li, Lingling Guo, and Lina Li. 2022. "NaCl-Templated Ultrathin 2D-Yttria Nanosheets Supported Pt Nanoparticles for Enhancing CO Oxidation Reaction" Nanomaterials 12, no. 13: 2306. https://doi.org/10.3390/nano12132306
APA StyleJiang, L., Tian, C., Li, Y., Si, R., Du, M., Li, X., Guo, L., & Li, L. (2022). NaCl-Templated Ultrathin 2D-Yttria Nanosheets Supported Pt Nanoparticles for Enhancing CO Oxidation Reaction. Nanomaterials, 12(13), 2306. https://doi.org/10.3390/nano12132306