
Investigation of the Stability and Hydrogen Evolution Activity of Dual-Atom Catalysts on Nitrogen-Doped Graphene


Abstract
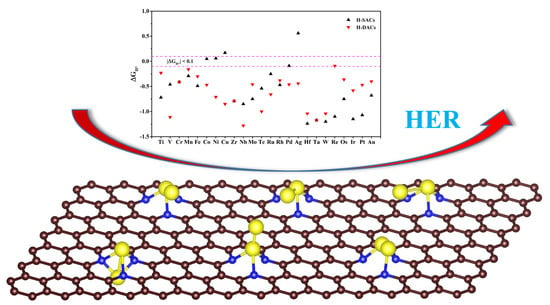
:
1. Introduction
2. Materials
3. Results and Discussion
3.1. Geometric Structures
3.1.1. Single-Atom
3.1.2. Dual-Atom
3.2. Stability
3.3. HER Activity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, L.; Corma, A. Metal Catalysts for Heterogeneous Catalysis: From Single Atoms to Nanoclusters and Nanoparticles. Chem. Rev. 2018, 118, 4981–5079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Wang, X.; Wang, J.; Cui, G.; Liu, D. Graphite carbon nitride/boron-doped graphene hybrid for efficient hydrogen generation reaction. Nanotechnology 2018, 29, 345705. [Google Scholar] [CrossRef] [PubMed]
- Jing, H.; Zhu, P.; Zheng, X.; Zhang, Z.; Wang, D.; Li, Y. Theory-oriented screening and discovery of advanced energy transformation materials in electrocatalysis. Adv. Powder Mater. 2021, 1, 100013. [Google Scholar] [CrossRef]
- Yang, J.; Li, W.H.; Tan, S.; Xu, K.; Wang, Y.; Wang, D.; Li, Y. The Electronic Metal-Support Interaction Directing the Design of Single Atomic Site Catalysts: Achieving High Efficiency Towards Hydrogen Evolution. Angew. Chem. Int. Ed. Engl. 2021, 60, 19085–19091. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, G.; Shi, L.; Ye, J. Single-Atom Catalysts: Emerging Multifunctional Materials in Heterogeneous Catalysis. Adv. Energy Mater. 2018, 8, 1701343. [Google Scholar] [CrossRef]
- Pan, T.; Wang, Y.; Xue, X.; Zhang, C. Rational design of allosteric switchable catalysts. Exploration 2022, 2, 20210095. [Google Scholar] [CrossRef]
- O’Neill, B.J.; Jackson, D.H.K.; Lee, J.; Canlas, C.; Stair, P.C.; Marshall, C.L.; Elam, J.W.; Kuech, T.F.; Dumesic, J.A.; Huber, G.W. Catalyst Design with Atomic Layer Deposition. ACS Catal. 2015, 5, 1804–1825. [Google Scholar] [CrossRef] [Green Version]
- Qiao, B.; Wang, A.; Yang, X.; Allard, L.F.; Jiang, Z.; Cui, Y.; Liu, J.; Li, J.; Zhang, T. Single-atom catalysis of CO oxidation using Pt1/FeOx. Nat. Chem. 2011, 3, 634–641. [Google Scholar] [CrossRef]
- Guo, X.; Fang, G.; Li, G.; Ma, H.; Fan, H.; Yu, L.; Ma, C.; Wu, X.; Deng, D.; Wei, M.; et al. Direct, nonoxidative conversion of methane to ethylene, aromatics, and hydrogen. Science 2014, 344, 616–619. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Jiao, M.; Mei, B.; Tong, Y.; Li, Y.; Ruan, M.; Song, P.; Sun, G.; Jiang, L.; Wang, Y.; et al. Carbon-Supported Divacancy-Anchored Platinum Single-Atom Electrocatalysts with Superhigh Pt Utilization for the Oxygen Reduction Reaction. Angew. Chem. Int. Ed. Engl. 2019, 58, 1163–1167. [Google Scholar] [CrossRef]
- Liu, M.; Zhang, R.; Chen, W. Graphene-supported nanoelectrocatalysts for fuel cells: Synthesis, properties, and applications. Chem. Rev. 2014, 114, 5117–5160. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Huo, J.; Liu, Y.; Wu, W.; Wang, Y.; Wu, M.; Liu, H.; Wang, G. Nitrogen-Doped Porous Carbon Supported Nonprecious Metal Single-Atom Electrocatalysts: From Synthesis to Application. Small Methods 2019, 3, 1900159. [Google Scholar] [CrossRef]
- Liu, Z.; Li, S.; Yang, J.; Tan, X.; Yu, C.; Zhao, C.; Han, X.; Huang, H.; Wan, G.; Liu, Y.; et al. Ultrafast Construction of Oxygen-Containing Scaffold over Graphite for Trapping Ni2+ into Single Atom Catalysts. ACS Nano 2020, 14, 11662–11669. [Google Scholar] [CrossRef] [PubMed]
- Pasti, I.A.; Jovanovic, A.; Dobrota, A.S.; Mentus, S.V.; Johansson, B.; Skorodumova, N.V. Atomic adsorption on graphene with a single vacancy: Systematic DFT study through the periodic table of elements. Phys. Chem. Chem. Phys. 2018, 20, 858–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Cui, X.; Zhao, J.; Jia, G.; Gu, L.; Zhang, Q.; Meng, L.; Shi, Z.; Zheng, L.; Wang, C.; et al. Rational Design of Fe–N/C Hybrid for Enhanced Nitrogen Reduction Electrocatalysis under Ambient Conditions in Aqueous Solution. ACS Catal. 2018, 9, 336–344. [Google Scholar] [CrossRef]
- Sa, Y.J.; Jung, H.; Shin, D.; Jeong, H.Y.; Ringe, S.; Kim, H.; Hwang, Y.J.; Joo, S.H. Thermal Transformation of Molecular Ni2+–N4 Sites for Enhanced CO2 Electroreduction Activity. ACS Catalysis 2020, 10, 10920–10931. [Google Scholar] [CrossRef]
- Zhong, L.; Li, S. Unconventional Oxygen Reduction Reaction Mechanism and Scaling Relation on Single-Atom Catalysts. ACS Catal. 2020, 10, 4313–4318. [Google Scholar] [CrossRef]
- Cheng, N.; Stambula, S.; Wang, D.; Banis, M.N.; Liu, J.; Riese, A.; Xiao, B.; Li, R.; Sham, T.K.; Liu, L.M.; et al. Platinum single-atom and cluster catalysis of the hydrogen evolution reaction. Nat. Commun. 2016, 7, 13638. [Google Scholar] [CrossRef]
- Fang, S.; Zhu, X.; Liu, X.; Gu, J.; Liu, W.; Wang, D.; Zhang, W.; Lin, Y.; Lu, J.; Wei, S.; et al. Uncovering near-free platinum single-atom dynamics during electrochemical hydrogen evolution reaction. Nat. Commun. 2020, 11, 1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hossain, M.D.; Liu, Z.; Zhuang, M.; Yan, X.; Xu, G.L.; Gadre, C.A.; Tyagi, A.; Abidi, I.H.; Sun, C.J.; Wong, H.; et al. Rational Design of Graphene-Supported Single Atom Catalysts for Hydrogen Evolution Reaction. Adv. Energy Mater. 2019, 9, 1803689. [Google Scholar] [CrossRef]
- Jung, E.; Shin, H.; Lee, B.H.; Efremov, V.; Lee, S.; Lee, H.S.; Kim, J.; Hooch Antink, W.; Park, S.; Lee, K.S.; et al. Atomic-level tuning of Co-N-C catalyst for high-performance electrochemical H2O2 production. Nat. Mater. 2020, 19, 436–442. [Google Scholar] [CrossRef] [PubMed]
- Zang, W.; Sun, T.; Yang, T.; Xi, S.; Waqar, M.; Kou, Z.; Lyu, Z.; Feng, Y.P.; Wang, J.; Pennycook, S.J. Efficient Hydrogen Evolution of Oxidized Ni-N3 Defective Sites for Alkaline Freshwater and Seawater Electrolysis. Adv. Mater. 2021, 33, e2003846. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Zhang, X.; Kang, Y.; Ye, C.; Jin, R.; Yan, H.; Lin, R.; Yang, J.; Xu, Q.; Wang, Y.; et al. A Supported Pd2 Dual-Atom Site Catalyst for Efficient Electrochemical CO2 Reduction. Angew. Chem. Int. Ed. Engl. 2021, 60, 13388–13393. [Google Scholar] [CrossRef] [PubMed]
- Tian, S.; Wang, B.; Gong, W.; He, Z.; Xu, Q.; Chen, W.; Zhang, Q.; Zhu, Y.; Yang, J.; Fu, Q.; et al. Dual-atom Pt heterogeneous catalyst with excellent catalytic performances for the selective hydrogenation and epoxidation. Nat. Commun. 2021, 12, 3181. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.W.; Chen, L.X.; Yang, C.C.; Jiang, Q. Atomic (single, double, and triple atoms) catalysis: Frontiers, opportunities, and challenges. J. Mater. Chem. A 2019, 7, 3492–3515. [Google Scholar] [CrossRef]
- Guo, X.; Gu, J.; Lin, S.; Zhang, S.; Chen, Z.; Huang, S. Tackling the Activity and Selectivity Challenges of Electrocatalysts toward the Nitrogen Reduction Reaction via Atomically Dispersed Biatom Catalysts. J. Am. Chem. Soc. 2020, 142, 5709–5721. [Google Scholar] [CrossRef]
- Li, H.; Zhao, Z.; Cai, Q.; Yin, L.; Zhao, J. Nitrogen electroreduction performance of transition metal dimers embedded into N-doped graphene: A theoretical prediction. J. Mater. Chem. A 2020, 8, 4533–4543. [Google Scholar] [CrossRef]
- Yang, Y.; Qian, Y.; Li, H.; Zhang, Z.; Mu, Y.; Do, D.; Zhou, B.; Dong, J.; Yan, W.; Qin, Y. O-coordinated W-Mo dual-atom catalyst for pH-universal electrocatalytic hydrogen evolution. Sci. Adv. 2020, 6, eaba6586. [Google Scholar] [CrossRef] [PubMed]
- Nørskov, J.K.; Bligaard, T.; Logadottir, A.; Kitchin, J.R.; Chen, J.G.; Pandelov, S.; Stimming, U. Trends in the Exchange Current for Hydrogen Evolution. J. Electrochem. Soc. 2005, 152, J23. [Google Scholar] [CrossRef] [Green Version]
- Greeley, J.; Jaramillo, T.F.; Bonde, J.; Chorkendorff, I.B.; Norskov, J.K. Computational high-throughput screening of electrocatalytic materials for hydrogen evolution. Nat. Mater. 2006, 5, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Fung, V.; Hu, G.; Wu, Z.; Jiang, D.-e. Descriptors for Hydrogen Evolution on Single Atom Catalysts in Nitrogen-Doped Graphene. J. Phys. Chem. C 2020, 124, 19571–19578. [Google Scholar] [CrossRef]
- Henkelman, G.; Uberuaga, B.P.; Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef] [Green Version]
- Carlsson, J.M.; Scheffler, M. Structural, electronic, and chemical properties of nanoporous carbon. Phys. Rev. Lett. 2006, 96, 046806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Y.; Lu, Z.; Chen, W.; Li, W.; Dai, X. Geometric stability and reaction activity of Pt clusters adsorbed graphene substrates for catalytic CO oxidation. Phys. Chem. Chem. Phys. 2015, 17, 11598–11608. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Gao, G.; Li, Y.; Chu, W.; Wang, L.W. Transition-metal single atoms in nitrogen-doped graphenes as efficient active centers for water splitting: A theoretical study. Phys. Chem. Chem. Phys. 2019, 21, 3024–3032. [Google Scholar] [CrossRef] [PubMed]
- Wang, V.; Xu, N.; Liu, J.-C.; Tang, G.; Geng, W.-T. VASPKIT: A user-friendly interface facilitating high-throughput computing and analysis using VASP code. Comput. Phys. Commun. 2021, 267, 108033. [Google Scholar] [CrossRef]
- Pašti, I.A.; Jovanović, A.; Dobrota, A.S.; Mentus, S.V.; Johansson, B.; Skorodumova, N.V. Atomic adsorption on pristine graphene along the Periodic Table of Elements—From PBE to non-local functionals. Appl. Surf. Sci. 2018, 436, 433–440. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Wu, H. A first-principles study on the interaction of biogas with noble metal (Rh, Pt, Pd) decorated nitrogen doped graphene as a gas sensor: A DFT study. Appl. Surf. Sci. 2018, 435, 1199–1212. [Google Scholar] [CrossRef]
- Manadé, M.; Viñes, F.; Illas, F. Transition metal adatoms on graphene: A systematic density functional study. Carbon 2015, 95, 525–534. [Google Scholar] [CrossRef]
- Habenicht, B.F.; Teng, D.; Semidey-Flecha, L.; Sholl, D.S.; Xu, Y. Adsorption and Diffusion of 4d and 5d Transition Metal Adatoms on Graphene/Ru(0001) and the Implications for Cluster Nucleation. Top. Catal. 2013, 57, 69–79. [Google Scholar] [CrossRef]
- Jin, C.; Cheng, L.; Feng, G.; Ye, R.; Lu, Z.H.; Zhang, R.; Yu, X. Adsorption of Transition-Metal Clusters on Graphene and N-Doped Graphene: A DFT Study. Langmuir 2022, 38, 3694–3710. [Google Scholar] [CrossRef]
- Ruban, A.V.; Skriver, H.L.; Nørskov, J.K. Surface segregation energies in transition-metal alloys. Phys. Rev. B 1999, 59, 15990–16000. [Google Scholar] [CrossRef] [Green Version]
- Vitos, L.; Ruban, A.; Skriver, H.L.; Kollár, J. The surface energy of metals. Surf. Sci. 1998, 411, 186–202. [Google Scholar] [CrossRef]
- Lee, J.Y.; Punkkinen, M.P.J.; Schönecker, S.; Nabi, Z.; Kádas, K.; Zólyomi, V.; Koo, Y.M.; Hu, Q.M.; Ahuja, R.; Johansson, B.; et al. The surface energy and stress of metals. Surf. Sci. 2018, 674, 51–68. [Google Scholar] [CrossRef] [Green Version]
- Seh, Z.W.; Kibsgaard, J.; Dickens, C.F.; Chorkendorff, I.; Norskov, J.K.; Jaramillo, T.F. Combining theory and experiment in electrocatalysis: Insights into materials design. Science 2017, 355, eaad4998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talib, S.H.; Lu, Z.; Yu, X.; Ahmad, K.; Bashir, B.; Yang, Z.; Li, J. Theoretical Inspection of M1/PMA Single-Atom Electrocatalyst: Ultra-High Performance for Water Splitting (HER/OER) and Oxygen Reduction Reactions (OER). ACS Catal. 2021, 11, 8929–8941. [Google Scholar] [CrossRef]
- Santos, E.; Quaino, P.; Schmickler, W. Theory of electrocatalysis: Hydrogen evolution and more. Phys. Chem. Chem. Phys. 2012, 14, 11224–11233. [Google Scholar] [CrossRef]
- Jovanović, A.Z.; Mentus, S.V.; Skorodumova, N.V.; Pašti, I.A. Reactivity Screening of Single Atoms on Modified Graphene Surface: From Formation and Scaling Relations to Catalytic Activity. Adv. Mater. Interfaces 2020, 8, 2001814. [Google Scholar] [CrossRef]
- Lim, J.; Back, S.; Choi, C.; Jung, Y. Ultralow Overpotential of Hydrogen Evolution Reaction using Fe-Doped Defective Graphene: A Density Functional Study. ChemCatChem 2018, 10, 4450–4455. [Google Scholar] [CrossRef]
- Dobrota, A.S.; Skorodumova, N.V.; Mentus, S.V.; Pašti, I.A. Surface pourbaix plots of M@N4-graphene single-atom electrocatalysts from density functional theory thermodynamic modeling. Electrochim. Acta 2022, 412, 140155. [Google Scholar] [CrossRef]
- Zhang, X.; Yu, S.; Chen, H.; Zheng, W. TM atoms on B/N doped defective graphene as a catalyst for oxygen reduction reaction: A theoretical study. RSC Adv. 2015, 5, 82804–82812. [Google Scholar] [CrossRef]
- Liu, S.; Huang, S. Theoretical insights into the activation of O2 by Pt single atom and Pt4 nanocluster on functionalized graphene support: Critical role of Pt positive polarized charges. Carbon 2017, 115, 11–17. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Site | h/Å | ||
|---|---|---|---|
| Ti | H | −1.98 | 1.85 |
| V | H | −1.20 | 1.85 |
| Cr | B | −0.31 | 2.23 |
| Mn | T | −0.23 | 2.24 |
| Fe | H | −1.17 | 1.55 |
| Co | H | −1.24 | 1.54 |
| Ni | H | −1.52 | 1.56 |
| Cu | T | −0.29 | 2.08 |
| Zr | H | −2.38 | 1.98 |
| Nb | H | −1.55 | 1.71 |
| Mo | B | −0.22 | 2.32 |
| Tc | H | −1.19 | 1.66 |
| Ru | H | −3.02 | 1.73 |
| Rh | H | −1.86 | 1.78 |
| Pd | B | −1.09 | 2.06 |
| Ag | B | −0.01 | 3.47 |
| Hf | H | −1.83 | 1.93 |
| Ta | H | −1.76 | 1.85 |
| W | B | −0.52 | 2.22 |
| Re | T | 0.07 | 2.20 |
| Os | H | −1.01 | 1.71 |
| Ir | B | −1.51 | 1.94 |
| Pt | B | −1.55 | 1.97 |
| Au | T | −0.18 | 3.46 |
| Site | dM1−N1/Å | dM1−N2/Å | dM1−N3/Å | h/Å | ||
|---|---|---|---|---|---|---|
| Ti | S | −6.29 | 1.91 | 1.91 | 1.91 | 1.58 |
| V | S | −5.80 | 1.90 | 1.90 | 1.90 | 1.52 |
| Cr | S | −3.73 | 1.96 | 1.96 | 1.96 | 1.51 |
| Mn | S | −3.98 | 2.00 | 2.00 | 2.00 | 1.65 |
| Fe | S | −4.57 | 1.78 | 1.78 | 1.78 | 1.23 |
| Co | S | −4.96 | 1.83 | 1.83 | 1.83 | 1.39 |
| Ni | S | −4.48 | 1.84 | 1.85 | 1.85 | 1.37 |
| Cu | S | −3.12 | 1.89 | 1.89 | 1.89 | 1.51 |
| Zr | S | −6.55 | 2.07 | 2.06 | 2.06 | 1.84 |
| Nb | S | −5.84 | 1.99 | 1.99 | 1.99 | 1.76 |
| Mo | S | −4.30 | 1.94 | 1.94 | 1.94 | 1.68 |
| Tc | S | −5.10 | 1.91 | 1.91 | 1.91 | 1.59 |
| Ru | S | −6.23 | 1.93 | 1.93 | 1.93 | 1.55 |
| Rh | S | −4.32 | 2.04 | 2.04 | 2.05 | 1.66 |
| Pd | S | −2.33 | 2.27 | 2.11 | 2.11 | 1.78 |
| Ag | S | −1.68 | 2.31 | 2.31 | 2.31 | 2.05 |
| Hf | S | −6.49 | 2.00 | 2.00 | 1.99 | 1.75 |
| Ta | S | −6.41 | 1.96 | 1.95 | 1.95 | 1.70 |
| W | S | −5.35 | 1.91 | 1.91 | 1.91 | 1.65 |
| Re | S | −4.36 | 1.91 | 1.91 | 1.91 | 1.60 |
| Os | S | −4.90 | 1.91 | 1.91 | 1.91 | 1.55 |
| Ir | S | −4.32 | 2.01 | 2.01 | 2.01 | 1.63 |
| Pt | S | −2.86 | 2.09 | 2.24 | 2.09 | 1.73 |
| Au | S | −1.09 | 2.34 | 2.34 | 2.35 | 2.03 |
| Configuration | dM1−N1/Å | dM1−N2/Å | dM1−N3/Å | dM2−N1/Å | dM2−N2/Å | dM2−N3/Å | dM1−M2/Å | ||
|---|---|---|---|---|---|---|---|---|---|
| Ti | Ⅰ | −9.92 | 2.08 | 2.10 | 2.10 | 2.15 | 2.06 | 2.06 | 2.82 |
| V | Ⅱ | −7.79 | 1.95 | 1.95 | 2.04 | 3.48 | 3.48 | 1.98 | 2.03 |
| Cr | Ⅵ | −5.70 | 2.07 | 2.07 | 2.07 | 3.26 | 3.27 | 3.28 | 1.51 |
| Mn | Ⅵ | −5.75 | 1.96 | 1.96 | 1.96 | 3.82 | 3.78 | 3.81 | 2.28 |
| Fe | Ⅵ | −7.21 | 1.85 | 1.85 | 1.85 | 3.43 | 3.44 | 3.43 | 2.06 |
| Co | Ⅵ | −7.07 | 1.94 | 1.95 | 1.95 | 3.80 | 3.59 | 3.43 | 2.11 |
| Ni | Ⅵ | −6.58 | 1.93 | 1.93 | 1.92 | 3.65 | 3.65 | 3.64 | 2.16 |
| Cu | Ⅵ | −4.54 | 2.02 | 2.02 | 2.02 | 3.88 | 3.89 | 3.87 | 2.25 |
| Zr | Ⅰ | −11.08 | 2.20 | 2.19 | 2.25 | 2.23 | 2.18 | 2.17 | 3.11 |
| Nb | Ⅱ | −9.55 | 2.07 | 2.07 | 2.14 | 3.77 | 3.77 | 2.09 | 2.34 |
| Mo | Ⅵ | −9.44 | 2.27 | 2.27 | 2.27 | 3.68 | 3.68 | 3.69 | 1.68 |
| Tc | Ⅵ | −12.92 | 2.15 | 2.15 | 2.15 | 3.17 | 3.16 | 3.17 | 1.28 |
| Ru | Ⅵ | −11.00 | 2.01 | 2.01 | 2.01 | 3.69 | 3.71 | 3.68 | 2.10 |
| Rh | Ⅳ | −7.71 | 1.97 | 2.04 | 1.97 | 3.20 | 2.86 | 4.50 | 2.51 |
| Pd | Ⅳ | −4.04 | 2.20 | 2.12 | 2.13 | 3.39 | 4.62 | 2.96 | 2.64 |
| Ag | Ⅵ | −2.77 | 2.39 | 2.39 | 2.40 | 4.72 | 4.74 | 4.73 | 2.63 |
| Hf | Ⅰ | −10.71 | 2.16 | 2.16 | 2.19 | 2.19 | 2.16 | 2.16 | 3.07 |
| Ta | Ⅱ | −10.93 | 2.04 | 2.04 | 2.12 | 3.81 | 3.81 | 2.12 | 2.40 |
| W | Ⅴ | −9.77 | 2.04 | 2.04 | 2.82 | 3.25 | 3.25 | 2.19 | 2.11 |
| Re | Ⅵ | −9.22 | 2.08 | 2.08 | 2.09 | 3.79 | 3.79 | 3.74 | 2.07 |
| Os | Ⅵ | −10.41 | 2.02 | 2.02 | 2.02 | 3.75 | 3.75 | 3.75 | 2.14 |
| Ir | Ⅵ | −9.89 | 2.03 | 2.03 | 2.04 | 3.85 | 3.85 | 3.85 | 2.21 |
| Pt | Ⅵ | −7.03 | 2.08 | 2.08 | 2.16 | 4.05 | 4.07 | 4.07 | 2.33 |
| Au | Ⅵ | −3.52 | 2.38 | 2.23 | 2.52 | 4.52 | 4.60 | 4.60 | 2.52 |
| Non−H | System | QM1/e | QM2/e | QM1+M2/e | |
| Co-SVGN3 | +0.82 | − | +0.82 | ||
| Co2-SVGN3 | +0.73 | −0.13 | +0.60 | ||
| Ni-SVGN3 | +0.73 | − | +0.73 | ||
| Ni2-SVGN3 | +0.64 | −0.18 | +0.46 | ||
| Pd-SVGN3 | +0.52 | − | +0.52 | ||
| Pd2-SVGN3 | +0.47 | +0.11 | +0.58 | ||
| Re-SVGN3 | +1.00 | − | +1.00 | ||
| Re2-SVGN3 | +0.97 | −0.17 | +0.80 | ||
| H adsorption | System | QM1/e | QM2/e | QM1+M2/e | |
| Co-SVGN3 | +0.95 | − | +0.95 | +0.13 | |
| Co2-SVGN3 | +0.77 | +0.08 | +0.85 | +0.25 | |
| Ni-SVGN3 | +0.74 | − | +0.74 | +0.01 | |
| Ni2-SVGN3 | +0.69 | +0.08 | +0.77 | +0.31 | |
| Pd-SVGN3 | +0.54 | − | +0.54 | +0.02 | |
| Pd2-SVGN3 | +0.52 | +0.10 | +0.62 | +0.04 | |
| Re-SVGN3 | +1.35 | − | +1.35 | +0.35 | |
| Re2-SVGN3 | +1.06 | −0.11 | +0.95 | +0.15 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Q.; Zhang, M.; Zhu, B.; Gao, Y. Investigation of the Stability and Hydrogen Evolution Activity of Dual-Atom Catalysts on Nitrogen-Doped Graphene. Nanomaterials 2022, 12, 2557. https://doi.org/10.3390/nano12152557
Zhou Q, Zhang M, Zhu B, Gao Y. Investigation of the Stability and Hydrogen Evolution Activity of Dual-Atom Catalysts on Nitrogen-Doped Graphene. Nanomaterials. 2022; 12(15):2557. https://doi.org/10.3390/nano12152557
Chicago/Turabian StyleZhou, Qiansong, Meng Zhang, Beien Zhu, and Yi Gao. 2022. "Investigation of the Stability and Hydrogen Evolution Activity of Dual-Atom Catalysts on Nitrogen-Doped Graphene" Nanomaterials 12, no. 15: 2557. https://doi.org/10.3390/nano12152557
APA StyleZhou, Q., Zhang, M., Zhu, B., & Gao, Y. (2022). Investigation of the Stability and Hydrogen Evolution Activity of Dual-Atom Catalysts on Nitrogen-Doped Graphene. Nanomaterials, 12(15), 2557. https://doi.org/10.3390/nano12152557