
On the Electron-Induced Reactions of (CH3)AuP(CH3)3: A Combined UHV Surface Science and Gas-Phase Study

 , , ,
, , ,
Abstract
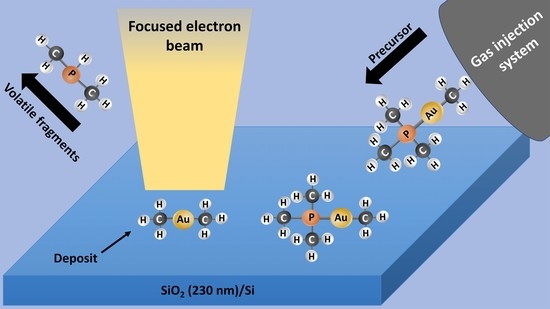
:
1. Introduction
2. Materials and Methods
3. Results and Discussion
3.1. UHV-FEBID Study
3.1.1. Promotion of the Intact Precursor into the Gas Phase and Its Stability
3.1.2. FEBID
3.2. HV Gas-Phase Study
Dissociative Ionization in the Gas Phase
3.3. Dissociative Ionization, UHV, and HV FEBID Composition
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Winkler, R.; Schmidt, F.P.; Haselmann, U.; Fowlkes, J.D.; Lewis, B.B.; Kothleitner, G.; Rack, P.D.; Plank, H. Direct-Write 3D Nanoprinting of Plasmonic Structures. ACS Appl. Mater. Interfaces 2017, 9, 8233–8240. [Google Scholar] [CrossRef] [PubMed]
- Drost, M.; Tu, F.; Berger, L.; Preischl, C.; Zhou, W.; Gliemann, H.; Wöll, C.; Marbach, H. Surface-Anchored Metal-Organic Frameworks as Versatile Resists for Gas-Assisted E-Beam Lithography: Fabrication of Sub-10 Nanometer Structures. ACS Nano 2018, 12, 3825–3835. [Google Scholar] [CrossRef] [PubMed]
- van Dorp, W.F.; Beyer, A.; Mainka, M.; Gölzhäuser, A.; Hansen, T.W.; Wagner, J.B.; Hagen, C.W.; De Hosson, J.T.M. Focused Electron Beam Induced Processing and the Effect of Substrate Thickness Revisited. Nanotechnology 2013, 24, 345301. [Google Scholar] [CrossRef]
- Prima, G.D.; Sachser, R.; Trompenaars, P.; Mulders, H.; Huth, M. Direct-Write Single Electron Transistors by Focused Electron Beam Induced Deposition. Nano Futur. 2019, 3, 25001. [Google Scholar] [CrossRef]
- Marbach, H. Electron Beam Induced Surface Activation: A Method for the Lithographic Fabrication of Nanostructures via Catalytic Processes. Appl. Phys. A Mater. Sci. Process. 2014, 117, 987–995. [Google Scholar] [CrossRef]
- Huth, M.; Porrati, F.; Schwalb, C.; Winhold, M.; Sachser, R.; Dukic, M.; Adams, J.; Fantner, G. Focused Electron Beam Induced Deposition: A Perspective. Beilstein J. Nanotechnol. 2012, 3, 597–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Utke, I.; Hoffmann, P.; Melngailis, J. Gas-Assisted Focused Electron Beam and Ion Beam Processing and Fabrication. J. Vac. Sci. Technol. B Microelectron. Nanom. Struct. 2008, 26, 1197–1276. [Google Scholar] [CrossRef] [Green Version]
- Heil, T.; Waldow, M.; Capelli, R.; Schneider, H.; Ahmels, L.; Tu, F.; Schöneberg, J.; Marbach, H. Pushing the Limits of EUV Mask Repair: Addressing Sub-10 Nm Defects with the next Generation e-Beam-Based Mask Repair Tool. J. Micro/Nanopatterning, Mater. Metrol. 2021, 20, 1–12. [Google Scholar] [CrossRef]
- Edinger, K.; Becht, H.; Bihr, J.; Boegli, V.; Budach, M.; Hofmann, T.; Koops, H.W.P.; Kuschnerus, P.; Oster, J.; Spies, P.; et al. Electron-Beam-Based Photomask Repair. J. Vac. Sci. Technol. B Microelectron. Nanom. Struct. 2004, 22, 2902–2906. [Google Scholar] [CrossRef]
- Liang, T.; Frendberg, E.; Lieberman, B.; Stivers, A. Advanced Photolithographic Mask Repair Using Electron Beams. J. Vac. Sci. Technol. B Microelectron. Nanom. Struct. 2005, 23, 3101–3105. [Google Scholar] [CrossRef]
- Fernández-Pacheco, A.; Serrano-Ramón, L.E.; Tyliszczak, T.; Chou, K.W.; Córdoba, R.; Szkudlarek, A.; Brien, L.O.; Kapusta, C.; Ibarra, M.R.; De Teresa, J.M. Correlation between the Magnetic Imaging of Cobalt Nanoconstrictions and Their Magnetoresistance Response. Nanotechnology 2012, 23, 105703. [Google Scholar] [CrossRef] [PubMed]
- Wendel, M.; Lorenz, H.; Kotthaus, J.P. Sharpened Electron Beam Deposited Tips for High Resolution Atomic Force Microscope Lithography and Imaging. Appl. Phys. Lett. 1995, 67, 3732–3734. [Google Scholar] [CrossRef]
- Utke, I.; Friedli, V.; Michler, J.; Bret, T.; Multone, X.; Hoffmann, P. Density Determination of Focused-Electron-Beam-Induced Deposits with Simple Cantilever-Based Method. Appl. Phys. Lett. 2006, 88, 31906. [Google Scholar] [CrossRef] [Green Version]
- Plank, H.; Winkler, R.; Schwalb, C.H.; Hütner, J.; Fowlkes, J.D.; Rack, P.D.; Utke, I.; Huth, M. Focused Electron Beam-Based 3D Nanoprinting for Scanning Probe Microscopy: A Review. Micromachines 2020, 11, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Teresa, J.M.; Fernández-Pacheco, A. Present and Future Applications of Magnetic Nanostructures Grown by FEBID. Appl. Phys. A 2014, 117, 1645–1658. [Google Scholar] [CrossRef]
- Höflich, K.; Yang, R.B.; Berger, A.; Leuchs, G.; Christiansen, S. The Direct Writing of Plasmonic Gold Nanostructures by Electron-Beam- Induced Deposition. Adv. Mater. 2011, 23, 2657–2661. [Google Scholar] [CrossRef]
- Utke, I.; Swiderek, P.; Höflich, K.; Madajska, K.; Jurczyk, J.; Martinović, P.; Szymańska, I.B. Coordination and Organometallic Precursors of Group 10 and 11: Focused Electron Beam Induced Deposition of Metals and Insight Gained from Chemical Vapour Deposition, Atomic Layer Deposition, and Fundamental Surface and Gas Phase Studies. Coord. Chem. Rev. 2022, 458, 213851. [Google Scholar] [CrossRef]
- Lukasczyk, T.; Schirmer, M.; Steinrück, H.P.; Marbach, H. Electron-Beam-Induced Deposition in Ultrahigh Vacuum: Lithographic Fabrication of Clean Iron Nanostructures. Small 2008, 4, 841–846. [Google Scholar] [CrossRef]
- Klein, K.L.; Randolph, S.J.; Fowlkes, J.D.; Allard, L.F.; Meyer III, H.M.; Simpson, M.L.; Rack, P.D. Single-Crystal Nanowires Grown via Electron-Beam-Induced Deposition. Nanotechnology 2008, 19, 345705. [Google Scholar] [CrossRef]
- Chien, M.H.; Shawrav, M.M.; Hingerl, K.; Taus, P.; Schinnerl, M.; Wanzenboeck, H.D.; Schmid, S. Analysis of Carbon Content in Direct-Write Plasmonic Au Structures by Nanomechanical Scanning Absorption Microscopy. J. Appl. Phys. 2021, 129. [Google Scholar] [CrossRef]
- Kuhness, D.; Gruber, A.; Winkler, R.; Sattelkow, J.; Fitzek, H.; Letofsky-Papst, I.; Kothleitner, G.; Plank, H. High-Fidelity 3D Nanoprinting of Plasmonic Gold Nanoantennas. ACS Appl. Mater. Interfaces 2021, 13, 1178–1191. [Google Scholar] [CrossRef] [PubMed]
- Marashdeh, A.; Tiesma, T.; van Velzen, N.J.C.; Harder, S.; Havenith, R.W.A.; De Hosson, J.T.M.; van Dorp, W.F. The Rational Design of a Au(I) Precursor for Focused Electron Beam Induced Deposition. Beilstein J. Nanotechnol. 2017, 8, 2753–2765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carden, W.G.; Thorman, R.M.; Unlu, I.; Abboud, K.A.; Fairbrother, D.H.; McElwee-White, L. Design, Synthesis, and Evaluation of CF3AuCNR Precursors for Focused Electron Beam-Induced Deposition of Gold. ACS Appl. Mater. Interfaces 2019, 11, 11976–11987. [Google Scholar] [CrossRef] [PubMed]
- Carden, W.G.; Lu, H.; Spencer, J.A.; Fairbrother, D.H.; McElwee-White, L. Mechanism-Based Design of Precursors for Focused Electron Beam-Induced Deposition. MRS Commun. 2018, 8, 343–357. [Google Scholar] [CrossRef] [Green Version]
- Van Dorp, W.F.; Wu, X.; Mulders, J.J.L.; Harder, S.; Rudolf, P.; De Hosson, J.T.M. Gold Complexes for Focused-Electron-Beam-Induced Deposition. Langmuir 2014, 30, 12097–12105. [Google Scholar] [CrossRef]
- Mulders, J.J.L.; Veerhoek, J.M.; Bosch, E.G.T.; Trompenaars, P.H.F. Fabrication of Pure Gold Nanostructures by Electron Beam Induced Deposition with Au(CO)Cl Precursor: Deposition Characteristics and Primary Beam Scattering Effects. J. Phys. D. Appl. Phys. 2012, 45, 475301. [Google Scholar] [CrossRef]
- Utke, I.; Hoffmann, P.; Dwir, B.; Leifer, K.; Kapon, E.; Doppelt, P. Focused Electron Beam Induced Deposition of Gold. J. Vac. Sci. Technol. B Microelectron. Nanom. Struct. 2000, 18, 3168–3171. [Google Scholar] [CrossRef]
- Wnuk, J.D.; Gorham, J.M.; Rosenberg, S.G.; van Dorp, W.F.; Madey, T.E.; Hagen, C.W.; Fairbrother, D.H. Electron Beam Irradiation of Dimethyl-(Acetylacetonate) Gold(III) Adsorbed onto Solid Substrates. J. Appl. Phys. 2010, 107, 54301. [Google Scholar] [CrossRef] [Green Version]
- Jenke, M.G.; Lerose, D.; Niederberger, C.; Michler, J.; Christiansen, S.; Utke, I. Toward Local Growth of Individual Nanowires on Three-Dimensional Microstructures by Using a Minimally Invasive Catalyst Templating Method. Nano Lett. 2011, 11, 4213–4217. [Google Scholar] [CrossRef]
- Koops, H.W.P.; Kretz, J.; Rudolph, M.; Weber, M.; Dahm, G.; Lee, K.L. Characterization and Application of Materials Grown by Electron-Beam-Induced Deposition. Jpn. J. Appl. Phys. 1994, 33, 7099–7107. [Google Scholar] [CrossRef]
- Folch, A.; Tejada, J.; Peters, C.H.; Wrighton, M.S. Electron Beam Deposition of Gold Nanostructures in a Reactive Environment. Appl. Phys. Lett. 1995, 66, 2080–2082. [Google Scholar] [CrossRef] [Green Version]
- Botman, A.; Mulders, J.J.L.; Weemaes, R.; Mentink, S. Purification of Platinum and Gold Structures after Electron-Beam-Induced Deposition. Nanotechnology 2006, 17, 3779–3785. [Google Scholar] [CrossRef]
- Shawrav, M.M.; Taus, P.; Wanzenboeck, H.D.; Schinnerl, M.; Stöger-Pollach, M.; Schwarz, S.; Steiger-Thirsfeld, A.; Bertagnolli, E. Highly Conductive and Pure Gold Nanostructures Grown by Electron Beam Induced Deposition. Sci. Rep. 2016, 6, 34003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorman, R.M.; Ragesh Kumar, T.P.; Howard Fairbrother, D.; Ingólfsson, O. The Role of Low-Energy Electrons in Focused Electron Beam Induced Deposition: Four Case Studies of Representative Precursors. Beilstein J. Nanotechnol. 2015, 6, 1904–1926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unlu, I.; Spencer, J.A.; Johnson, K.R.; Thorman, R.M.; Ingólfsson, O.; McElwee-White, L.; Fairbrother, D.H. Electron Induced Surface Reactions of (η5-C5H5)Fe(CO)2Mn(CO)5, a Potential Heterobimetallic Precursor for Focused Electron Beam Induced Deposition (FEBID). Phys. Chem. Chem. Phys. 2018, 20, 7862–7874. [Google Scholar] [CrossRef]
- Ragesh Kumar, T.P.; Weirich, P.; Hrachowina, L.; Hanefeld, M.; Bjornsson, R.; Hrodmarsson, H.R.; Barth, S.; Howard Fairbrother, D.; Huth, M.; Ingólfsson, O. Electron Interactions with the Heteronuclear Carbonyl Precursor H2FeRu3(CO)13 and Comparison with HFeCo3(CO)12: From Fundamental Gas Phase and Surface Science Studies to Focused Electron Beam Induced Deposition. Beilstein J. Nanotechnol. 2018, 9, 555–579. [Google Scholar] [CrossRef]
- Ingólfsson, O. Low-Energy Electrons Fundamentals and Applications; Pan Stanford Publishing Pte. Ltd.: Singapore, 2019. [Google Scholar]
- Märk, T.D.; Dunn, G.H. Electron Impact Ionization; Springer Science & Business Media: Berlin/Heidelberg, Germany, 1985. [Google Scholar]
- Cipriani, M.; Thorman, R.M.; Brewer, C.R.; McElwee-White, L.; Ingólfsson, O. Dissociative Ionization of the Potential Focused Electron Beam Induced Deposition Precursor π-Allyl Ruthenium(II) Tricarbonyl Bromide, a Combined Theoretical and Experimental Study. Eur. Phys. J. D 2019, 73, 227. [Google Scholar] [CrossRef]
- Thorman, R.M.; Unlu, I.; Johnson, K.; Bjornsson, R.; McElwee-White, L.; Fairbrother, D.H.; Ingólfsson, O. Low Energy Electron-Induced Decomposition of (η5-Cp)Fe(CO)2Mn(CO)5, a Potential Bimetallic Precursor for Focused Electron Beam Induced Deposition of Alloy Structures. Phys. Chem. Chem. Phys. 2018, 20, 5644–5656. [Google Scholar] [CrossRef]
- Bjarnason, E.H.; Ómarsson, B.; Jónsdóttir, N.R.; Árnason, I.; Ingólfsson, O. Dissociative Electron Attachment and Dissociative Ionization of 1,1-Dichloro-1-Silacyclohexane and Silacyclohexane. Int. J. Mass Spectrom. 2014, 370, 39–43. [Google Scholar] [CrossRef]
- Vollnhals, F.; Drost, M.; Tu, F.; Carrasco, E.; Späth, A.; Fink, R.H.; Steinrück, H.P.; Marbach, H. Electron-Beam Induced Deposition and Autocatalytic Decomposition of Co(CO)3NO. Beilstein J. Nanotechnol. 2014, 5, 1175–1185. [Google Scholar] [CrossRef] [Green Version]
- Gavagnin, M.; Wanzenboeck, H.D.; Shawrav, M.M.; Belic, D.; Wachter, S.; Waid, S.; Stoeger-Pollach, M.; Bertagnolli, E. Focused Electron Beam-Induced CVD of Iron: A Practical Guide for Direct Writing. Chem. Vap. Depos. 2014, 20, 243–250. [Google Scholar] [CrossRef]
- Gazzadi, G.C.; Mulders, H.; Trompenaars, P.; Ghirri, A.; Affronte, M.; Grillo, V.; Frabboni, S. Focused Electron Beam Deposition of Nanowires from Cobalt Tricarbonyl Nitrosyl (Co(CO)3NO) Precursor. J. Phys. Chem. C 2011, 115, 19606–19611. [Google Scholar] [CrossRef]
- Gazzadi, G.C.; Mulders, J.J.L.; Trompenaars, P.; Ghirri, A.; Rota, A.; Affronte, M.; Frabboni, S. Characterization of a New Cobalt Precursor for Focused Beam Deposition of Magnetic Nanostructures. Microelectron. Eng. 2011, 88, 1955–1958. [Google Scholar] [CrossRef]
- Mulders, J.J.L.; Belova, L.M.; Riazanova, A. Electron Beam Induced Deposition at Elevated Temperatures: Compositional Changes and Purity Improvement. Nanotechnology 2010, 22, 55302. [Google Scholar] [CrossRef]
- Schmidbaur, H.; Shiotani, A. Organogold-Chemie, VI Darstellung Komplexer Organogold-Verbindungen Durch Liganden-Substitutionsreaktionen. Chem. Ber. 1971, 104, 2821–2830. [Google Scholar] [CrossRef]
- Vollnhals, F. Exploring Electron Beam Induced Surface Activation for the Fabrication of Well-Defined Nanostructures: On the Role of Catalytic Processes, Substrates and Precursors. Master’s Thesis, Friedrich-Alexander-Universität Erlangen-Nürnberg, Erlangen, Germany, 2015. [Google Scholar]
- Friedli, V.; Utke, I. Optimized Molecule Supply from Nozzle-Based Gas Injection Systems for Focused Electron- and Ion-Beam Induced Deposition and Etching: Simulation and Experiment. J. Phys. D. Appl. Phys. 2009, 42, 125305. [Google Scholar] [CrossRef]
- Bjarnason, E.H.; Ómarsson, B.; Engmann, S.; Ómarsson, F.H.; Ingólfsson, O. Dissociative Electron Attachment to Titatinum Tetrachloride and Titanium Tetraisopropoxide. Eur. Phys. J. D 2014, 68, 121. [Google Scholar] [CrossRef]
- Wetzel, R.C.; Baiocchi, F.A.; Hayes, T.R.; Freund, R.S. Absolute Cross Sections for Electron-Impact Ionization of the Rare-Gas Atoms by the Fast-Neutral-Beam Method. Phys. Rev. A 1987, 35, 559–577. [Google Scholar] [CrossRef]
- Fiegele, T.; Hanel, G.; Torres, I.; Lezius, M.; Märk, T.D. Threshold Electron Impact Ionization of Carbon Tetrafluoride, Trifluoromethane, Methane and Propane. J. Phys. B At. Mol. Opt. Phys. 2000, 33, 4263–4283. [Google Scholar] [CrossRef]
- Neese, F. Software Update: The ORCA Program System, Version 4.0. WIREs Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Kepp, K.P. Benchmarking Density Functionals for Chemical Bonds of Gold. J. Phys. Chem. A 2017, 121, 2022–2034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goel, S.; A. Velizhanin, K.; Piryatinski, A.; Tretiak, S.; Ivanov, S.A. DFT Study of Ligand Binding to Small Gold Clusters Satyende. J. Phys. Chem. Lett. 2010, 1, 927–931. [Google Scholar] [CrossRef]
- Riplinger, C.; Neese, F. An Efficient and near Linear Scaling Pair Natural Orbital Based Local Coupled Cluster Method. J. Chem. Phys. 2013, 138, 34106. [Google Scholar] [CrossRef] [PubMed]
- Riplinger, C.; Sandhoefer, B.; Hansen, A.; Neese, F. Natural Triple Excitations in Local Coupled Cluster Calculations with Pair Natural Orbitals. J. Chem. Phys. 2013, 139, 134101. [Google Scholar] [CrossRef]
- Riplinger, C.; Pinski, P.; Becker, U.; Valeev, E.F.; Neese, F. Sparse Maps—A Systematic Infrastructure for Reduced-Scaling Electronic Structure Methods. II. Linear Scaling Domain Based Pair Natural Orbital Coupled Cluster Theory. J. Chem. Phys. 2016, 144, 24109. [Google Scholar] [CrossRef]
- Griffiths, M.B.E.; Dubrawski, Z.S.; Bačić, G.; Japahuge, A.; Masuda, J.D.; Zeng, T.; Barry, S.T. Controlling the Thermal Stability and Volatility of Organogold(I) Compounds for Vapor Deposition with Complementary Ligand Design. Eur. J. Inorg. Chem. 2019, 2019, 4927–4938. [Google Scholar] [CrossRef]
- Wallace, W.E. Mass Spectra. In NIST Chemistry WebBook, NIST Standard Reference Database Number 69; Linstrom, P.J., Mallard, W.G., Eds.; National Institute of Standards and Technology: Gaithersburg, MD, USA, 2021; p. 20899. [Google Scholar] [CrossRef]
- Banaszak Holl, M.M.; Seidler, P.F.; Kowalczyk, S.P.; McFeely, F.R. Surface Reactivity of Alkylgold(I) Complexes: Substrate-Selective Chemical Vapor Deposition of Gold from RAuP(CH3)3 (R = CH2CH3, CH3) at Remarkably Low Temperatures. Inorg. Chem. 1994, 33, 510–517. [Google Scholar] [CrossRef]
- Schirmer, M.; Walz, M.M.; Vollnhals, F.; Lukasczyk, T.; Sandmann, A.; Chen, C.; Steinrück, H.P.; Marbach, H. Electron-Beam-Induced Deposition and Post-Treatment Processes to Locally Generate Clean Titanium Oxide Nanostructures on Si(100). Nanotechnology 2011, 22, 085301. [Google Scholar] [CrossRef]
- Preischl, C.; Le, L.H.; Bilgilisoy, E.; Vollnhals, F.; Gölzhäuser, A.; Marbach, H. Controlled Electron-Induced Fabrication of Metallic Nanostructures on 1 Nm Thick Membranes. Small 2020, 16, 2003947. [Google Scholar] [CrossRef]
- van Staden, M.J.; Roux, J.P. The Superposition of Carbon and Ruthenium Auger Spectra. Appl. Surf. Sci. 1990, 44, 259–262. [Google Scholar] [CrossRef]
- Davis, L.E.; MacDonald, N.C.; Palmberg, P.W.; Riach, G.E.; Weber, R.E. Handbook of Auger Electron Spectroscopy; Perkin·Elmer Corporation, Physical Electronics Division: Eden Prairie, MN, USA, 1976. [Google Scholar]
- Mathieu, H.J. Auger Electron Spectroscopy. In Surface Analysis—The Principal Techniques; Vickerman, J.C., Gilmore, I.S., Eds.; Wiley Online Books; John Wiley & Sons: Hoboken, NJ, USA, 2009; pp. 9–45. [Google Scholar]
- van Dorp, W.F.; Hagen, C.W. A Critical Literature Review of Focused Electron Beam Induced Deposition. J. Appl. Phys. 2008, 104, 81301. [Google Scholar] [CrossRef] [Green Version]
- Böhler, E.; Warneke, J.; Swiderek, P. Control of Chemical Reactions and Synthesis by Low-Energy Electrons. Chem. Soc. Rev. 2013, 42, 9219–9231. [Google Scholar] [CrossRef] [PubMed]
- Bodi, A.; Kercher, J.P.; Baer, T.; Sztáray, B. On the Parallel Mechanism of the Dissociation of Energy-Selected P(CH3)3+ Ions. J. Phys. Chem. B 2005, 109, 8393–8399. [Google Scholar] [CrossRef]
- Mahgoub, A.; Lu, H.; Thorman, R.M.; Preradovic, K.; Jurca, T.; McElwee-White, L.; Fairbrother, H.; Hagen, C.W. Electron Beam-Induced Deposition of Platinum from Pt(CO)2Cl2 and Pt(CO)2Br2. Beilstein J. Nanotechnol. 2020, 11, 1789–1800. [Google Scholar] [CrossRef] [PubMed]
- Warneke, J.; Rohdenburg, M.; Zhang, Y.; Orszagh, J.; Vaz, A.; Utke, I.; De Hosson, J.T.M.; van Dorp, W.F.; Swiderek, P. Role of NH3 in the Electron-Induced Reactions of Adsorbed and Solid Cisplatin. J. Phys. Chem. C 2016, 120, 4112–4120. [Google Scholar] [CrossRef]
- Rohdenburg, M.; Boeckers, H.; Brewer, C.R.; McElwee-White, L.; Swiderek, P. Efficient NH3-Based Process to Remove Chlorine from Electron Beam Deposited Ruthenium Produced from (η3-C3H5)Ru(CO)3Cl. Sci. Rep. 2020, 10, 10901. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| m/z | Product | TPSS-TZVP | DLPNO-CCSD-QZVPP | AE (eV) |
|---|---|---|---|---|
| 288 | [(CH3)AuP(CH3)3]+ | 7.45 | 7.58 | 7.5 ± 0.2 |
| 273 | [AuP(CH3)3]+ + (CH3) | 8.59 | 8.38 | 8.1 ± 0.2 |
| [(CH3)AuP(CH3)2]+ + (CH3) | 10.20 | 10.44 | ||
| 76 | [P(CH3)3]+ + (CH3)Au | 9.38 | 9.17 | 8.6 ± 0.2 |
| [HP(CH3)2(CH2)]+ + (CH3)Au | 9.73 | 9.61 | ||
| [P(CH3)3]+ + (CH3) + Au | 12.04 | 11.54 | ||
| 75 | [P(CH3)2(CH2)]+ + (CH3)AuH | 10.64 | 10.68 | 10.5 ± 0.2 |
| [P(CH3)2(CH2)]+ + (CH3)Au + H | 12.12 | 11.86 | ||
| [HP(CH3)(CH2)2]+ + (CH3)AuH | 12.62 | 12.90 | ||
| 61 | [HP(CH3)(CH2)]+ + (CH3)2Au | 11.16 | 11.20 | 11.1 ± 0.2 |
| [HP(CH3)(CH2)]+ + C2H6 + Au | 11.22 | 10.43 | ||
| [P(CH3)2]+ + (CH3)2Au | 11.64 | 11.62 | ||
| 59 | [P(CH2)2]+ + (CH3)2Au + H2 | 13.46 | 13.45 | 13.4 ± 0.3 |
| [P(CH2)2]+ + (CH3)AuH + CH4 | 13.18 | 12.93 | ||
| [P(CH2)2]+ + H2Au + C2H6 | 13.56 | 13.07 | ||
| [HP(CH2)CH]+ + (CH3)2Au + H2 | 15.22 | 15.61 | ||
| 45 | [PCH2]+ + (CH3)AuH + 2(CH3) | 17.50 | 17.04 | 13.6 ± 0.4 |
| [PCH2]+ + (CH3)AuH + C2H6 | 13.84 | 13.20 | ||
| [PCH2]+ + Au(CH3) + C2H6 + H | 15.32 | 14.38 | ||
| [PCH2]+ + (CH3)Au(CH3) + CH4 | 13.50 | 13.06 | ||
| [PCH2]+ + Au + (CH3) + C2H6 + H | 17.98 | 16.76 | ||
| [HPCH]+ + (CH3)AuH + C2H6 | 16.19 | 15.97 | ||
| [PCH2]+ + AuH + (CH3) + C2H6 | 14.85 | 13.75 | ||
| [HPCH]+ + AuH + (CH3) + C2H6 | 17.19 | 16.52 |
| Fragment | m/z | Relative DI Yield (Intensity) | Relative DI Yield (Integration) | |
|---|---|---|---|---|
| [AuP(CH3)3]+ | 273 | 78.41 | 79.07 | |
| [AuP(CH3)2]+ | 258 | 5.64 | 13.30 | |
| [AuP(CH3)]+ | 243 | 2.36 | 0.36 | |
| [Au]+ | 197 | 0.51 | 0.29 | |
| [P(CH3)3]+ | 76 | 64.78 | 78.98 | |
| [P(CH3)3 − H] | 75 | 33.12 | 28.47 | |
| [HP(CH3)(CH2)]+ | 61 | 100 | 100 | |
| [P(CH3)2 − 2H]+ | 59 | 68.8 | 68 | |
| [P(CH3)2 − 4H]+ | 57 | 26.19 | 17.98 | |
| [P(CH3)]+ | 46 | 14.1 | 2.54 | |
| [P(CH3) − H]+ | 45 | 33.7 | 24.7 | |
| Avrg. C loss per incident | 1.94 | 2.01 | ||
| Avrg. P loss per incident | 0.80 | 0.76 | ||
| UHV deposit composition | 31–34 at.% Au | 65–67 at.% C | 1–2 at.% P | |
| HV deposit composition | 19–25 at.% Au | 54–62 at.% C | 12–16 at.% P | 2–7 at.% O |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kamali, A.; Bilgilisoy, E.; Wolfram, A.; Gentner, T.X.; Ballmann, G.; Harder, S.; Marbach, H.; Ingólfsson, O. On the Electron-Induced Reactions of (CH3)AuP(CH3)3: A Combined UHV Surface Science and Gas-Phase Study. Nanomaterials 2022, 12, 2727. https://doi.org/10.3390/nano12152727
Kamali A, Bilgilisoy E, Wolfram A, Gentner TX, Ballmann G, Harder S, Marbach H, Ingólfsson O. On the Electron-Induced Reactions of (CH3)AuP(CH3)3: A Combined UHV Surface Science and Gas-Phase Study. Nanomaterials. 2022; 12(15):2727. https://doi.org/10.3390/nano12152727
Chicago/Turabian StyleKamali, Ali, Elif Bilgilisoy, Alexander Wolfram, Thomas Xaver Gentner, Gerd Ballmann, Sjoerd Harder, Hubertus Marbach, and Oddur Ingólfsson. 2022. "On the Electron-Induced Reactions of (CH3)AuP(CH3)3: A Combined UHV Surface Science and Gas-Phase Study" Nanomaterials 12, no. 15: 2727. https://doi.org/10.3390/nano12152727
APA StyleKamali, A., Bilgilisoy, E., Wolfram, A., Gentner, T. X., Ballmann, G., Harder, S., Marbach, H., & Ingólfsson, O. (2022). On the Electron-Induced Reactions of (CH3)AuP(CH3)3: A Combined UHV Surface Science and Gas-Phase Study. Nanomaterials, 12(15), 2727. https://doi.org/10.3390/nano12152727