
Stability and Bandgap Engineering of In1−xGaxSe Monolayer

 ,
,  , and
, and 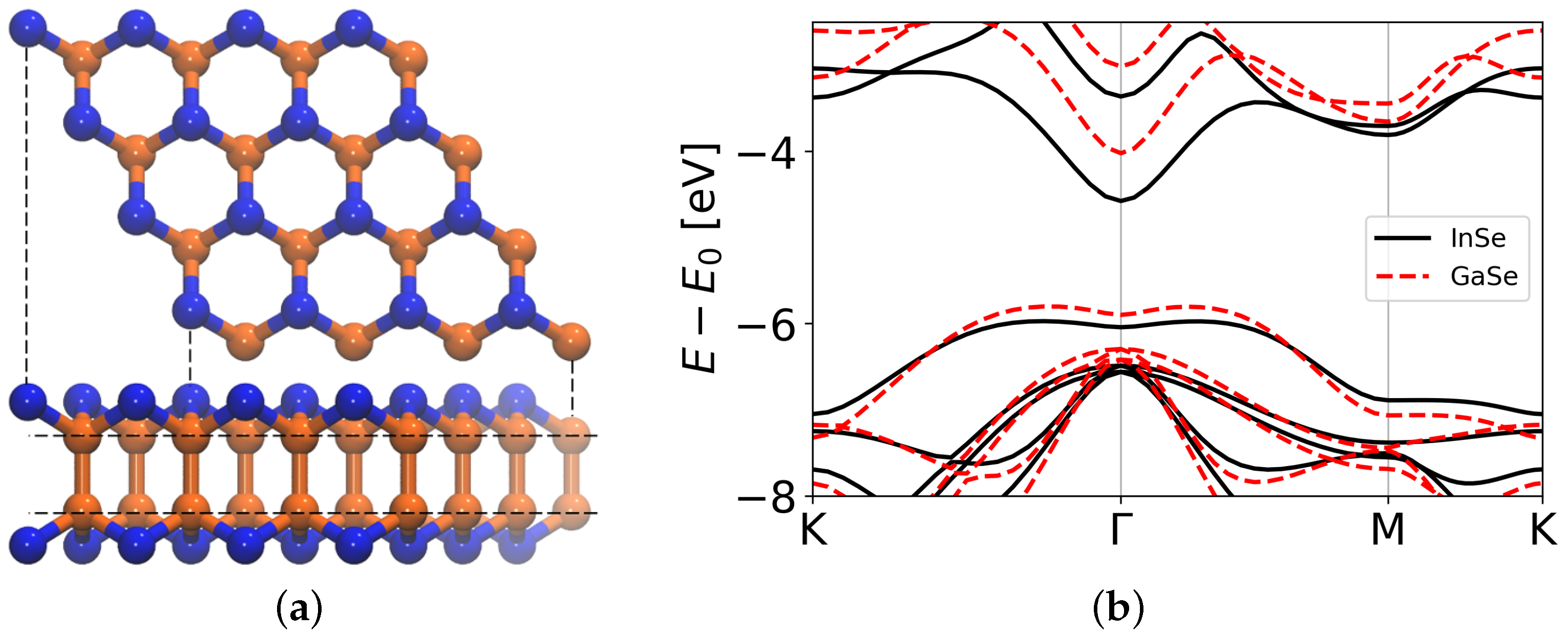{kind=link}
{kind=link}
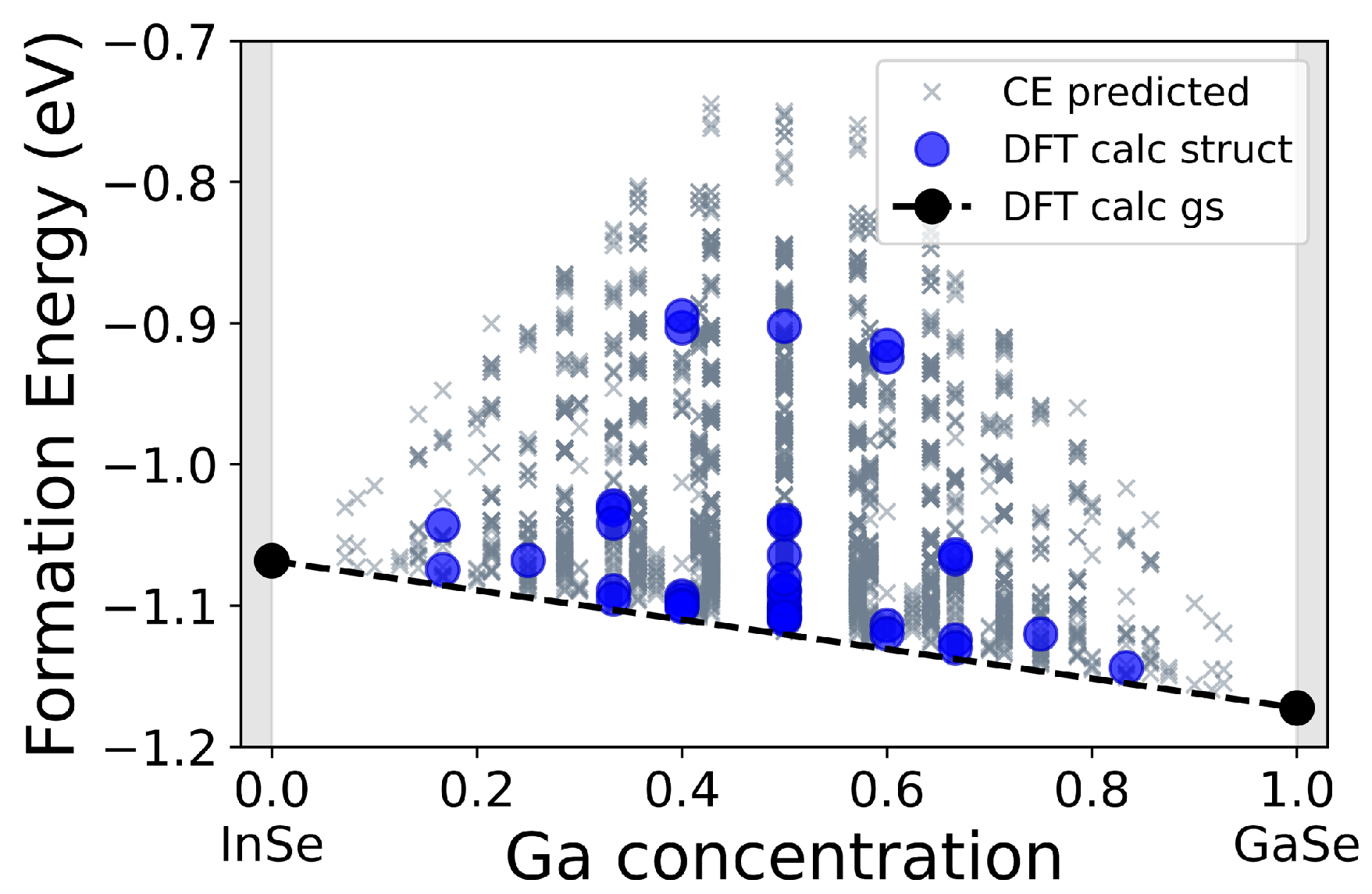
{kind=link}
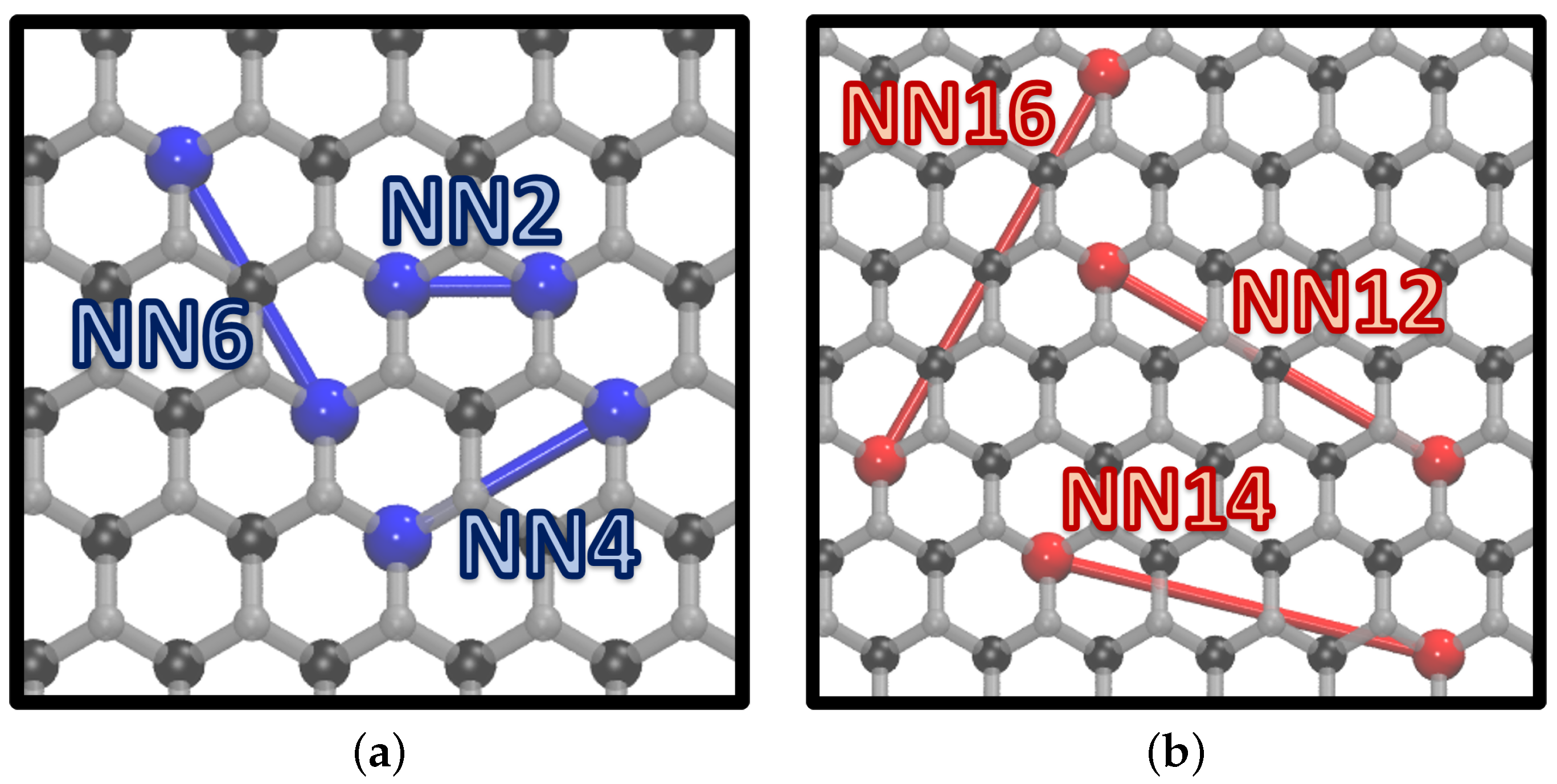
{kind=link}
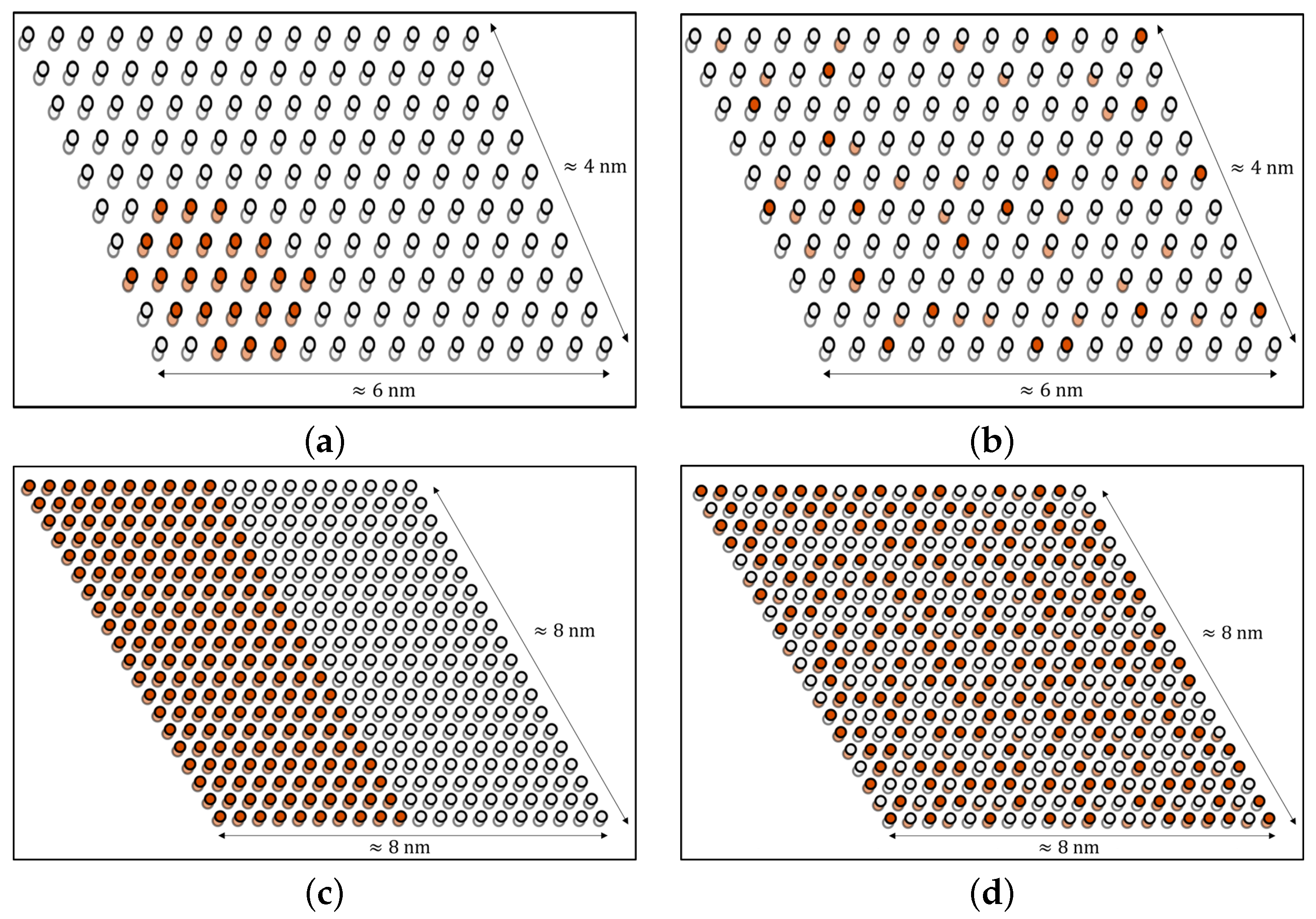
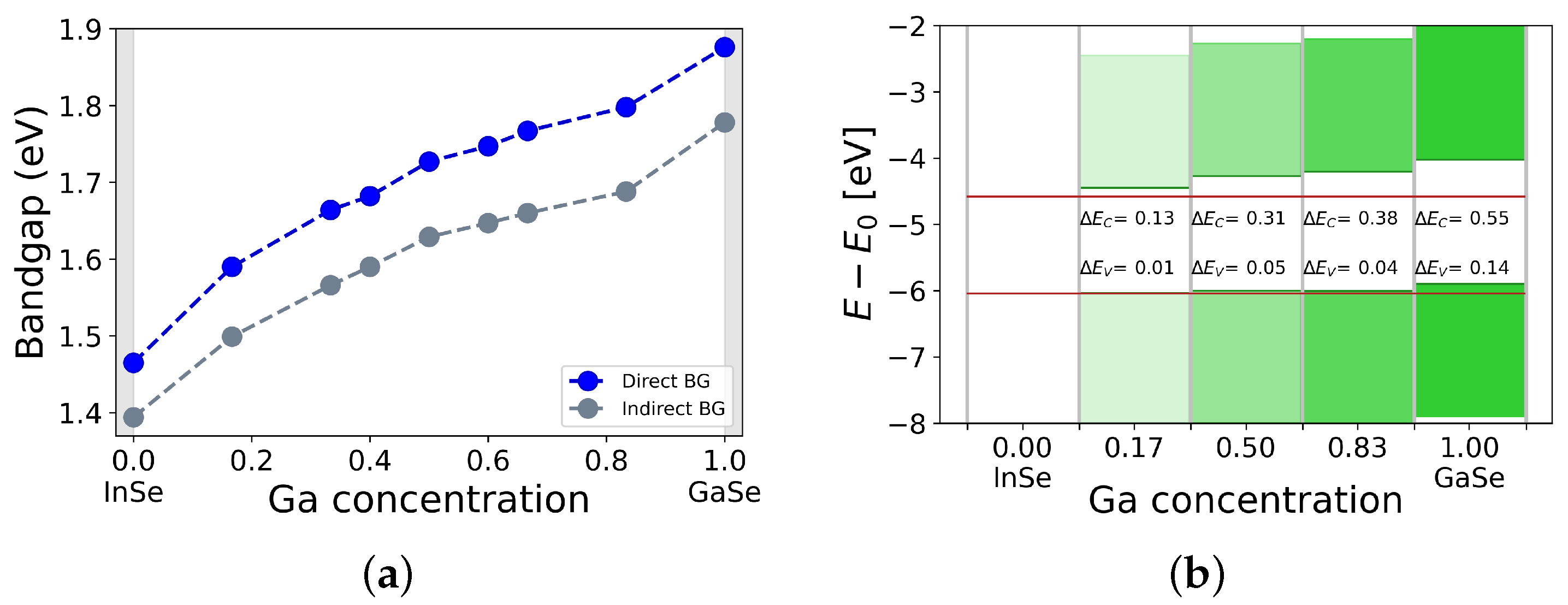{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hu, H.; Tang, B.; Wan, H.; Sun, H.; Zhou, S.; Dai, J.; Chen, C.; Liu, S.; Guo, L.J. Boosted ultraviolet electroluminescence of InGaN/AlGaN quantum structures grown on high-index contrast patterned sapphire with silica array. Nano Energy 2020, 69, 104427. [Google Scholar] [CrossRef]
- Zhao, X.; Tang, B.; Gong, L.; Bai, J.; Ping, J.; Zhou, S. Rational construction of staggered InGaN quantum wells for efficient yellow light-emitting diodes. Appl. Phys. Lett. 2021, 118, 182102. [Google Scholar] [CrossRef]
- Zhou, S.; Liu, X.; Yan, H.; Chen, Z.; Liu, Y.; Liu, S. Highly efficient GaN-based high-power flipchip light-emitting diodes. Opt. Express 2019, 27, A669–A692. [Google Scholar] [CrossRef] [PubMed]
- Chitraleema, C.; Nick, V.; Dirk, E. Advances in quantum light emission from 2D materials. Nanophotonics 2019, 8, 2017–2032. [Google Scholar] [CrossRef]
- Lodahl, P.; Mahmoodian, S.; Stobbe, S. Interfacing single photons and single quantum dots with photonic nanostructures. Rev. Mod. Phys. 2015, 87, 347–400. [Google Scholar] [CrossRef] [Green Version]
- Aspuru-Guzik, A.; Walther, P. Photonic quantum simulators. Nat. Phys. 2012, 8, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Lo, H.; Curty, M.; Tamaki, K. Secure quantum key distribution. Nat. Photonics 2014, 8, 595–604. [Google Scholar] [CrossRef] [Green Version]
- Xia, F.; Wang, H.; Xiao, D.; Dubey, M.; Ramasubramaniam, A. Two-dimensional material nanophotonics. Nat. Photonics 2014, 8, 899–907. [Google Scholar] [CrossRef]
- Gan, X.; Shiue, R.J.; Gao, Y.; Meric, I.; Heinz, T.F.; Shepard, K.; Hone, J.; Assefa, S.; Englund, D. Chip-integrated ultrafast graphene photodetector with high responsivity. Nat. Photonics 2013, 7, 883–887. [Google Scholar] [CrossRef]
- Furchi, M.; Urich, A.; Pospischil, A.; Lilley, G.; Unterrainer, K.; Detz, H.; Klang, P.; Andrews, A.M.; Schrenk, W.; Strasser, G.; et al. Microcavity-integrated graphene photodetector. Nano Lett. 2012, 12, 2773–2777. [Google Scholar] [CrossRef]
- Gan, X.; Mak, K.F.; Gao, Y.; You, Y.; Hatami, F.; Hone, J.; Heinz, T.F.; Englund, D. Strong enhancement of light-matter interaction in graphene coupled to a photonic crystal nanocavity. Nano Lett. 2012, 12, 5626–5631. [Google Scholar] [CrossRef]
- Dean, C.R.; Young, A.F.; Meric, I.; Lee, C.; Wang, L.; Sorgenfrei, S.; Watanabe, K.; Taniguchi, T.; Kim, P.; Shepard, K.L.; et al. Boron nitride substrates for high-quality graphene electronics. Nat. Nanotechnol. 2010, 5, 722–726. [Google Scholar] [CrossRef]
- Mak, K.F.; Lee, C.; Hone, J.; Shan, J.; Heinz, T.F. Atomically thin MoS2: A New Direct-Gap Semiconductor. Phys. Rev. Lett. 2010, 105, 136805. [Google Scholar] [CrossRef] [Green Version]
- Eda, G.; Maier, S.A. Two-dimensional crystals: Managing light for optoelectronics. ACS Nano 2013, 7, 5660–5665. [Google Scholar] [CrossRef] [PubMed]
- Gul, H.Z.; Sakong, W.; Ji, H.; Torres, J.; Yi, H.; Ghimire, M.K.; Yoon, J.H.; Yun, M.H.; Hwang, H.R.; Lee, Y.H.; et al. Semimetallic Graphene for Infrared Sensing. ACS Appl. Mater. Interfaces 2019, 11, 19565–19571. [Google Scholar] [CrossRef]
- Yang, Z.; Jie, W.; Mak, C.H.; Lin, S.; Lin, H.; Yang, X.; Yan, F.; Lau, S.P.; Hao, J. Wafer-Scale Synthesis of High-Quality Semiconducting Two-Dimensional Layered InSe with Broadband Photoresponse. ACS Nano 2017, 11, 4225–4236. [Google Scholar] [CrossRef]
- Lei, S.; Ge, L.; Najmaei, S.; George, A.; Kappera, R.; Lou, J.; Chhowalla, M.; Yamaguchi, H.; Gupta, G.; Vajtai, R.; et al. Evolution of the Electronic Band Structure and Efficient Photo-Detection in Atomic Layers of InSe. ACS Nano 2014, 8, 1263–1272. [Google Scholar] [CrossRef] [PubMed]
- Bandurin, D.A.; Tyurnina, A.V.; Geliang, L.Y.; Mishchenko, A.; Zólyomi, V.; Morozov, S.V.; Kumar, R.K.; Gorbachev, R.V.; Kudrynskyi, Z.R.; Pezzini, S.; et al. High electron mobility, quantum Hall effect and anomalous optical response in atomically thin InSe. Nat. Nanotechnol. 2016, 12, 223–227. [Google Scholar] [CrossRef]
- Susarla, S.; Kutana, A.; Hachtel, J.A.; Kochat, V.; Apte, A.; Vajtai, R.; Idrobo, J.C.; Yakobson, B.I.; Tiwary, C.S.; Ajayan, P.M. Quaternary 2D transition metal dichalcogenides (TMDS) with tunable bandgap. Adv. Mater. 2017, 29, 1702457. [Google Scholar] [CrossRef] [PubMed]
- Raffone, F.; Ataca, C.; Grossman, J.C.; Cicero, G. MoS2 Enhanced T-Phase Stabilization and Tunability through Alloying. J. Phys. Chem. Lett. 2016, 7, 2304–2309. [Google Scholar] [CrossRef]
- Verma, A.K.; Raffone, F.; Cicero, G. Prediction of the structural and electronic properties of MoxTi1-xS2 monolayers via first principle simulations. Nanomater. Nanotechnol. 2020, 10, 1–6. [Google Scholar] [CrossRef]
- Salomone, M.; Fiorentin, M.R.; Cicero, G.; Risplendi, F. Point Defects in Two-Dimensional Indium Selenide as Tunable Single-Photon Sources. J. Phys. Chem. Lett. 2021, 12, 10947–10952. [Google Scholar] [CrossRef]
- Eisaman, M.D.; Fan, J.; Migdall, A.; Polyakov, S.V. Invited Review Article: Single-photon sources and detectors. Rev. Sci. Instrum. 2011, 82, 071101. [Google Scholar] [CrossRef] [Green Version]
- Darquie, B. Controlled Single-Photon Emission from a Single Trapped Two-Level Atom. Science 2005, 309, 454–456. [Google Scholar] [CrossRef] [Green Version]
- Tonndorf, P.; Schwarz, S.; Kern, J.; Niehues, I.; Pozo-Zamudio, O.D.; Dmitriev, A.I.; Bakhtinov, A.P.; Borisenko, D.N.; Kolesnikov, N.N.; Tartakovskii, A.I.; et al. Single-photon emitters in GaSe. 2D Mater. 2017, 4, 021010. [Google Scholar] [CrossRef]
- Zhou, J.; Shi, J.; Zeng, Q.; Chen, Y.; Niu, L.; Liu, F.; Yu, T.; Suenaga, K.; Liu, X.; Lin, J.; et al. InSe monolayer: Synthesis, structure and ultra-high second-harmonic generation. 2D Mater. 2018, 5, 025019. [Google Scholar] [CrossRef]
- Zhou, X.; Cheng, J.; Zhou, Y.; Cao, T.; Hong, H.; Liao, Z.; Wu, S.; Peng, H.; Liu, K.; Yu, D. Strong Second-Harmonic Generation in Atomic Layered GaSe. J. Am. Chem. Soc. 2015, 137, 7994–7997. [Google Scholar] [CrossRef]
- Balakrishnan, N.; Kudrynskyi, Z.R.; Smith, E.F.; Fay, M.W.; Makarovsky, O.; Kovalyuk, Z.D.; Eaves, L.; Beton, P.H.; Patanè, A. Engineering p – n junctions and bandgap tuning of InSe nanolayers by controlled oxidation. 2D Mater. 2017, 4, 025043. [Google Scholar] [CrossRef]
- Li, Y.; Wang, T.; Wu, M.; Cao, T.; Chen, Y.; Sankar, R.; Ulaganathan, R.K.; Chou, F.; Wetzel, C.; Xu, C.Y.; et al. Ultrasensitive tunability of the direct bandgap of 2D InSe flakes via strain engineering. 2D Mater. 2018, 5, 021002. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; He, B.; Song, T.; Gao, J.; Shi, S. Cluster expansion method and its application in computational materials science. Comput. Mater. Sci. 2016, 125, 243–254. [Google Scholar] [CrossRef]
- Raffone, F.; Savazzi, F.; Cicero, G. Controlled Pore Generation in Single-Layer Graphene Oxide for Membrane Desalination. J. Phys. Chem. Lett. 2019, 10, 7492–7497. [Google Scholar] [CrossRef]
- Connolly, J.W.D.; Williams, A.R. Density-functional theory applied to phase transformations in transition-metal alloys. Phys. Rev. B Condens. Matter Mater. Phys. 1983, 27, 5169. [Google Scholar] [CrossRef]
- van de Walle, A.; Ceder, G. Automating first-principles phase diagram calculations. J. Phase Equilib. 2002, 23, 348. [Google Scholar] [CrossRef] [Green Version]
- van de Walle, A.; Asta, M.; Ceder, G. The alloy theoretic automated toolkit: A user guide. Calphad 2002, 26, 539–553. [Google Scholar] [CrossRef] [Green Version]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys.-Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef]
- Giannozzi, P.; Andreussi, O.; Brumme, T.; Bunau, O.; Nardelli, M.B.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Cococcioni, M.; et al. Advanced capabilities for materials modelling with quantum espresso. J. Phys.-Condens. Matter 2017, 29, 465901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlipf, M.; Gygi, F. Optimization algorithm for the generation of ONCV pseudopotentials. Comput. Phys. Commun. 2015, 196, 36–44. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.; Burke, K.; Wang, Y. Generalized gradient approximation for the echange-correlation hole of many electron system. Phys. Rev. B 1996, 54, 16533–16539. [Google Scholar] [CrossRef] [Green Version]
- Monkhorst, H.J.; Pack, J.D. Special points for brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]
- Magorrian, S.J.; Zolyomi, V.; Fal’ko, V.I. Electronic and optical properties of two-dimensional InSe from a DFT-parametrized tight-binding model. Phys. Rev. B 2016, 94, 245431. [Google Scholar] [CrossRef] [Green Version]
- Ghalouci, L.; Benbahi, B.; Hiadsi, S.; Abidri, B.; Vergoten, G.; Ghalouci, F. First principle investigation into hexagonal and cubic structures of Gallium Selenide. Comput. Mater. Sci. 2013, 67, 73–82. [Google Scholar] [CrossRef]
- Jansen, A.P.J. An Introduction to Kinetic Monte Carlo Simulations of Surface Reactions; Springer: Berlin, Germany, 2012; p. 856. [Google Scholar]
- Raffone, F.; Cicero, G. Unveiling the fundamental role of temperature in RRAM switching mechanism by multiscale simulations. ACS Appl. Mater. Interfaces 2018, 10, 7512–7519. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J.; D’Avezac, M.; Hetherington, J.; Stamatakis, M. Parallel kinetic Monte Carlo simulation framework incorporating accurate models of adsorbate lateral interactions. J. Chem. Phys. 2013, 224706, 139. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, M.; Vlachos, D.G. A graph-theoretical kinetic Monte Carlo framework for on-lattice chemical kinetics. J. Chem. Phys. 2011, 134, 214115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Han, X.; Pan, D.; Yan, X.; Wang, H.W.; Wu, C.; Cheng, G.; Zhang, H.; Yang, S.; Li, B.; et al. Bandgap engineering of InSe Single Crystals through S substitution. Cryst. Growth Des. 2018, 18, 2899–2904. [Google Scholar] [CrossRef]
- Ma, Y.; Dai, Y.; Yu, L.; Liu, C.; Huang, B. Engineering a topological phase transition in β-InSe via strain. New J. Phys. 2013, 15, 073008. [Google Scholar] [CrossRef]
- Zolyomi, V.; Drummond, N.D.; Falko, V.I. Electrons and phonons in single layers of hexagonal indium chalcogenides from ab initio calculations. Phys. Rev. B Condens. Matter Mater. Phys. 2014, 89, 205416. [Google Scholar] [CrossRef] [Green Version]
- Nahory, R.E.; Pollack, M.A.; Johnston, W.D., Jr.; Barns, R.L. Band gap versus composition and demonstration of Vegard’s law for In1−xGaxAsyP1−y lattice matched to InP. Appl. Phys. Lett. 2008, 33, 659–661. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salomone, M.; Raffone, F.; Re Fiorentin, M.; Risplendi, F.; Cicero, G. Stability and Bandgap Engineering of In1−xGaxSe Monolayer. Nanomaterials 2022, 12, 515. https://doi.org/10.3390/nano12030515
Salomone M, Raffone F, Re Fiorentin M, Risplendi F, Cicero G. Stability and Bandgap Engineering of In1−xGaxSe Monolayer. Nanomaterials. 2022; 12(3):515. https://doi.org/10.3390/nano12030515
Chicago/Turabian StyleSalomone, Mattia, Federico Raffone, Michele Re Fiorentin, Francesca Risplendi, and Giancarlo Cicero. 2022. "Stability and Bandgap Engineering of In1−xGaxSe Monolayer" Nanomaterials 12, no. 3: 515. https://doi.org/10.3390/nano12030515
APA StyleSalomone, M., Raffone, F., Re Fiorentin, M., Risplendi, F., & Cicero, G. (2022). Stability and Bandgap Engineering of In1−xGaxSe Monolayer. Nanomaterials, 12(3), 515. https://doi.org/10.3390/nano12030515