
Role of Surface Energy of Nanoparticle Stabilizers in the Synthesis of Microspheres via Pickering Emulsion Polymerization


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of Silica Nanoparticles
2.3. Surface Modification of Silica NPs with (3-Glycidoxypropyl)trimethoxysilane (NPs-Gly)
2.4. Pickering Emulsion Preparation and Polymerization
2.5. Measurement of the Contact Angles for NP-Gly
3. Results
3.1. Pickering Emulsion Formation and Polymerization
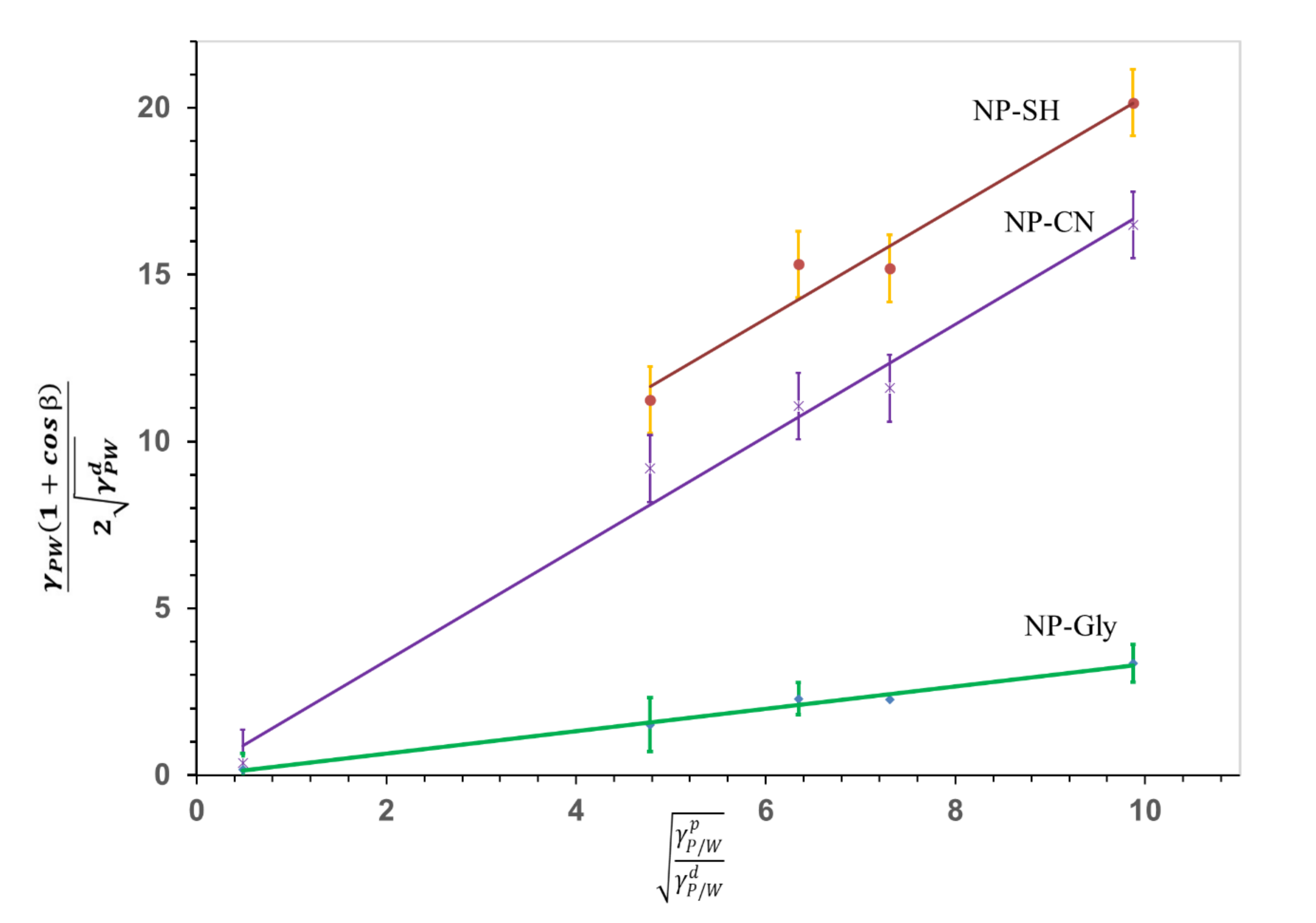
3.2. Determining the Interfacial and Surface Energy of NPs
4. Discussion
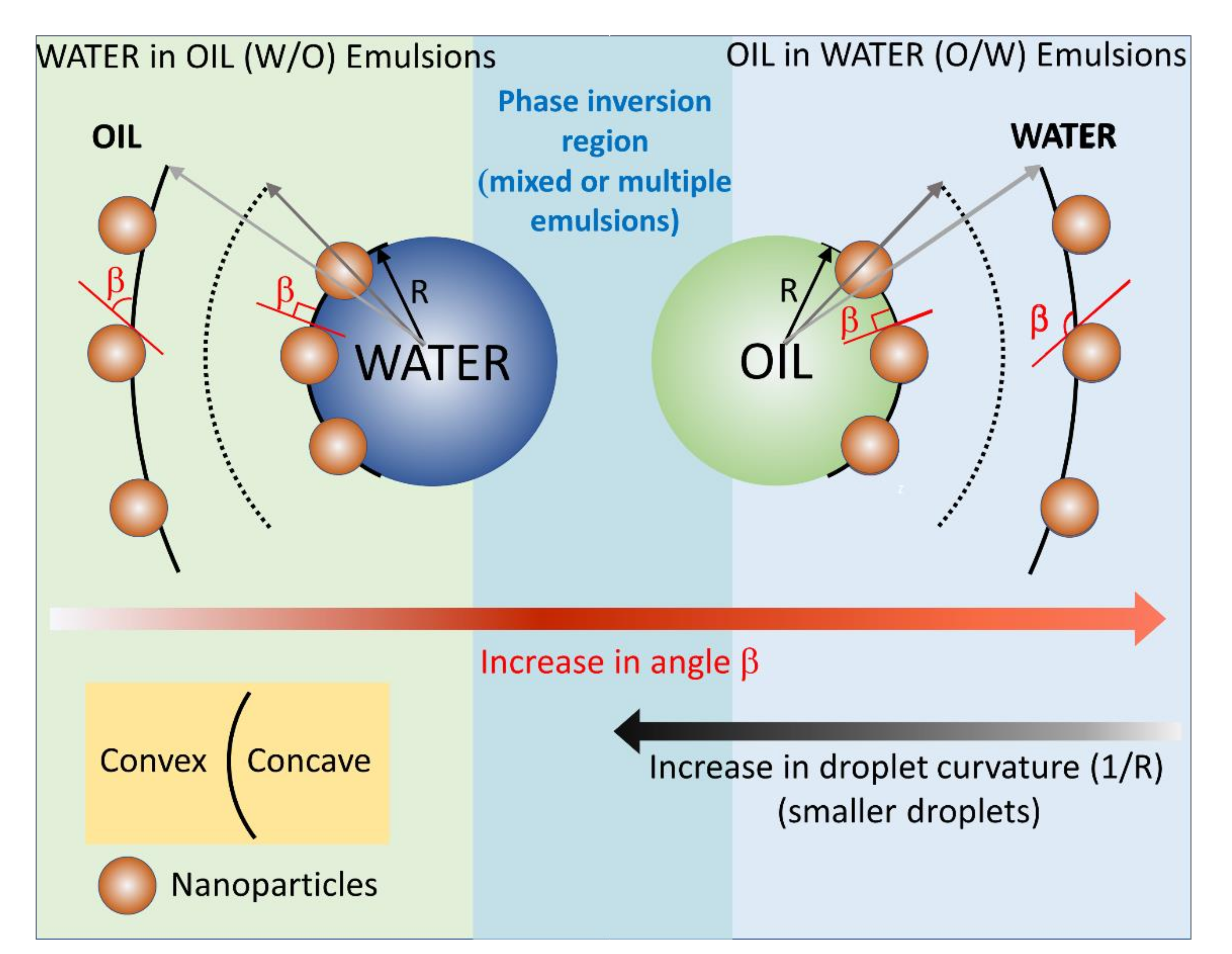
4.1. Role of Interfaces in Pickering Emulsion Polymerization
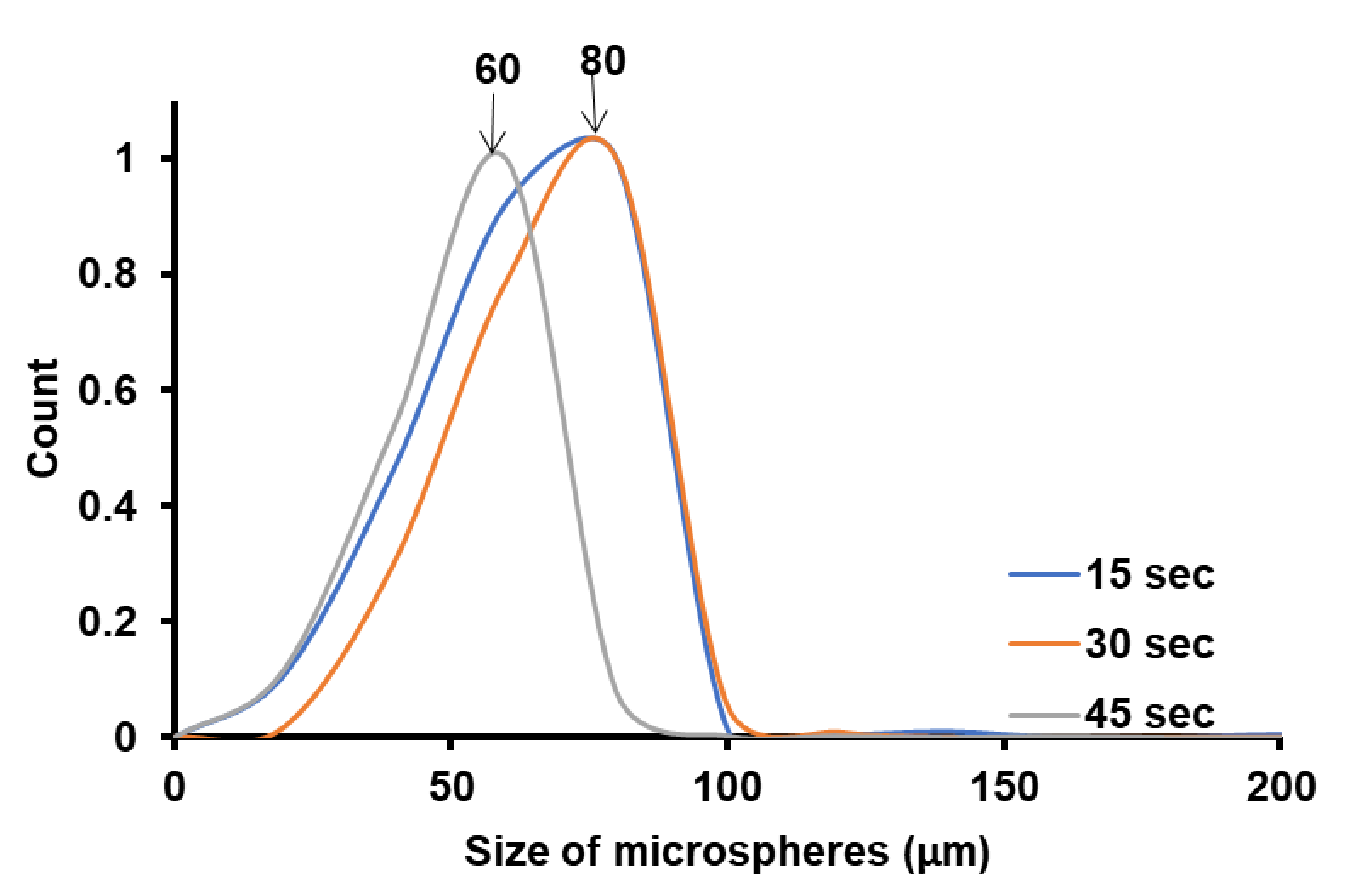
4.2. Microsphere Size Function of the Pickering Emulsions Preparation Conditions
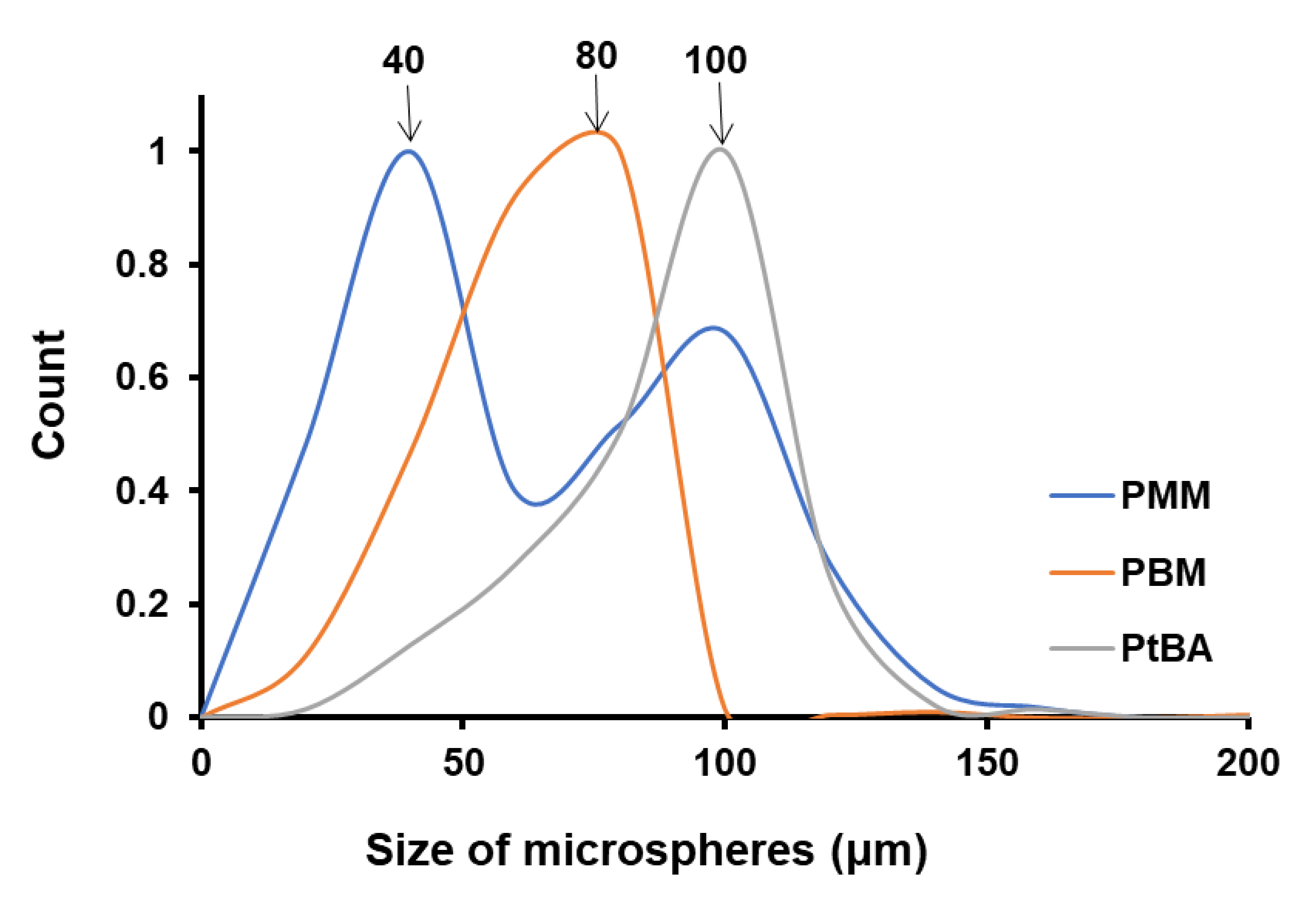
4.3. Size of the Microspheres Function of Monomer Type for the Same NP-Gly
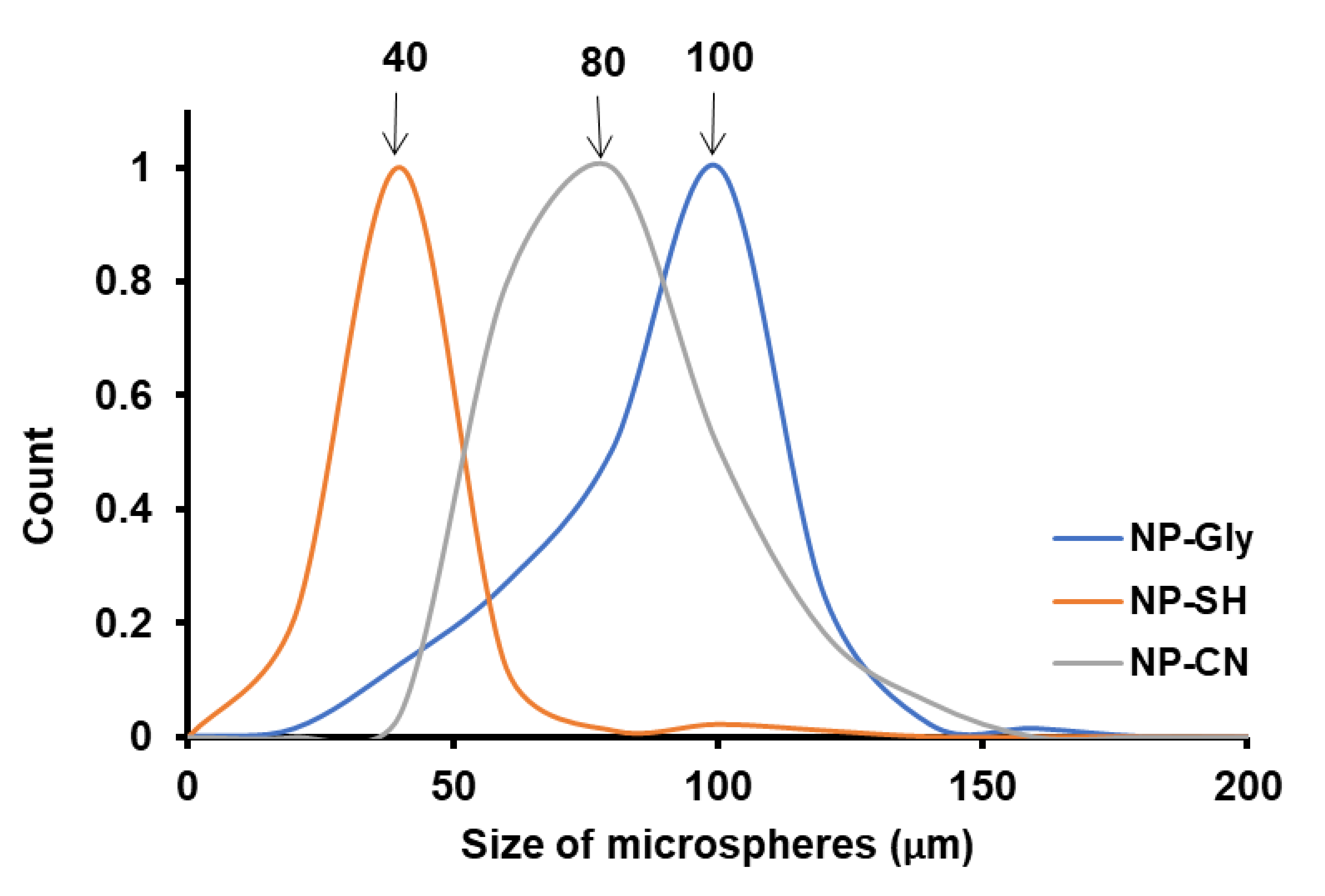
4.4. Size of Microspheres Function of NP Type for the Same Monomer
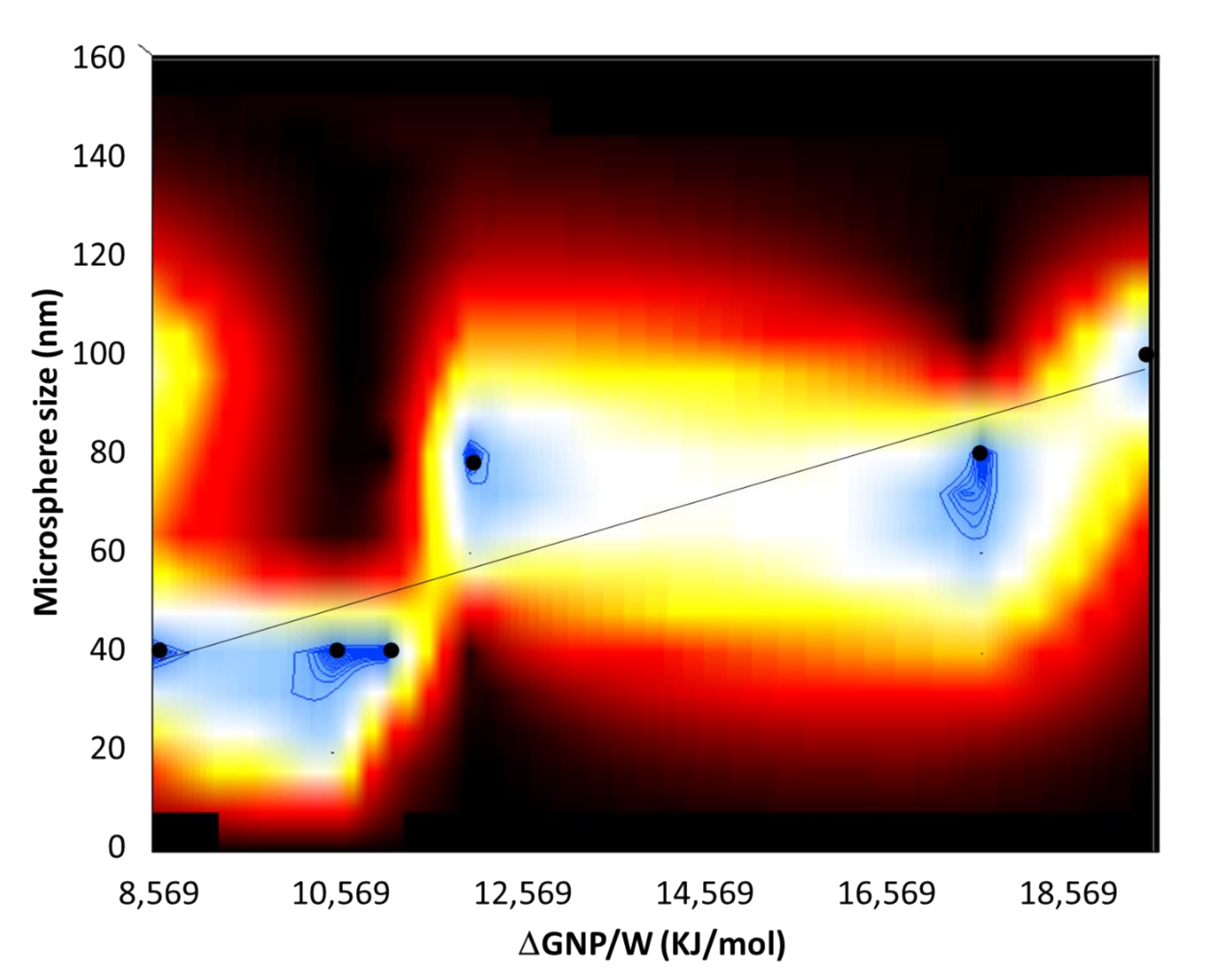
4.5. Energy of Attachment of NPs at Interface vs. Size of the Microspheres
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Yang, Y.; Fang, Z.; Chen, X.; Zhang, W.; Xie, Y.; Chen, Y.; Liu, Z.; Yuan, W. An Overview of Pickering Emulsions: Solid-Particle Materials, Classification, Morphology, and Applications. Front. Pharmacol. 2017, 8, 287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chevalier, Y.; Bolzinger, M.-A. Emulsions Stabilized with Solid Nanoparticles: Pickering Emulsions. Colloids Surf. A Physicochem. Eng. Asp. 2013, 439, 23–34. [Google Scholar] [CrossRef]
- Xie, B.; Zhang, X.; Luo, X.; Wang, Y.; Li, Y.; Li, B.; Liu, S. Edible Coating Based on Beeswax-in-Water Pickering Emulsion Stabilized by Cellulose Nanofibrils and Carboxymethyl Chitosan. Food Chem. 2020, 331, 127108. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Zhou, Z.; Yang, J.; Zhang, M.; Cai, F.; Lu, P. ZnO Nanoparticles Stabilized Oregano Essential Oil Pickering Emulsion for Functional Cellulose Nanofibrils Packaging Films with Antimicrobial and Antioxidant Activity. Int. J. Biol. Macromol. 2021, 190, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Hussein, M.A.; Mohammed, A.A.; Atiya, M.A. Application of Emulsion and Pickering Emulsion Liquid Membrane Technique for Wastewater Treatment: An Overview. Environ. Sci. Pollut. Res. 2019, 26, 36184–36204. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Wang, W.; Yu, H.; Wang, A. Preparation of Porous Adsorbent via Pickering Emulsion Template for Water Treatment: A Review. J. Environ. Sci. 2020, 88, 217–236. [Google Scholar] [CrossRef] [PubMed]
- Zhai, W.; Li, G.; Yu, P.; Yang, L.; Mao, L. Silver Phosphate/Carbon Nanotube-Stabilized Pickering Emulsion for Highly Efficient Photocatalysis. J. Phys. Chem. C 2013, 117, 15183–15191. [Google Scholar] [CrossRef]
- Frelichowska, J.; Bolzinger, M.-A.; Valour, J.-P.; Mouaziz, H.; Pelletier, J.; Chevalier, Y. Pickering w/o Emulsions: Drug Release and Topical Delivery. Int. J. Pharm. 2009, 368, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Marku, D.; Wahlgren, M.; Rayner, M.; Sjöö, M.; Timgren, A. Characterization of Starch Pickering Emulsions for Potential Applications in Topical Formulations. Int. J. Pharm. 2012, 428, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Honciuc, A. Contrasting Mechanisms of Spontaneous Adsorption at Liquid-Liquid Interfaces of Nanoparticles “Constituted of” and “Grafted with” PH-Responsive Polymers. Langmuir 2018, 34, 6170–6182. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Mihali, V.; Honciuc, A. PH-Responsive Pickering Foams Generated by Surfactant-Free Soft Hydrogel Particles. Langmuir 2019, 35, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Honciuc, A. Design of Janus Nanoparticles with PH-Triggered Switchable Amphiphilicity for Interfacial Applications. ACS Appl. Nano Mater. 2018, 34, 1225–1233. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.; Chew, J.W.; Honciuc, A. Polarity Reversal in Homologous Series of Surfactant-Free Janus Nanoparticles: Toward the Next Generation of Amphiphiles. Langmuir 2016, 32, 6376–6386. [Google Scholar] [CrossRef]
- Jalilian, R.; Shahmari, M.; Taheri, A.; Gholami, K. Ultrasonic-Assisted Micro Solid Phase Extraction of Arsenic on a New Ion-Imprinted Polymer Synthesized from Chitosan-Stabilized Pickering Emulsion in Water, Rice and Vegetable Samples. Ultrason. Sonochemistry 2020, 61, 104802. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Zhu, Y.; Cui, Z.; Binks, B.P. Switchable Pickering Emulsions Stabilized by Silica Nanoparticles Hydrophobized In Situ with a Switchable Surfactant. Angew. Chem. Int. Ed. 2013, 52, 12373–12376. [Google Scholar] [CrossRef]
- Kim, I.; Worthen, A.J.; Johnston, K.P.; DiCarlo, D.A.; Huh, C. Size-Dependent Properties of Silica Nanoparticles for Pickering Stabilization of Emulsions and Foams. J. Nanopart. Res. 2016, 18, 82–93. [Google Scholar] [CrossRef]
- Ashby, N.P.; Binks, B.P. Pickering Emulsions Stabilised by Laponite Clay Particles. Phys. Chem. Chem. Phys. 2000, 2, 5640–5646. [Google Scholar] [CrossRef]
- Liu, X.; Okada, M.; Maeda, H.; Fujii, S.; Furuzono, T. Hydroxyapatite/Biodegradable Poly(l-Lactide–Co-ε-Caprolactone) Composite Microparticles as Injectable Scaffolds by a Pickering Emulsion Route. Acta Biomater. 2011, 7, 821–828. [Google Scholar] [CrossRef]
- Jin Ahn, W.; Seung Jung, H.; Jin Choi, H. Pickering Emulsion Polymerized Smart Magnetic Poly(Methyl Methacrylate)/Fe2O3 Composite Particles and Their Stimulus-Response. RSC Adv. 2015, 5, 23094–23100. [Google Scholar] [CrossRef]
- Gao, Z.-M.; Yang, X.-Q.; Wu, N.-N.; Wang, L.-J.; Wang, J.-M.; Guo, J.; Yin, S.-W. Protein-Based Pickering Emulsion and Oil Gel Prepared by Complexes of Zein Colloidal Particles and Stearate. J. Agric. Food Chem. 2014, 62, 2672–2678. [Google Scholar] [CrossRef]
- Wongkongkatep, P.; Manopwisedjaroen, K.; Tiposoth, P.; Archakunakorn, S.; Pongtharangkul, T.; Suphantharika, M.; Honda, K.; Hamachi, I.; Wongkongkatep, J. Bacteria Interface Pickering Emulsions Stabilized by Self-Assembled Bacteria–Chitosan Network. Langmuir 2012, 28, 5729–5736. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wu, Y.; Fang, R.; Lei, C.; Li, Y.; Li, B.; Pei, Y.; Luo, X. ShilinLiu Application of Nanocellulose as Particle Stabilizer in Food Pickering Emulsion: Scope, Merits and Challenges. Trends Food Sci. Technol. 2021, 110, 573–583. [Google Scholar] [CrossRef]
- Tarimala, S.; Dai, L.L. Structure of Microparticles in Solid-Stabilized Emulsions. Langmuir 2004, 20, 3492–3494. [Google Scholar] [CrossRef] [PubMed]
- Tarimala, S.; Ranabothu, S.R.; Vernetti, J.P.; Dai, L.L. Mobility and In Situ Aggregation of Charged Microparticles at Oil−Water Interfaces. Langmuir 2004, 20, 5171–5173. [Google Scholar] [CrossRef] [PubMed]
- Tarimala, S.; Wu, C.; Dai, L.L. Dynamics and Collapse of Two-Dimensional Colloidal Lattices. Langmuir 2006, 22, 7458–7461. [Google Scholar] [CrossRef] [PubMed]
- Rozynek, Z.; Kaczmarek-Klinowska, M.; Magdziarz, A. Assembly and Rearrangement of Particles Confined at a Surface of a Droplet, and Intruder Motion in Electro-Shaken Particle Films. Materials 2016, 9, 679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Wang, Q.; Dai, F.; Gu, Y.; Qian, G.; Chen, C.; Yu, Y. Novel TiO2 Nanoparticles/Polysulfone Composite Hollow Microspheres for Photocatalytic Degradation. Polymers 2021, 13, 336. [Google Scholar] [CrossRef] [PubMed]
- Dai, F.; Zhang, S.; Wang, Q.; Chen, H.; Chen, C.; Qian, G.; Yu, Y. Preparation and Characterization of Reduced Graphene Oxide/TiO2 Blended Polyphenylene Sulfone Antifouling Composite Membrane With Improved Photocatalytic Degradation Performance. Front. Chem. 2021, 9, 753741. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Dai, F.; Zhang, S.; Wang, M.; Chen, C.; Yu, Y. Design of a Novel Poly(Aryl Ether Nitrile)-Based Composite Ultrafiltration Membrane with Improved Permeability and Antifouling Performance Using Zwitterionic Modified Nano-Silica. RSC Adv. 2021, 11, 15231–15244. [Google Scholar] [CrossRef]
- Zhang, S.; Dai, F.; Wang, Q.; Qian, G.; Chen, C.; Yu, Y. The Fabrication of Porous Hollow Polysulfone Microspheres with PEG as a Porogen for Methylene Blue Adsorption. Colloids Surf. A Physicochem. Eng. Asp. 2022, 634, 127949. [Google Scholar] [CrossRef]
- Fan, T.; Yang, W.; Wang, N.; Ni, X.; Wen, J.; Xu, W. Molecularly Imprinted Polymer Microspheres Derived from Pickering Emulsions Polymerization in Determination of Di(2-Ethylhexyl) Phthalate in Bottled Water Samples. J. Appl. Polym. Sci. 2016, 133, 43484–43495. [Google Scholar] [CrossRef]
- Kim, S.-H.; Yi, G.-R.; Kim, K.H.; Yang, S.-M. Photocurable Pickering Emulsion for Colloidal Particles with Structural Complexity. Langmuir 2008, 24, 2365–2371. [Google Scholar] [CrossRef]
- Mihali, V.; Honciuc, A. Evolution of Self-Organized Microcapsules with Variable Conductivities from Self-Assembled Nanoparticles at Interfaces. ACS Nano 2019, 13, 3483–3491. [Google Scholar] [CrossRef]
- Ma, H.; Luo, M.; Sanyal, S.; Rege, K.; Dai, L. The One-Step Pickering Emulsion Polymerization Route for Synthesizing Organic-Inorganic Nanocomposite Particles. Materials 2010, 3, 1186–1202. [Google Scholar] [CrossRef] [Green Version]
- Honciuc, A.; Negru, O.-I. NanoTraPPED—A New Method for Determining the Surface Energy of Nanoparticles via Pickering Emulsion Polymerization. Nanomaterials 2021, 11, 3200. [Google Scholar] [CrossRef] [PubMed]
- Bon, S.A.F. Chapter The Phenomenon of Pickering Stabilization: A Basic Introduction. In Particle-Stabilized Emulsions and Colloids: Formation and Applications; Ngai, T., Bon, S., Eds.; RSC Soft Matter Series: Cambridge, UK, 2014. [Google Scholar]
- Finkle, P.; Draper, H.D.; Hildebrand, J.H. The Theory of Emulsification. J. Am. Chem. Soc. 1923, 45, 2780–2788. [Google Scholar] [CrossRef]
- Binks, B.P.; Clint, J.H. Solid Wettability from Surface Energy Components: Relevance to Pickering Emulsions. Langmuir 2002, 18, 1270–1273. [Google Scholar] [CrossRef]
- Aveyard, R.; Binks, B.P.; Clint, J.H. Emulsions Stabilised Solely by Colloidal Particles. Adv. Colloid Interface Sci. 2003, 100, 503–546. [Google Scholar] [CrossRef]
- Nowak, E.; Combes, G.; Stitt, E.H.; Pacek, A.W. A Comparison of Contact Angle Measurement Techniques Applied to Highly Porous Catalyst Supports. Powder Technol. 2013, 233, 52–64. [Google Scholar] [CrossRef]
- Paunov, V.N. Novel Method for Determining the Three-Phase Contact Angle of Colloid Particles Adsorbed at Air−Water and Oil−Water Interfaces. Langmuir 2003, 19, 7970–7976. [Google Scholar] [CrossRef]
- Tzirakis, M.D.; Zambail, R.; Tan, Y.Z.; Chew, J.W.; Adlhart, C.; Honciuc, A. Surfactant-Free Synthesis of Sub-100 Nm Poly(Styrene-Co-Divinylbenzene) Nanoparticles by One-Step Ultrasonic Assisted Emulsification/Polymerization. RSC Adv. 2015, 5, 103218–103228. [Google Scholar] [CrossRef] [Green Version]
- Honciuc, A. Chemistry of Functional Materials Surfaces and Interfaces: Fundamentals and Applications, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2021. [Google Scholar]
- Harkins, W.D. A General Theory of the Mechanism of Emulsion Polymerization. J. Am. Chem. Soc. 1947, 69, 1428–1444. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.H.; Fan, X.D.; Tian, W.; Fan, W.W. Polystyrene/Nano-SiO2 Composite Microspheres Fabricated by Pickering Emulsion Polymerization: Preparation, Mechanisms and Thermal Properties. Express Polym. Lett. 2012, 6, 532–542. [Google Scholar] [CrossRef]
- Lotierzo, A.; Bon, S.A.F. A Mechanistic Investigation of Pickering Emulsion Polymerization. Polym. Chem. 2017, 8, 5100–5111. [Google Scholar] [CrossRef] [Green Version]
- Owens, D.K.; Wendt, R.C. Estimation of the Surface Free Energy of Polymers. J. Appl. Polym. Sci. 1969, 13, 1741–1747. [Google Scholar] [CrossRef]
- Rabel, W. Einige Aspekte Der Benetzungstheorie Und Ihre Anwendung Auf Die Untersuchung Und Veränderung Der Oberflächeneigenschaften von Polymeren. Farbe Und Lack 1971, 77, 997–1005. [Google Scholar]
- Mansel, D. Some Electrical and Optical Aspects of Molecular Behaviour, 1st ed.; Pergamon Press, Ltd.: London, UK, 1965. [Google Scholar]
- Yoneda, H. Effect of Substitution on Dipole Moments of Molecules. BCSJ 1958, 31, 708–714. [Google Scholar] [CrossRef] [Green Version]
- Racles, C.; Cozan, V.; Bele, A.; Dascalu, M. Polar Silicones: Structure-Dielectric Properties Relationship. Des. Monomers Polym. 2016, 19, 496–507. [Google Scholar] [CrossRef] [Green Version]
- Shi, B.; Peng, P.; Jia, L. Relationship Between Total Surface Tension of Monomer and Its Homopolymer. J. Macromol. Sci. Part B 2011, 50, 952–955. [Google Scholar] [CrossRef]
- Luck, R.M.; Sadhir, R.K. Shrinkage in Conventional Monomers during Polymerization. In Expanding Monomers; Synthesis, Characterization and Applications; CRC Press: Boca Raton, FL, USA, 1992. [Google Scholar]
- Bancroft, W.D. Applied Colloid Chemistry; General Theory; McGraw-Hill Book Company, Inc.: New York, NY, USA, 1921. [Google Scholar]
- Mihali, V.; Honciuc, A. Semiconductive Materials with Tunable Electrical Resistance and Surface Polarity Obtained by Asymmetric Functionalization of Janus Nanoparticles. Adv. Mater. Interfaces 2017, 4, 1700914. [Google Scholar] [CrossRef]
- Wu, D.; Binks, B.P.; Honciuc, A. Modeling the Interfacial Energy of Surfactant-Free Amphiphilic Janus Nanoparticles from Phase Inversion in Pickering Emulsions. Langmuir 2018, 34, 1225–1233. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nanoparticle | γdNP/water (mN/m) | γpNP/water (mN/m) | γNP/water (mN/m) |
|---|---|---|---|
| NP-SH | 0.21 | 3.10 | 3.10 |
| NP-CN | 0.00 | 2.77 | 2.77 |
| NP-Gly | 0.00 | 0.11 | 0.11 |
| Nanoparticle | γdNP (mN/m) | γpNP (mN/m) | γNP (mN/m) |
|---|---|---|---|
| NP-SH | 20.96 | 26.60 | 47.56 |
| NP-CN | 25.40 | 26.94 | 52.34 |
| NP-Gly | 25.40 | 42.56 | 67.96 |
| Nanoparticle | ||||
|---|---|---|---|---|
| NP-SH | 116.9 | 76.3 | 64.5 | 57.9 |
| NP-CN | 122.0 | 81.7 | 70.5 | 63.6 |
| NP-Gly | 140.2 | 87.9 | 72.0 | 64.1 |
| NP-SH | NP-CN | NP-Gly | ||||
|---|---|---|---|---|---|---|
| Polymer | Microsphere Diameter [μm] | β° | Microsphere Diameter [μm] | β° | Microsphere Diameter [μm] | β° |
| P(MM) | - | 106.0 ± 5.2 | - | 108.1 ± 2.9 | 40, 100 | 149.9 ± 0.5 |
| P(BM) | 40 | 118.2 ± 1.9 | - | 114.4 ± 2.2 | 80 | 154.6 ± 0.5 |
| P(tBA) | 40 | 115.7 ± 2.6 | 80 | 119.4 ± 1.8 | 100 | 153.5 ± 0.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Honciuc, A.; Negru, O.-I. Role of Surface Energy of Nanoparticle Stabilizers in the Synthesis of Microspheres via Pickering Emulsion Polymerization. Nanomaterials 2022, 12, 995. https://doi.org/10.3390/nano12060995
Honciuc A, Negru O-I. Role of Surface Energy of Nanoparticle Stabilizers in the Synthesis of Microspheres via Pickering Emulsion Polymerization. Nanomaterials. 2022; 12(6):995. https://doi.org/10.3390/nano12060995
Chicago/Turabian StyleHonciuc, Andrei, and Oana-Iuliana Negru. 2022. "Role of Surface Energy of Nanoparticle Stabilizers in the Synthesis of Microspheres via Pickering Emulsion Polymerization" Nanomaterials 12, no. 6: 995. https://doi.org/10.3390/nano12060995
APA StyleHonciuc, A., & Negru, O. -I. (2022). Role of Surface Energy of Nanoparticle Stabilizers in the Synthesis of Microspheres via Pickering Emulsion Polymerization. Nanomaterials, 12(6), 995. https://doi.org/10.3390/nano12060995