
Oxide Derived Copper for Electrochemical Reduction of CO2 to C2+ Products



Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Section
2.1.1. Chemicals
2.1.2. Preparation of CuO Nanoparticles
2.1.3. Preparation of Glassy Carbon Plate/Electrodes
2.1.4. Instruments for SEM and XRD
2.1.5. Electrochemical Instrumentation and Procedures
2.1.6. Analysis of the Electrolysis Products
2.1.7. Preparation of H2 and C2H4 Calibration Curves
- Electrolyte solution and head space were saturated with pure CO2 into the same H-cell that was used for bulk electrolysis;
- Standard gases of C2H4 and H2 of known amounts were injected into the electrolyte solution and head space of the same volume;
- The mixture of gases of the same volume was then injected to GC to obtain the area of the peak corresponding to H2 and C2H4;
- Range of different amounts of C2H4 and H2 were tested and repeated the experiments to obtain the calibration plots of C2H4 and H2;
- Calibration curves of C2H4 and H2 were obtained by plotting the concentration of gases against the peak intensity of the respective gases.
2.1.8. Faradaic Efficiency
3. Results and Discussion
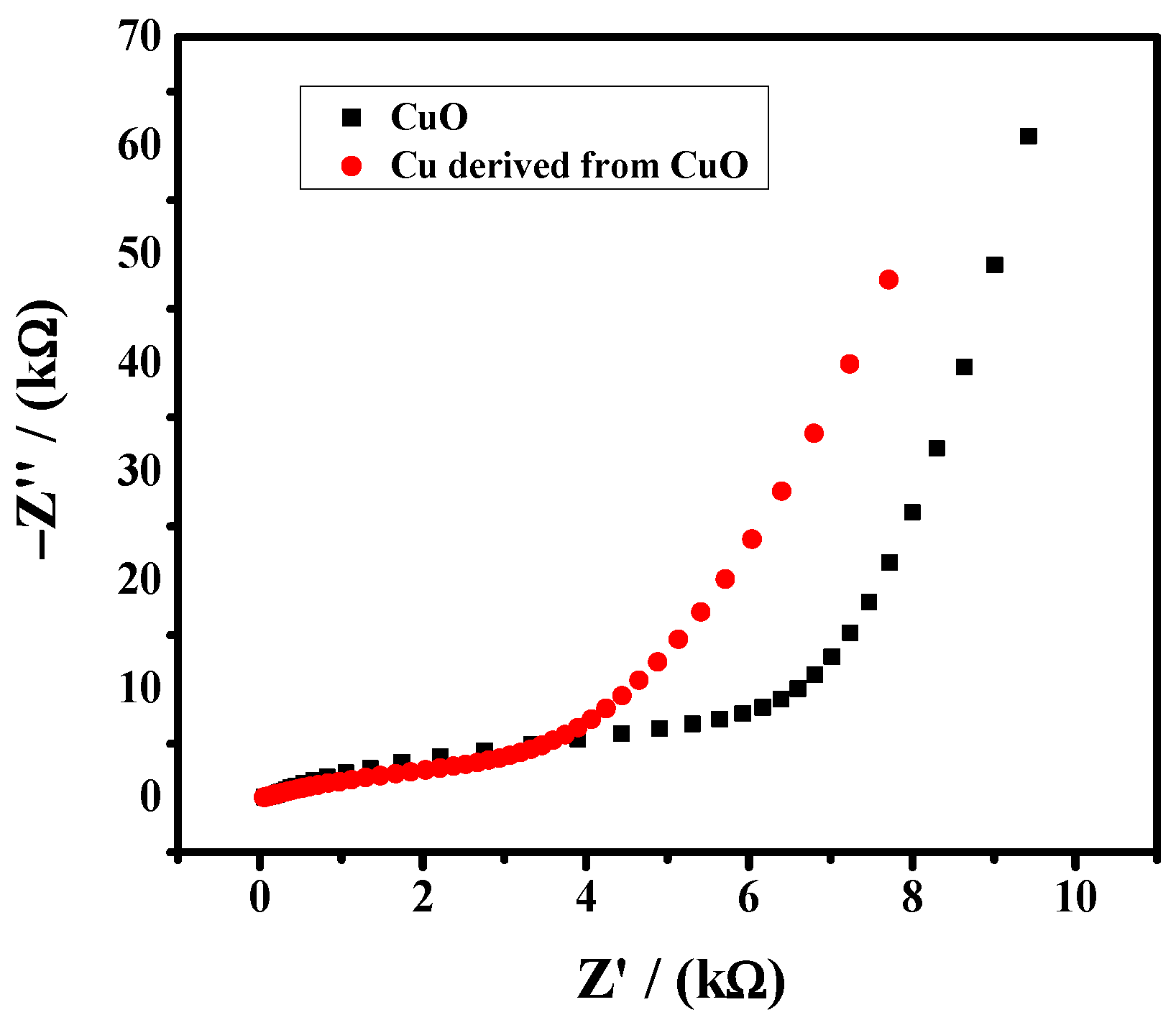
3.1. Physical Characterization
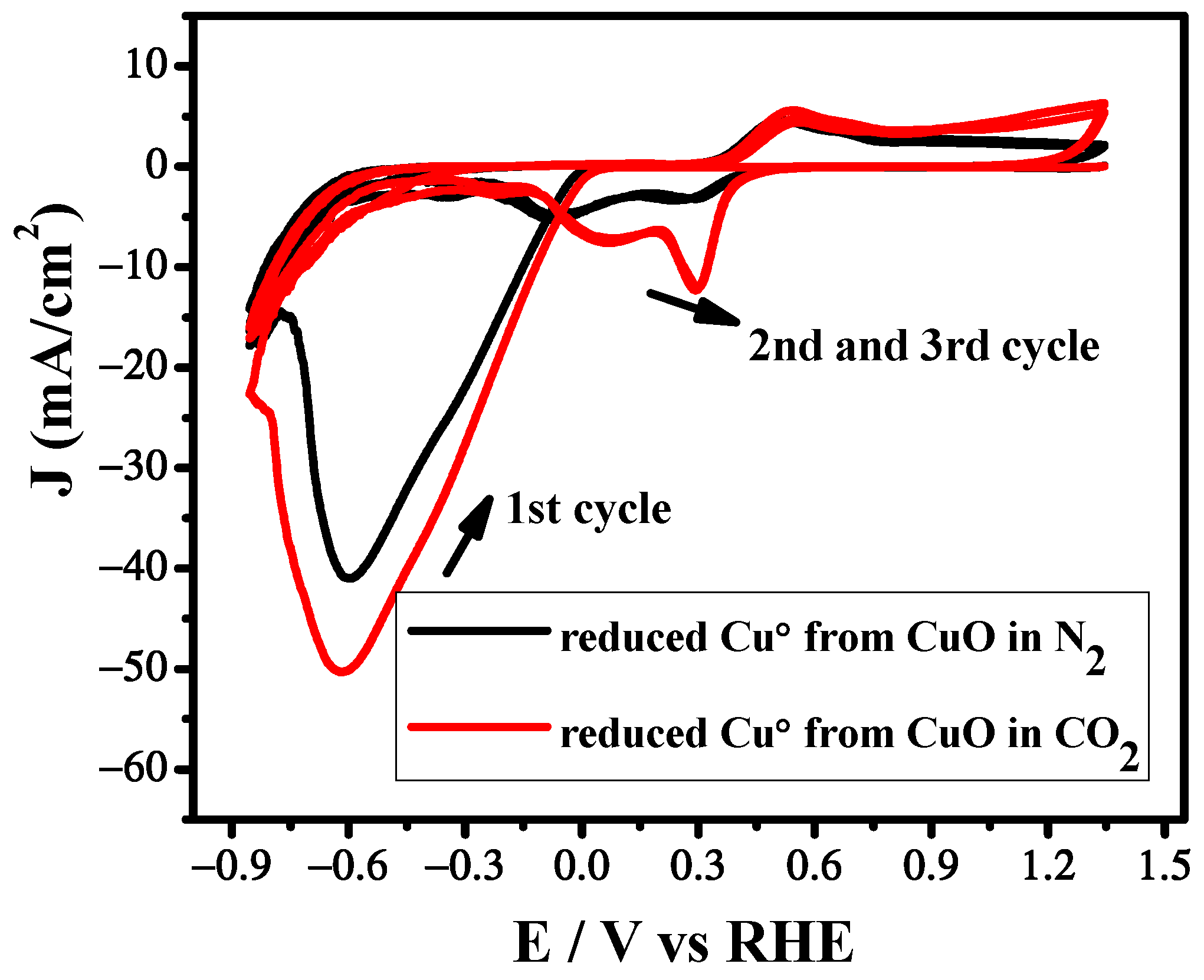
3.2. Cyclic Voltammetry
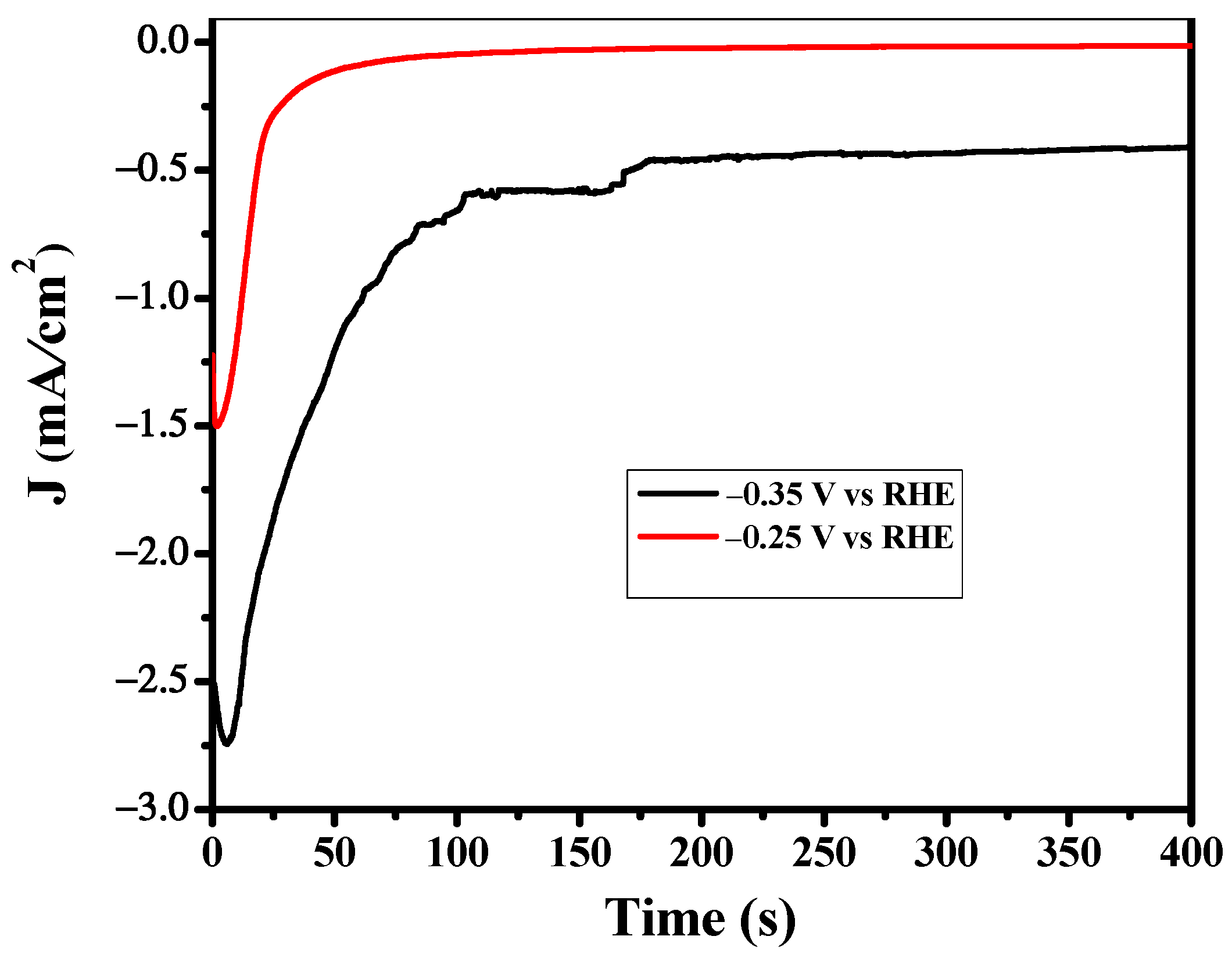
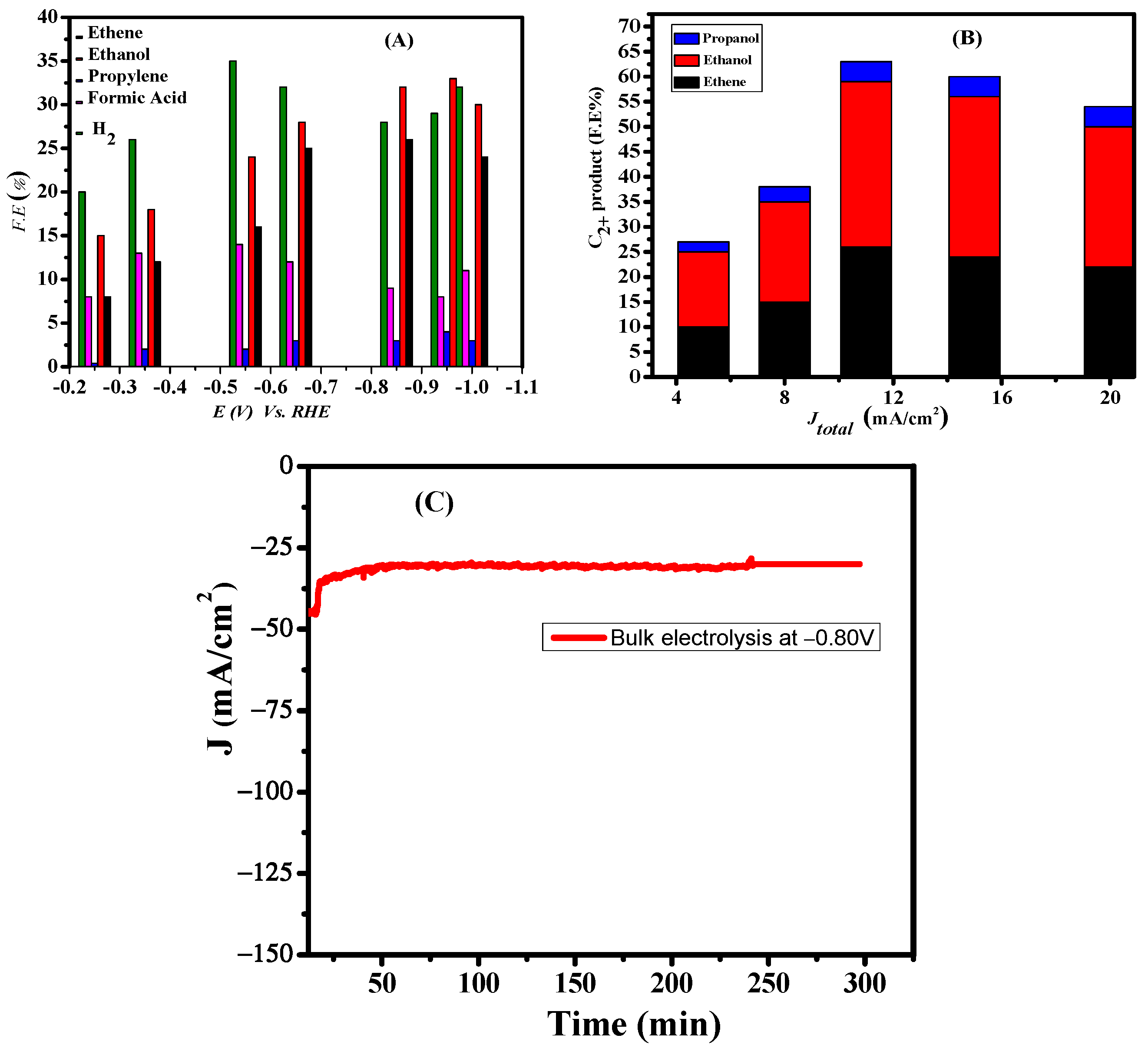
3.3. Bulk Electrolysis
3.4. Effect of Electrolyte
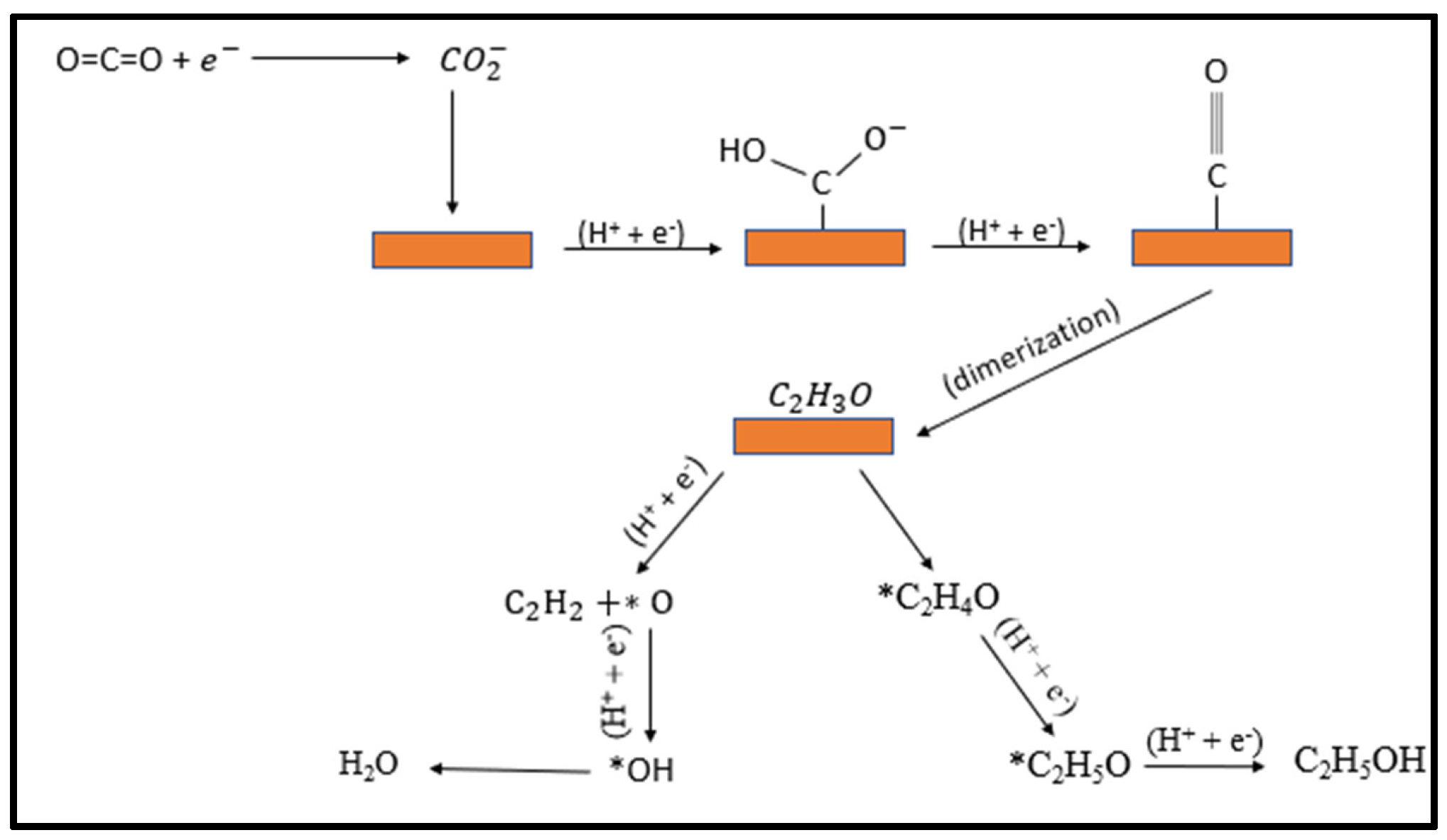
4. Origin of Selectivity
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fan, Q.; Zhang, M.; Jia, M.; Liu, S.; Qiu, J.; Sun, Z. Electrochemical CO2 reduction to C2+ species: Heterogeneous electrocatalysts, reaction pathways, and optimization strategies. Mater. Today Energy 2018, 10, 280–301. [Google Scholar] [CrossRef]
- Mi, Y.; Peng, X.; Liu, X.; Luo, J. Selective formation of C2 products from electrochemical CO2 reduction over Cu1.8 Se nanowires. ACS Appl. Energy Mater. 2018, 10, 5119–5123. [Google Scholar]
- Zhuang, T.-T.; Liang, Z.-Q.; Seifitokaldani, A.; Li, Y.; De Luna, P.; Burdyny, T.; Che, F.; Meng, F.; Min, Y.; Quintero-Bermudez, R. Steering post-C–C coupling selectivity enables high efficiency electroreduction of carbon dioxide to multi-carbon alcohols. Nat. Catal. 2018, 6, 421. [Google Scholar] [CrossRef]
- Ren, D.; Deng, Y.; Handoko, A.D.; Chen, C.S.; Malkhandi, S.; Yeo, B.S. Selective electrochemical reduction of carbon dioxide to ethylene and ethanol on copper (I) oxide catalysts. ACS Catal. 2015, 5, 2814–2821. [Google Scholar] [CrossRef]
- Zhang, X.; Sun, X.; Guo, S.-X.; Bond, A.M.; Zhang, J. Formation of lattice-dislocated bismuth nanowires on copper foam for enhanced electrocatalytic CO2 reduction at low overpotential. Energy Environ. Sci. 2019, 12, 1334–1340. [Google Scholar] [CrossRef]
- Ma, M.; Djanashvili, K.; Smith, W.A. Selective electrochemical reduction of CO2 to CO on CuO-derived Cu nanowires. Phys. Chem. Chem. Phys. 2015, 17, 20861–20867. [Google Scholar] [CrossRef] [PubMed]
- Garza, A.J.; Bell, A.T.; Head-Gordon, M. Is subsurface oxygen necessary for the electrochemical reduction of CO2 on copper? J. Phys. Chem. Lett. 2018, 9, 601–606. [Google Scholar] [CrossRef] [Green Version]
- Torelli, D.A.; Francis, S.A.; Crompton, J.C.; Javier, A.; Thompson, J.R.; Brunschwig, B.S.; Soriaga, M.P.; Lewis, N.S. Nickel–gallium-catalyzed electrochemical reduction of CO2 to highly reduced products at low overpotentials. ACS Catal. 2016, 6, 2100–2104. [Google Scholar] [CrossRef] [Green Version]
- Kuhl, K.P.; Cave, E.R.; Abram, D.N.; Jaramillo, T.F. New insights into the electrochemical reduction of carbon dioxide on metallic copper surfaces. Energy Environ. Sci. 2012, 5, 7050–7059. [Google Scholar]
- Zhang, Y.; Li, F.; Zhang, X.; Williams, T.; Easton, C.D.; Bond, A.M.; Zhang, J. Electrochemical reduction of CO2 on defect-rich Bi derived from Bi2S3 with enhanced formate selectivity. J. Mater. Chem. A 2018, 6, 4714–4720. [Google Scholar] [CrossRef]
- Qiao, J.; Fan, M.; Fu, Y.; Bai, Z.; Ma, C.; Liu, Y.; Zhou, X.-D. Highly-active copper oxide/copper electrocatalysts induced from hierarchical copper oxide nanospheres for carbon dioxide reduction reaction. Electrochim. Acta 2015, 153, 559–565. [Google Scholar] [CrossRef]
- Han, L.; Zhou, W.; Xiang, C. High-rate electrochemical reduction of carbon monoxide to ethylene using Cu-nanoparticle-based gas diffusion electrodes. ACS Energy Lett. 2018, 3, 855–860. [Google Scholar] [CrossRef] [Green Version]
- Lu, Q.; Jiao, F. Electrochemical CO2 reduction: Electrocatalyst, reaction mechanism, and process engineering. Nano Energy 2016, 29, 439–456. [Google Scholar] [CrossRef] [Green Version]
- Hori, Y.; Takahashi, I.; Koga, O.; Hoshi, N. Selective formation of C2 compounds from electrochemical reduction of CO2 at a series of copper single crystal electrodes. J. Phys. Chem. B 2002, 106, 15–17. [Google Scholar] [CrossRef]
- Jiang, K.; Huang, Y.; Zeng, G.; Toma, F.M.; Goddard, W.A., III; Bell, A.T. Effects of surface roughness on the electrochemical reduction of CO2 over Cu. ACS Energy Lett. 2020, 5, 1206–1214. [Google Scholar] [CrossRef] [Green Version]
- Vasileff, A.; Zhu, Y.; Zhi, X.; Zhao, Y.; Ge, L.; Chen, H.M.; Qiao, S.Z. Electrochemical reduction of CO2 to ethane through stabilization of an ethoxy intermediate. Angew. Chem. 2020, 132, 19817–19821. [Google Scholar] [CrossRef]
- Cao, S.M.; Chen, H.B.; Dong, B.X.; Zheng, Q.H.; Ding, Y.X.; Liu, M.J.; Liu, W.L. Nitrogen-rich metal-organic framework mediated Cu–N–C composite catalysts for the electrochemical reduction of CO2. J. Energy Chem. 2021, 54, 555–563. [Google Scholar] [CrossRef]
- Yano, J.; Morita, T.; Shimano, K.; Nagami, Y.; Yamasaki, S. Selective ethylene formation by pulse-mode electrochemical reduction of carbon dioxide using copper and copper-oxide electrodes. J. Solid State Electrochem. 2007, 11, 554–557. [Google Scholar] [CrossRef]
- Kovalenko, A.; Neburchilov, V. Response to Comment on Density Functional Theory and 3D-RISM-KH molecular theory of solvation studies of CO2 reduction on Cu-, Cu2O-, Fe-, and Fe3O4-based nanocatalysts. J. Mol. Model. 2022, 28, 33. [Google Scholar] [CrossRef]
- Dinh, C.T.; Burdyny, T.; Kibria, M.G.; Seifitokaldani, A.; Gabardo, C.M.; Kiani, A.; Edwards, J.P.; De Luna, P.; Bushuyev, O.S. CO2 electroreduction to ethylene via hydroxide-mediated copper catalysis at an abrupt interface. Science 2018, 360, 783–787. [Google Scholar] [CrossRef] [Green Version]
- Lamy, E.; Nadjo, L.; Saveant, J.M. Standard. potential and kinetic parameters of the electrochemical reduction of carbon dioxide in dimethylformamide. J. Electroanal. Chem. Interf. Electrochem. 1977, 78, 403–407. [Google Scholar] [CrossRef]
- Velasco-Vélez, J.J.; Jones, T.; Gao, D.; Carbonio, E.; Arrigo, R.; Hsu, C.J.; Huang, Y.-C.; Dong, C.L.; Chen, J.M.; Lee, J.F. The role of the copper oxidation state in the electrocatalytic reduction of CO2 into valuable hydrocarbon. ACS Sustain. Chem. Eng. 2018, 7, 1485–1492. [Google Scholar] [CrossRef]
- Calvinho, K.U.; Laursen, A.B.; Yap, K.M.; Goetjen, T.A.; Hwang, S.; Murali, N.; Mejia-Sosa, B.; Lubarski, A.; Teeluck, K.M.; Hall, E.S. Selective CO2 reduction to C3 and C4 oxyhydrocarbons on nickel phosphides at overpotentials as low as 10 mV. Energy Environ. Sci. 2018, 11, 2550–2559. [Google Scholar] [CrossRef]
- Khan, Y.; Durrani, S.; Mehmood, M.; Ahmad, J.; Khan, M.R.; Firdous, S. Low temperature synthesis of fluorescent ZnO nanoparticles. Appl. Surf. Sci. 2010, 257, 1756–1761. [Google Scholar] [CrossRef]
- Akhade, S.A.; McCrum, I.T.; Janik, M.J. The impact of specifically adsorbed ions on the copper-catalyzed electroreduction of CO2. J. Electrochem. Soc. 2016, 163, F477–F484. [Google Scholar] [CrossRef]
- Yuan, J.; Zhang, J.J.; Yang, M.P.; Meng, W.J.; Wang, H.; Lu, J.X. CuO nanoparticles supported on TiO2 with high efficiency for CO2 electrochemical reduction to ethanol. Catalysts 2018, 8, 171. [Google Scholar] [CrossRef] [Green Version]
- Mandal, L.; Yang, K.R.; Motapothula, M.R.; Ren, D.; Lobaccaro, P.; Patra, A.; Venkatesan, T. Investigating the role of copper oxide in electrochemical CO2 reduction in real time. ACS Appl. Mater. Interface. 2018, 10, 8574–8584. [Google Scholar] [CrossRef] [Green Version]
- Jeong, H.M.; Kwon, Y.; Won, J.H.; Lum, Y.; Cheng, M.J.; Kim, K.H.; Kang, J.K. Atomic-Scale Spacing between Copper Facets for the Electrochemical Reduction of Carbon Dioxide. Adv. Energy Mater 2020, 10, 1903423. [Google Scholar] [CrossRef] [Green Version]
- Le, M.; Ren, M.; Zhang, Z.; Sprunger, P.T.; Kurtz, R.L.; Flake, J.C. Electrochemical reduction of CO2 to CH3OH at copper oxide surfaces. J. Electrochem. Soc. 2011, 5, E45. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Sethuraman, V.; Michalsky, R.; Peterson, A.A. Competition between CO2 reduction and H2 evolution on transition-metal electrocatalysts. ACS Catal. 2014, 4, 3742–3748. [Google Scholar] [CrossRef]
- Lan, Y.; Gai, C.; Kenis, P.J.; Lu, J. Electrochemical reduction of carbon dioxide on Cu/CuO core/shell catalysts. ChemElectroChem 2014, 1, 1577–1582. [Google Scholar] [CrossRef]
- Lan, Y.; Ma, S.; Lu, J.; Kenis, P.J. Investigation of a Cu (core)/CuO (shell) catalyst for electrochemical reduction of CO2 in aqueous soultion. Int. J. Electrochem. Sci. 2014, 9, 7300–7308. [Google Scholar]
- Kumar, B.; Asadi, M.; Pisasale, D.; Sinha-Ray, S.; Rosen, B.A.; Haasch, R.; Abiade, J.; Yarin, A.L.; Salehi-Khojin, A. Renewable and metal-free carbon nanofibre catalysts for carbon dioxide reduction. Nat. Commun. 2013, 4, 2819. [Google Scholar] [CrossRef]
- Xiao, H.; Cheng, T.; Goddard, W.A., III. Atomistic mechanisms underlying selectivities in C1 and C2 products from electrochemical reduction of CO on Cu (111). J. Am. Chem. Soc. 2016, 1, 130–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Hong, S.; Lee, J. Bulk pH contribution to CO/HCOO−production from CO2 on oxygen-evacuated Cu2O electrocatalyst. Catal. Today 2017, 288, 11–17. [Google Scholar] [CrossRef]
- Angamuthu, R.; Byers, P.; Lutz, M.; Spek, A.L.; Bouwman, E. Electrocatalytic CO2 conversion to oxalate by a copper complex. Science 2010, 327, 313–315. [Google Scholar] [CrossRef] [Green Version]
- Schouten, K.; Kwon, Y.; Van der Ham, C.; Qin, Z.; Koper, M. A new mechanism for the selectivity to C1 and C2 species in the electrochemical reduction of carbon dioxide on copper electrodes. Chem. Sci. 2011, 2, 1902–1909. [Google Scholar] [CrossRef]
- Pang, Y.; Burdyny, T.; Dinh, C.T.; Kibria, M.G.; Fan, J.Z.; Liu, M.; Sargent, E.H.; Sinton, D. Joint tuning of nanostructured Cu-oxide morphology and local electrolyte programs high-rate CO2 reduction to C2H4. Green Chem. 2017, 19, 4023–4030. [Google Scholar] [CrossRef]
- Abhijit Rahaman, D.; Mohos, M.; AZanetti, P. Electrochemical CO2 conversion using skeleton (sponge) type of Cu catalysts. ACS Catal. 2017, 7, 5431–5437. [Google Scholar]
- Rahaman, M.; Dutta, A.; Zanetti, A.; Broekmann, P. Electrochemical reduction of CO2 into multicarbon alcohols on activated Cu mesh catalysts: An identical location (IL) study. ACS Catal. 2017, 7, 7946–7956. [Google Scholar] [CrossRef]
- Shi, G.; Yu, L.; Ba, X.; Zhang, X.; Zhou, J.; Yu, Y. Copper nanoparticle interspersed MoS2 nanoflowers with enhanced efficiency for CO2 electrochemical reduction to fuel. Dalton Trans 2017, 46, 10569–10577. [Google Scholar] [CrossRef] [PubMed]
- Jiang, K.; Sandberg, R.B.; Akey, A.J.; Liu, X.; Bell, D.C.; Nørskov, J.K.; Chan, K.; Wang, H. Metal ion cycling of Cu foil for selective C–C coupling in electrochemical CO2 reduction. Nat. Catal. 2018, 1, 111. [Google Scholar] [CrossRef]
- Kusama, S.; Saito, T.; Hashiba, H.; Sakai, A.; Yotsuhashi, S. Crystalline copper(II) phthalocyanine catalysts for electrochemical reduction of carbon dioxide in aqueous media. ACS Catal. 2017, 7, 8382–8385. [Google Scholar] [CrossRef]
- Ma, M.; Djanashvili, K.; Smith, W.A. Controllable hydrocarbon formation from the electrochemical reduction of CO2 over Cu nanowire arrays. Angew. Chem. Int. Ed. 2016, 55, 6680–6684. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.D.; Ko, W.R.; Lee, J.H.; Kim, S.J.; Lee, H.; Lee, M.H.; Nam, K.T. Morphology-directed selective production of ethylene or ethane from CO2 on a Cu mesopore electrode. Angew. Chem. Int. Ed. 2017, 56, 796–800. [Google Scholar] [CrossRef] [PubMed]
- Weng, Z.; Jiang, J.; Wu, Y.; Wu, Z.; Guo, X.; Materna, K.L.; Liu, W.; Batista, V.S.; Brudvig, G.W. Electrochemical CO2 reduction to hydrocarbons on a heterogeneous molecular Cu catalyst in aqueous solution. J. Am. Chem. Soc. 2016, 138, 8076–8079. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Zheng, Y.; Chen, P.; Jaroniec, M.; Qiao, S.-Z. Molecular scaffolding strategy with synergistic active centers to facilitate electrocatalytic CO2 reduction to hydrocarbon/alcohol. J. Am. Chem. Soc. 2017, 139, 18093–18100. [Google Scholar] [CrossRef]
- Gattrell, M.; Gupta, N.; Co, A. A review of the aqueous electrochemical reduction of CO2 to hydrocarbons at copper. J. Electroanal. Chem. 2006, 594, 1–19. [Google Scholar] [CrossRef]
- Montoya, H.; Shi CChan KNørskov, J.K. Theoretical insights into a CO dimerization mechanism in CO2 electroreduction. J. Phys. Chem. Lett. 2015, 6, 2032–2037. [Google Scholar] [CrossRef]
- Singh, M.R.; Goodpaster, J.D.; Weber, A.Z.; Head-Gordon, M.; Bell, A.T. Mechanistic insights into electrochemical reduction of CO2 over Ag using density functional theory and transport models. Proc. Natl. Acad. Sci. USA 2017, 114, E8812–E8821. [Google Scholar] [CrossRef] [Green Version]
- Hahn, C.; Hatsukade, T.; Kim, Y.G.; Vailionis, A.; Baricuatro, J.H.; Higgins, D.C.; Nitopi, S.A.; Soriaga, M.P.; Jaramillo, T.F. Engineering Cu surfaces for the electrocatalytic conversion of CO2: Controlling selectivity toward oxygenates and hydrocarbons. Proc. Natl. Acad. Sci. USA 2017, 114, 5918–5923. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Experimental Condition | Onset Potential | Products | Faradic Efficiency | Refs. |
|---|---|---|---|---|---|
| Copper oxide derived catalyst | 0.2 M KHCO3 @ −1.6 V (vs. RHE) | N/A | C2H4 | 29.7% | [38] |
| Cu-porphyrin complex | −0.976 V (vs. RHE) | −0.976 V (vs. RHE) | C2H4 | 17% | [46] |
| Cu skeletons | 0.5 M NaHCO3 −1.1 V (vs. RHE) | −0.25 V vs. RHE | C2+ products: | 32.2% | [39] |
| Cu NWs | 0.1 M KHCO3 −1.1 V (vs. RHE) | N/A | C2H4 | 17.4% | [44] |
| Cu meshes | 0.5 M KHCO3 @ −1.1 V (vs. RHE) | −0.7 V (vs RHE) | C2H4 | 34.3% | [40] |
| Cu/C3N4 | ~7.5 mA/cm2 @ −1.6 V (vs. Ag/AgCl) | −0.75 V vs. RHE | C2H4 | ~18% | [47] |
| Nanoporous Cu film | 14.3 mA/cm2 −1.7 V (vs. NHE) | −0.96 V vs. NHE | C2H6 | 46% | [45] |
| Cu(II) Phthalocyanine/C | 2.8 mA/cm2 @−1.6 V (vs. Ag/AgCl) | N/A | C2H4: 25% | 25% | [43] |
| Cu/MoS2 | 0.1 M KHCO3 | N/A | C2H5OH | 42.4% | [41] |
| Cu nanocube | 0.25 M KHCO3 68 mA/cm2 @ 0.963 V | −0.7 V (vs. RHE) | C2H4 | 32% | [42] |
| ODCu | −0.95 V vs. RHE | −0.10 vs. RHE | C2+ products Ethylene Ethanol Propanol | 57% 20% 33% 4% | Present work |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zahid, A.; Shah, A.; Shah, I. Oxide Derived Copper for Electrochemical Reduction of CO2 to C2+ Products. Nanomaterials 2022, 12, 1380. https://doi.org/10.3390/nano12081380
Zahid A, Shah A, Shah I. Oxide Derived Copper for Electrochemical Reduction of CO2 to C2+ Products. Nanomaterials. 2022; 12(8):1380. https://doi.org/10.3390/nano12081380
Chicago/Turabian StyleZahid, Anum, Afzal Shah, and Iltaf Shah. 2022. "Oxide Derived Copper for Electrochemical Reduction of CO2 to C2+ Products" Nanomaterials 12, no. 8: 1380. https://doi.org/10.3390/nano12081380
APA StyleZahid, A., Shah, A., & Shah, I. (2022). Oxide Derived Copper for Electrochemical Reduction of CO2 to C2+ Products. Nanomaterials, 12(8), 1380. https://doi.org/10.3390/nano12081380