

Bifunctional P-Containing RuO2 Catalysts Prepared from Surplus Ru Co-Ordination Complexes and Applied to Zn/Air Batteries

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
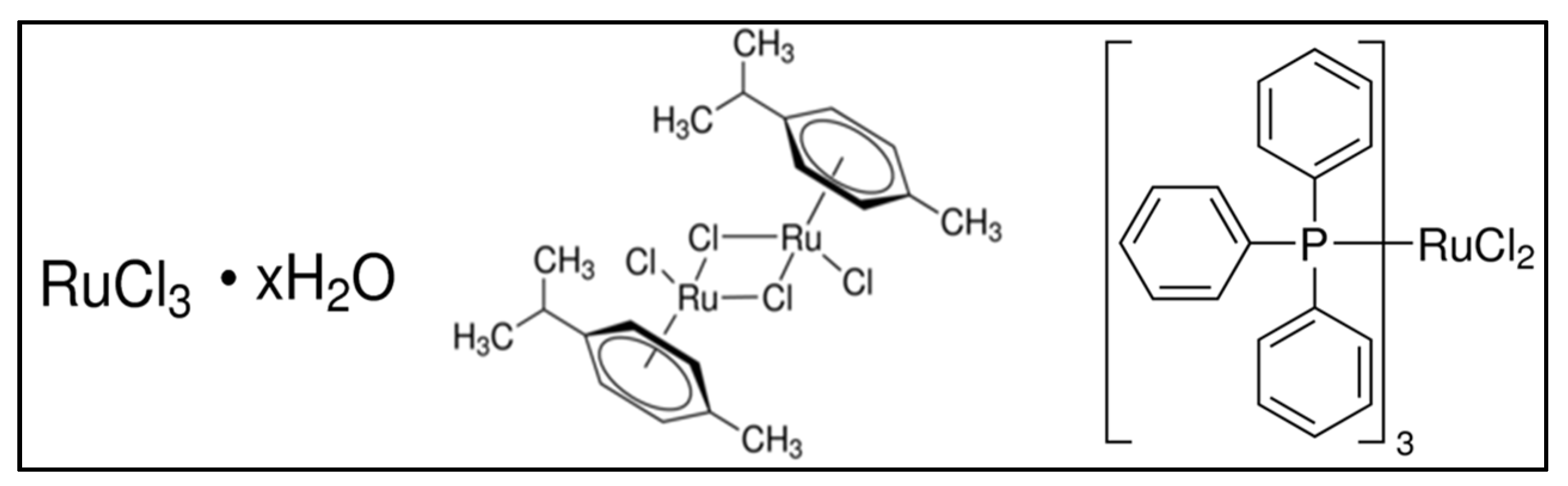
2.2. Synthesis Methods
2.3. Structural Characterization
2.4. Electrochemical Characterization
3. Results
3.1. Structural Characterization
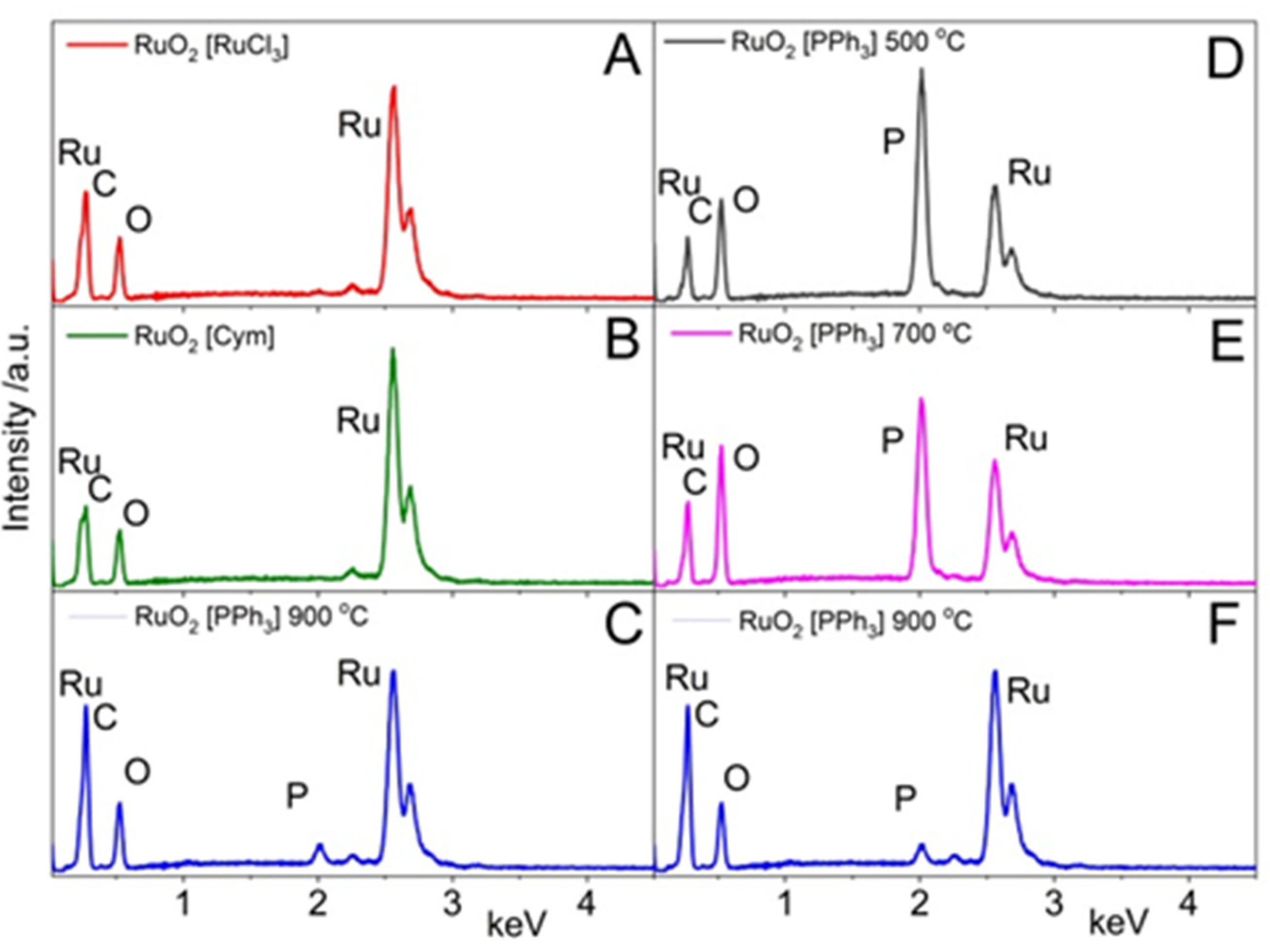
3.1.1. EDX and SEM
3.1.2. XRD
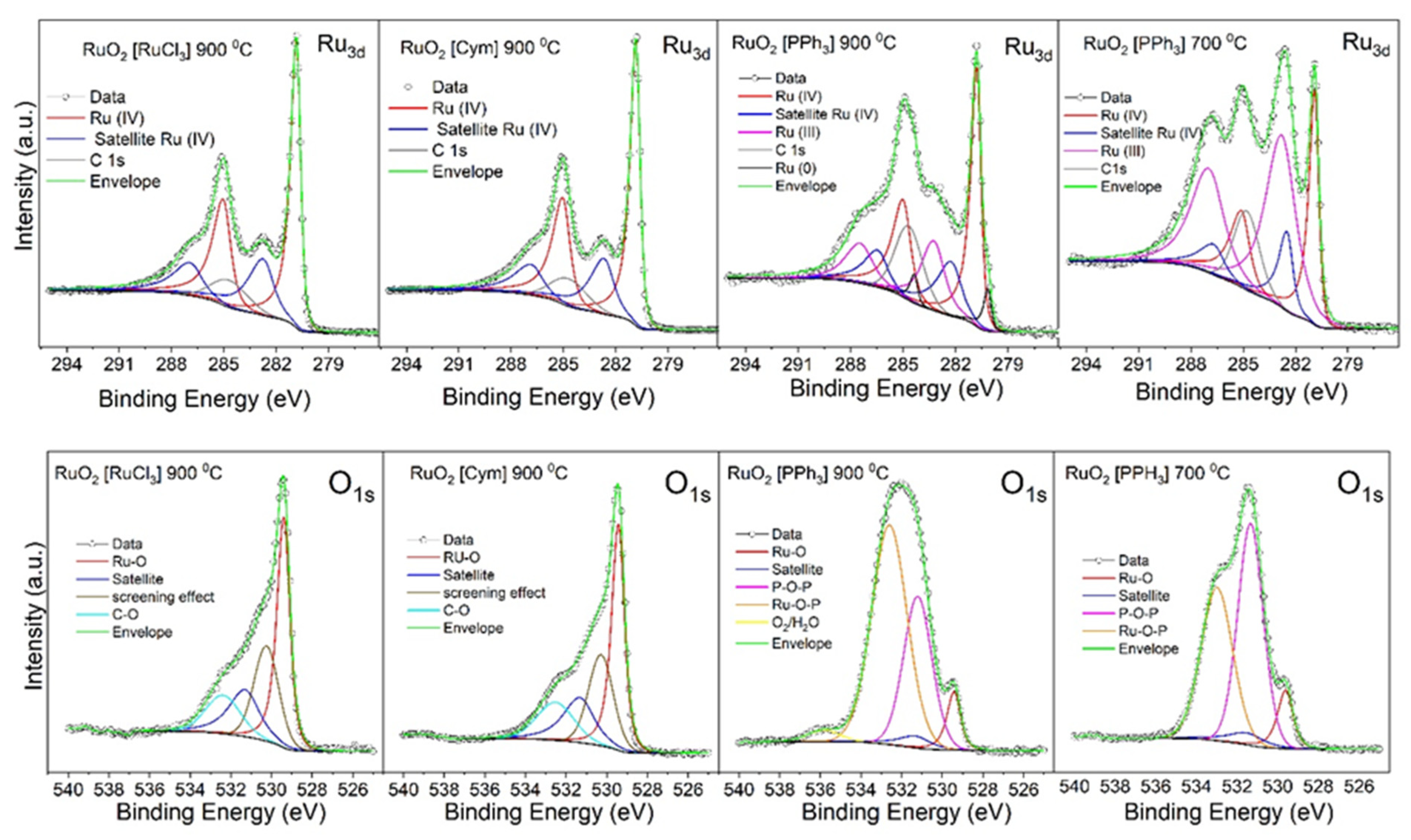
3.1.3. XPS
3.2. Electrochemical Measurements
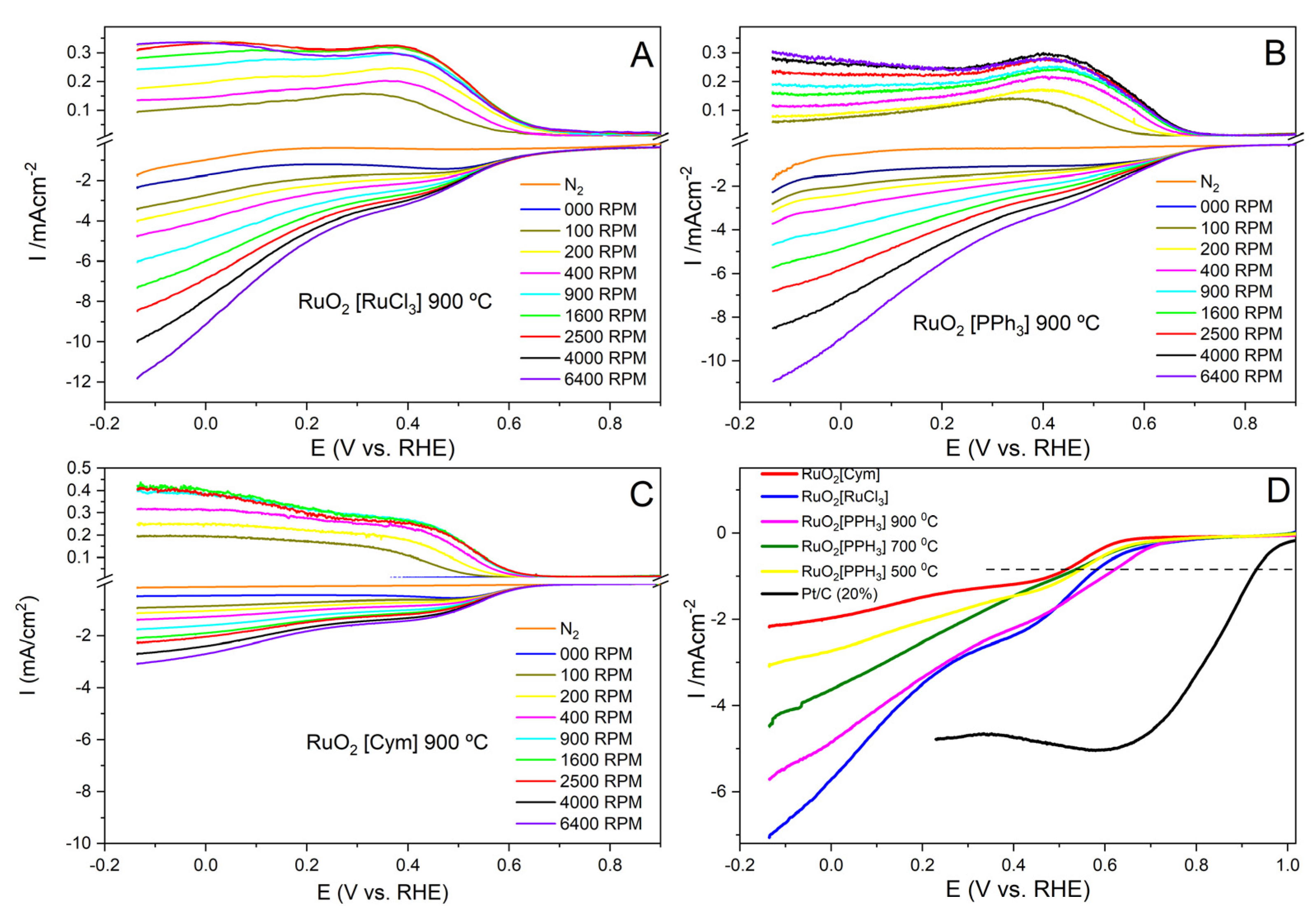
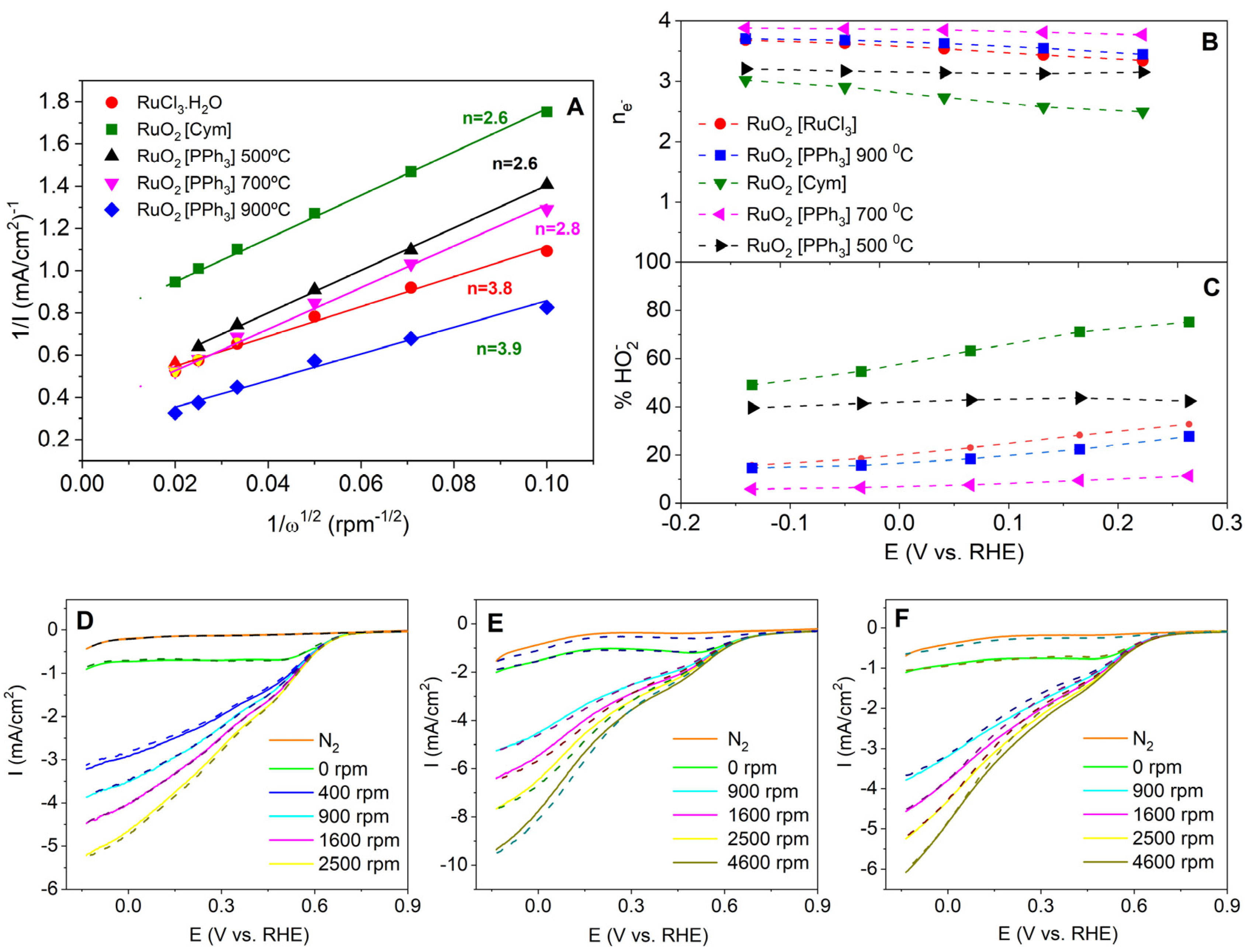
3.2.1. ORR Activity of the Different Materials
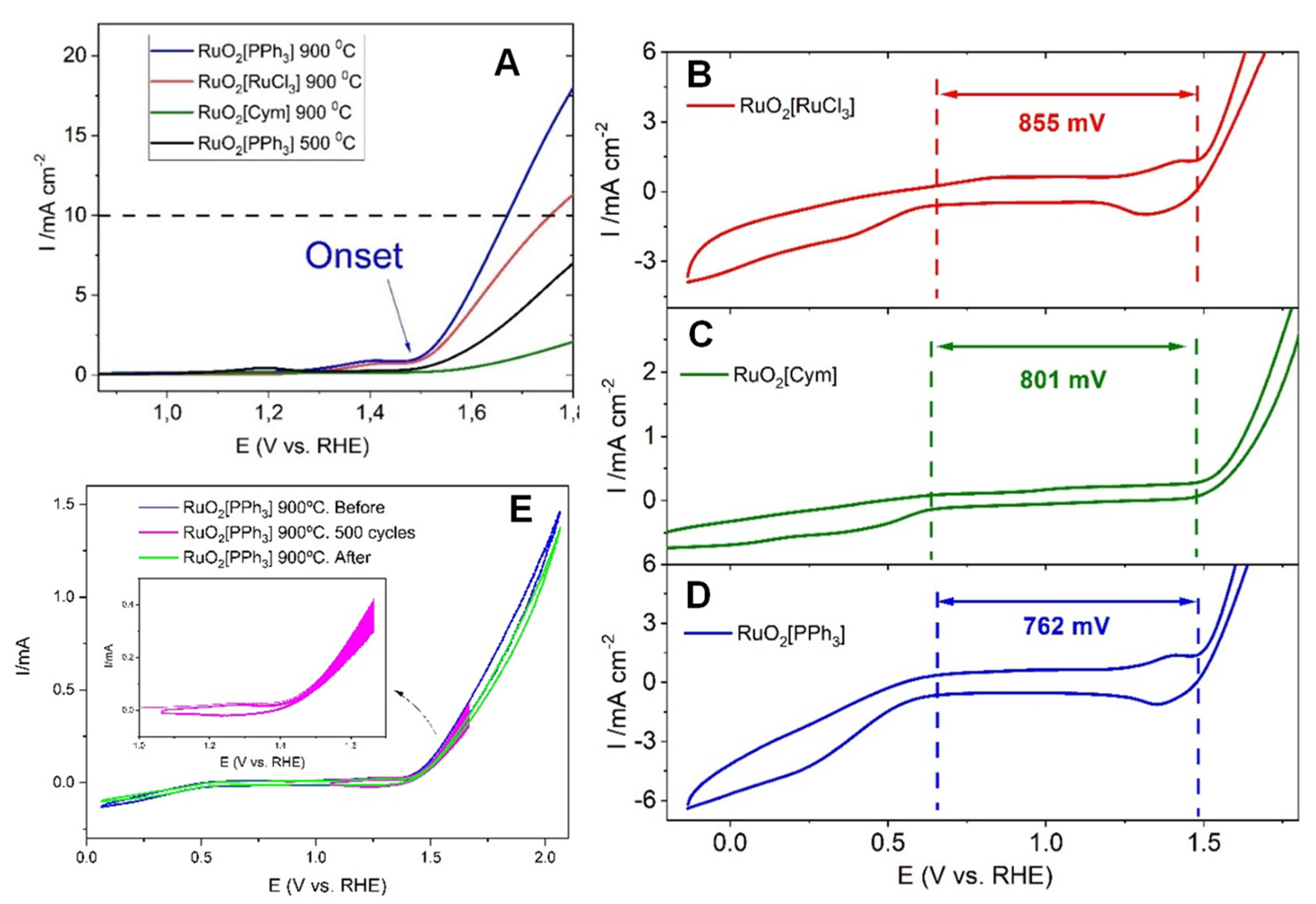
3.2.2. QER Activity of the RuO2−Based Materials

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst [a] | Eonset [b] (mV) | E [b],[c] (mV) | ΔEonset [d] (mV) | ΔE [e] (mV) | Tafel (mV dec−1) |
|---|---|---|---|---|---|
| RuO2 [RuCl3] | 1509 ± 10 | 1755 ± 7 | 855 ± 18 | 1200 ± 14 | 147 ± 7 |
| RuO2 [Cym] | 1498 ± 12 | -- | 801 ± 22 | -- | 145 ± 10 |
| RuO2 [PPh3] | 1505 ± 8 | 1670 ± 10 | 762 ± 18 | 1068 ± 15 | 153 ± 8 |
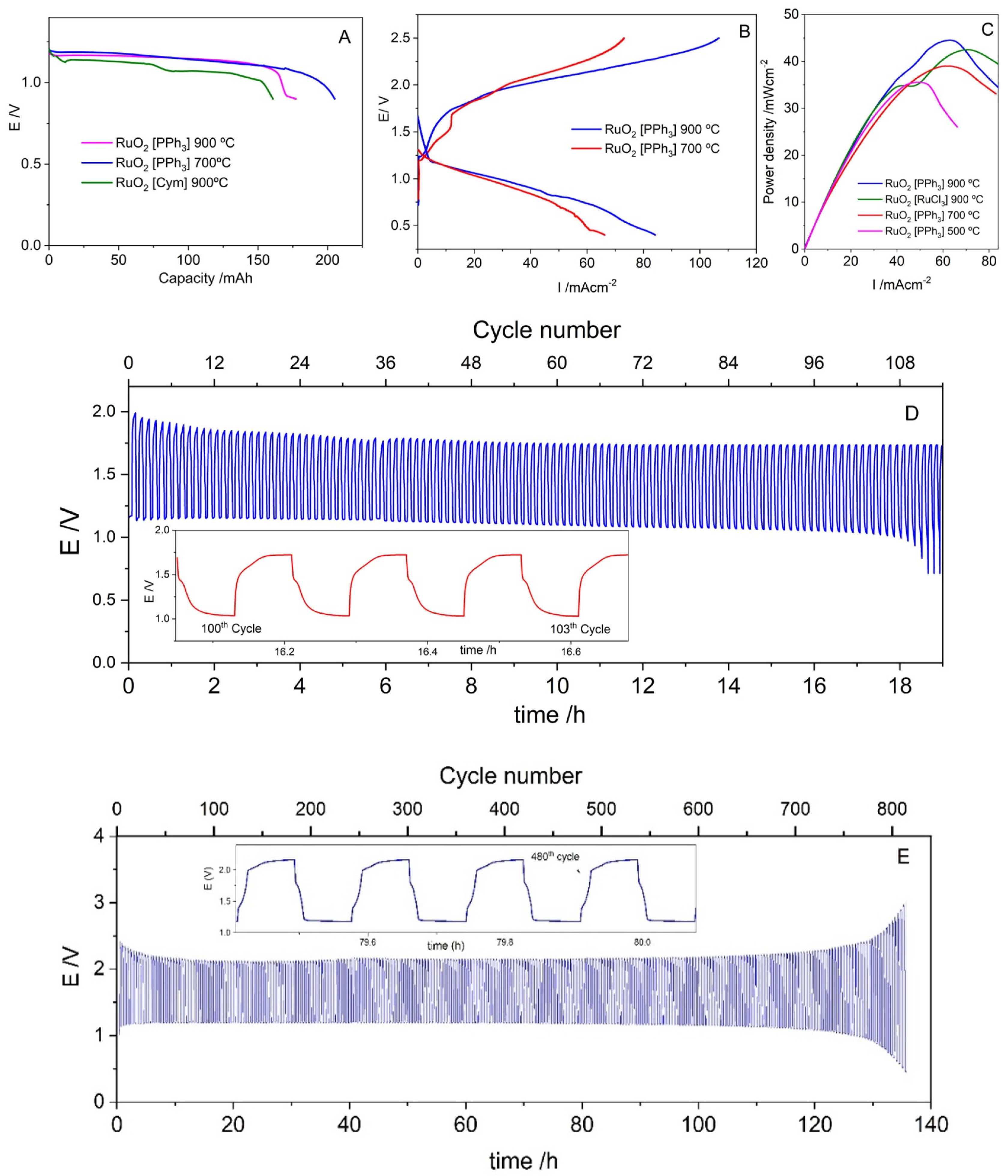
3.2.3. Zn/Battery Discharge/Charge
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dromantien, R.Ė.; Tripolskaja, L. European Association for Storage Energy; The European Association for Storage of Energy (EASE): Brussels, Belgium, 2009; Volume 2, pp. 97–110. [Google Scholar]
- Santos, F.; Romero, A.J.F. Hydration as a solution to zinc batteries. Nat. Sustain. 2022, 5, 179–180. [Google Scholar] [CrossRef]
- Santos, F.; Urbina, A.; Abad, J.; López, R.; Toledo, C.; Romero, A.J.F. Environmental and economical assessment for a sustainable Zn/air battery. Chemosphere 2020, 250, 126273. [Google Scholar] [CrossRef]
- Ren, S.; Duan, X.; Liang, S.; Zhang, M.; Zheng, H. Bifunctional electrocatalysts for Zn–air batteries: Recent developments and future perspectives. J. Mater. Chem. A Mater. 2020, 8, 6144–6182. [Google Scholar] [CrossRef]
- Si, F.; Zhang, Y.; Yan, L.; Zhu, J.; Xiao, M.; Liu, C.; Xing, W.; Zhang, J. Electrochemical Oxygen Reduction Reaction; Elsevier: Amsterdam, The Netherlands, 2014. [Google Scholar] [CrossRef]
- Keersemaker, M. Critical Raw Materials. In Suriname Revisited: Economic Potential of Its Mineral Resources; Springer Briefs in Earth Sciences; Springer: Cham, Switzerland, 2020; pp. 69–82. [Google Scholar] [CrossRef]
- Yang, C.; Liu, Z. Bifunctional OER-ORR Electrodes for Metal-Air Batteries; Elsevier Inc.: Amsterdam, The Netherlands, 2021; Volume 2. [Google Scholar] [CrossRef]
- Ma, R.; Lin, G.; Zhou, Y.; Liu, Q.; Zhang, T.; Shan, G.; Yang, M.; Wang, J. A review of oxygen reduction mechanisms for metal-free carbon-based electrocatalysts. NPJ Comput. Mater. 2019, 5, 78. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Li, G.; Gao, Y.; Xiao, Z.; Zhang, J.; Wang, Q.; Zhang, X.; Wang, L. Doping carbon nanotubes with N, S, and B for electrocatalytic oxygen reduction: A systematic investigation on single, double, and triple doped modes. Catal. Sci. Technol. 2017, 7, 4007–4016. [Google Scholar] [CrossRef]
- Almodóvar, P.; Santos, F.; González, J.; Ramírez-Castellanos, J.; González-Calbet, J.M.; Díaz-Guerra, C.; Romero, A.J.F. Study of Cr2O3 nanoparticles supported on carbonaceous materials as catalysts for O2 reduction reaction. J. Electroanal. Chem. 2021, 895, 115441. [Google Scholar] [CrossRef]
- Jung, S.H.; Kim, D.H.; Brüner, P.; Lee, H.; Hah, H.J.; Kim, S.K.; Jung, Y.S. Extremely conductive RuO2-coated LiNi0.5Mn1.5O4 for lithium-ion batteries. Electrochim. Acta 2017, 232, 236–243. [Google Scholar] [CrossRef]
- Jiao, Y.; Jiang, H.; Chen, F. RuO2/TiO2/Pt Ternary Photocatalysts with Epitaxial Heterojunction and Their Application in CO Oxidation. ACS Catal. 2014, 4, 2249–2257. [Google Scholar] [CrossRef]
- Gobal, F.; Faraji, M. RuO2/MWCNT/stainless steel mesh as a novel positive electrode in vanadium redox flow batteries. RSC Adv. 2015, 5, 68378–68384. [Google Scholar] [CrossRef]
- Park, H.-S.; Seo, E.; Yang, J.; Lee, Y.; Kim, B.-S.; Song, H.-K. Bifunctional hydrous RuO2 nanocluster electrocatalyst embedded in carbon matrix for efficient and durable operation of rechargeable zinc–air batteries. Sci. Rep. 2017, 7, 7150. [Google Scholar] [CrossRef] [Green Version]
- Cruz, J.C.; Baglio, V.; Siracusano, S.; Antonucci, V.; Aricò, A.S.; Ornelas, R.; Ortiz-Frade, L.; Osorio-Monreal, G.; Durón-Torres, S.M.; Arriaga, L.G. Preparation and Characterization of Ruo2 Catalysts for Oxygen Evolution in a Solid Polymer Electrolyte—Tags: PHOTOSYNTHETIC oxygen evolution POLYELECTROLYTES. Int. J. Electrochem. Sci. 2011, 6, 6607–6619. [Google Scholar]
- Cherevko, S.; Geiger, S.; Kasian, O.; Kulyk, N.; Grote, J.-P.; Savan, A.; Shrestha, B.R.; Merzlikin, S.; Breitbach, B.; Ludwig, A.; et al. Oxygen and hydrogen evolution reactions on Ru, RuO2, Ir, and IrO2 thin film electrodes in acidic and alkaline electrolytes: A comparative study on activity and stability. Catal. Today 2016, 262, 170–180. [Google Scholar] [CrossRef]
- Doyle, R.L.; Godwin, I.J.; Brandon, M.P.; Lyons, M.E.G. Redox and electrochemical water splitting catalytic properties of hydrated metal oxide modified electrodes. Phys. Chem. Chem. Phys. 2013, 15, 13737. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Shang, Y.; Lu, Y.; Liu, C.; Li, Z.; Liu, Q. A free-standing biomass-derived RuO2/N-doped porous carbon cathode towards highly performance lithium-oxygen batteries. J. Power Sources 2020, 471, 228444. [Google Scholar] [CrossRef]
- Cherevko, S.; Geiger, S.; Kasian, O.; Kulyk, N.; Grote, J.-P.; Savan, A.; Shrestha, B.R.; Merzlikin, S.; Breitbach, B.; Ludwig, A.; et al. Ruthenium Phosphide Synthesis and Electroactivity toward Oxygen Reduction in Acid Solutions. ACS Catal. 2015, 5, 4260–4267. [Google Scholar] [CrossRef]
- Anastasijević, N.A.; Dimitrijević, Z.M.; Adžić, R.R. Oxygen reduction on a ruthenium electrode in alkaline electrolytes. J. Electroanal. Chem. 1986, 199, 351–364. [Google Scholar] [CrossRef]
- Majumdar, D.; Maiyalagan, T.; Jiang, Z. Recent Progress in Ruthenium Oxide-Based Composites for Supercapacitor Applications. ChemElectroChem 2019, 6, 4343–4372. [Google Scholar] [CrossRef]
- Izumizaki, Y.; Park, K.C.; Tachibana, Y.; Tomiyasu, H.; Fujii, Y. Organic decomposition in supercritical water by an aid of ruthenium (iv) oxide as a catalyst-exploitation of biomass resources for hydrogen production. Prog. Nucl. Energy 2005, 47, 544–552. [Google Scholar] [CrossRef]
- Iqbal, M.N.; Abdel-Magied, A.F.; Abdelhamid, H.N.; Olsén, P.; Shatskiy, A.; Zou, X.; Åkermark, B.; Kärkäs, M.D.; Johnston, E.V. Mesoporous Ruthenium Oxide: A Heterogeneous Catalyst for Water Oxidation. ACS Sustain. Chem. Eng. 2017, 5, 9651–9656. [Google Scholar] [CrossRef]
- Musić, S.; Popović, S.; Maljković, M.; Furić, K.; Gajović, A. Influence of synthesis procedure on the formation of RuO2. Mater. Lett. 2002, 56, 806–811. [Google Scholar] [CrossRef]
- Nguyen, T.D.; Scherer, G.G.; Xu, Z.J. A Facile Synthesis of Size-Controllable IrO2 and RuO2 Nanoparticles for the Oxygen Evolution Reaction. Electrocatalysis 2016, 7, 420–427. [Google Scholar] [CrossRef]
- Zhang, Y.; Yue, L.; Teng, K.; Yuan, S.; Ma, H. Synthesis and Characterization of RuO2 Anode Materials with Large Surface Areas for Oxygen Evolution Reaction. J. New Mater. Electrochem. Syst. 2012, 15, 271–276. [Google Scholar] [CrossRef]
- Refat, M.S.; Saad, H.A.; Gobouri, A.A.; Alsawat, M.; Belgacem, K.; Majrashi, B.M.; Adam, A.M.A. RuO2 Nanostructures from Ru(III) Complexes As a New Smart Nanomaterials for Using in the Recycling and Sustainable Wastewater Treatment: Synthesis, Characterization, and Catalytic Activity in the Hydrogen Peroxide Decomposition. Russ. J. Phys. Chem. A 2021, 95, S346–S351. [Google Scholar] [CrossRef]
- Zhang, N.-N.; Bigdeli, F.; Miao, Q.; Hu, M.-L.; Morsali, A. Ultrasonic-assisted synthesis, characterization and DNA binding studies of Ru(II) complexes with the chelating N-donor ligand and preparing of RuO2 nanoparticles by the easy method of calcination. J. Organomet. Chem. 2018, 878, 11–18. [Google Scholar] [CrossRef]
- Pérez, J.; Serrano, J.L.; Granados, J.E.; Alcolea, L.A. Recovering palladium from its surplus complexes in research laboratories by solid state thermal treatment. RSC Adv. 2013, 3, 4558. [Google Scholar] [CrossRef]
- Pérez, J.; Serrano, J.L.; Sánchez, G.; Lozano, P.; da Silva, I.; Alcolea, A. From Coordination Complexes to Potential Heterogeneous Catalysts via Solid-State Thermal Decomposition: Precursor, Atmosphere and Temperature as Tuning Variables. ChemistrySelect 2019, 4, 8365–8371. [Google Scholar] [CrossRef]
- Ding, Y.; Zhang, S.; Liu, B.; Zheng, H.; Chang, C.; Ekberg, C. Recovery of precious metals from electronic waste and spent catalysts: A review. Resour. Conserv. Recycl. 2019, 141, 284–298. [Google Scholar] [CrossRef]
- Ceurstemont, S. Urban Mining to Reduce Environmental Footprint of Consumer Goods. 2018. Available online: https://ec.europa.eu/research-and-innovation/en/horizon-magazine/urban-mining-reduce-environmental-footprint-consumer-goods (accessed on 16 December 2021).
- Astrand, C.; Zackrisson, M.; Bengtsson, G. Waste and recycling. In Measuring Your Company’s Environmental Impact; Earthsca: London, UK, 2020; pp. 39–42. [Google Scholar] [CrossRef]
- Santos, F.; Tafur, J.P.; Abad, J.; Romero, A.J.F. Structural modifications and ionic transport of PVA-KOH hydrogels applied in Zn/Air batteries. J. Electroanal. Chem. 2019, 850, 113380. [Google Scholar] [CrossRef]
- Morgan, D.J. Resolving ruthenium: XPS studies of common ruthenium materials. Surf. Interface Anal. 2015, 47, 1072–1079. [Google Scholar] [CrossRef]
- Garsany, Y.; Ge, J.; St-Pierre, J.; Rocheleau, R.; Swider-Lyons, K. ORR Measurements Reproducibility Using a RRDE. ECS Trans. 2013, 58, 1233–1241. [Google Scholar] [CrossRef] [Green Version]
- Näslund, L.-Å.; Ingason, Á.S.; Holmin, S.; Rosen, J. Formation of RuO(OH)2 on RuO2—Based Electrodes for Hydrogen Production. J. Phys. Chem. C 2014, 118, 15315–15323. [Google Scholar] [CrossRef] [Green Version]
- Ananth, A.; Gandhi, M.S.; Mok, Y.S. A dielectric barrier discharge (DBD) plasma reactor: An efficient tool to prepare novel RuO2 nanorods. J. Phys. D Appl. Phys. 2013, 46, 155202. [Google Scholar] [CrossRef]
- Rochefort, D.; Dabo, P.; Guay, D.; Sherwood, P.M.A. XPS investigations of thermally prepared RuO2 electrodes in reductive conditions. Electrochim. Acta 2003, 48, 4245–4252. [Google Scholar] [CrossRef]
- Wagner, C.; Naumkin, A.; Kraut-Vass, A.; Allison, J.; Powell, C.; Rumble, J.R., Jr. NIST Standard Reference Database 20. 2003. Available online: http:/srdata.nist.gov/xps/ (accessed on 12 May 2022).
- Swift, P. Adventitious carbon?the panacea for energy referencing? Surf. Interface Anal. 1982, 4, 47–51. [Google Scholar] [CrossRef]
- Fukuoka, H.; Imoto, H.; Saito, T. New polymorphs of RuIIIP3O9: Cyclo-hexaphosphate Ru2P6O18 and metaphosphate Ru(PO3)3 with a novel structure. J. Solid State Chem. 1995, 119, 107–114. [Google Scholar] [CrossRef]
- Imoto, H.; Fukuoka, H.; Tsunesawa, S.; Horiuchi, H.; Amemiya, T.; Koga, N. Preparation and Crystal Structure of Ruthenium Metaphosphate Ru(PO3)3 with an 8-fold Superstructure. Analysis of Structural Frustration with a Simple Model. Inorg. Chem. 1997, 36, 4172–4181. [Google Scholar] [CrossRef]
- Gardner, S.D.; Singamsetty, C.S.K.; Booth, G.L.; He, G.-R.; Pittman, C.U. Surface characterization of carbon fibers using angle-resolved XPS and ISS. Carbon N. Y. 1995, 33, 587–595. [Google Scholar] [CrossRef]
- Mainar, A.R.; Leonet, O.; Bengoechea, M.; Boyano, I.; de Meatza, I.; Kvasha, A.; Guerfi, A.; Blázquez, J.A. Alkaline aqueous electrolytes for secondary zinc-air batteries: An overview. Int. J. Energy Res. 2016, 40, 1032–1049. [Google Scholar] [CrossRef]
- Daems, N.; Breugelmans, T.; Vankelecom, I.F.J.; Pescarmona, P. Influence of the Composition and Preparation of the Rotating Disk Electrode on the Performance of Mesoporous Electrocatalysts in the Alkaline Oxygen Reduction Reaction. ChemElectroChem 2018, 5, 119–128. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.; Zheng, Y.; Jaroniec, M.; Qiao, S.Z. Determination of the Electron Transfer Number for the Oxygen Reduction Reaction: From Theory to Experiment. ACS Catal. 2016, 6, 4720–4728. [Google Scholar] [CrossRef]
- Chai, G.; Hou, Z.; Ikeda, T.; Terakura, K. Two-Electron Oxygen Reduction on Carbon Materials Catalysts: Mechanisms and Active Sites Two-Electron Oxygen Reduction on Carbon Materials Catalysts: Mechanisms and Active Sites. J. Phys. Chem. C 2017, 121, 14524–14533. [Google Scholar] [CrossRef]
- Di Noto, V.; Negro, E.; Nale, A.; Pagot, G.; Vezzù, K.; Atanassov, P. Hidden in plain sight: Unlocking the full potential of cyclic voltammetry with the thin-film rotating (ring) disk electrode studies for the investigation of oxygen reduction reaction electrocatalysts. Curr. Opin. Electrochem. 2021, 25, 100626. [Google Scholar] [CrossRef]
- Xu, S.; Kim, Y.; Higgins, D.; Yusuf, M.; Jaramillo, T.F.; Prinz, F.B. Building upon the Koutecky-Levich Equation for Evaluation of Next-Generation Oxygen Reduction Reaction Catalysts. Electrochim. Acta 2017, 255, 99–108. [Google Scholar] [CrossRef]
- Du, C.; Tan, Q.; Yin, G.; Zhang, J. Rotating Ring-Disk Electrode Method. In Rotating Electrode Methods and Oxygen Reduction Electrocatalysts; Elsevier: Amsterdam, The Netherlands, 2014; pp. 171–198. [Google Scholar] [CrossRef]
- Zhang, H.-J.; Li, H.; Li, X.; Qiu, H.; Yuan, X.; Zhao, B.; Ma, Z.-F.; Yang, J. Pyrolyzing cobalt diethylenetriamine chelate on carbon (CoDETA/C) as a family of non-precious metal oxygen reduction catalyst. Int. J. Hydrogen Energy 2014, 39, 267–276. [Google Scholar] [CrossRef]
- Mamlouk, M.; Kumar, S.M.S.; Gouerec, P.; Scott, K. Electrochemical and fuel cell evaluation of Co based catalyst for oxygen reduction in anion exchange polymer membrane fuel cells. J. Power Sources 2011, 196, 7594–7600. [Google Scholar] [CrossRef]
- Khotseng, L. Oxygen Reduction Reaction. In Electrocatalysts for Fuel Cells and Hydrogen Evolution—Theory to Design; IntechOpen: London, UK, 2018. [Google Scholar] [CrossRef]
- Briega-Martos, V.; Ferre-Vilaplana, A.; de la Peña, A.; Segura, J.L.; Zamora, F.; Feliu, J.M.; Herrero, E. An Aza-Fused π-Conjugated Microporous Framework Catalyzes the Production of Hydrogen Peroxide. ACS Catal. 2017, 7, 1015–1024. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.-W.; Su, D. Heterogeneous nanocarbon materials for oxygen reduction reaction. Energy Environ. Sci. 2014, 7, 576. [Google Scholar] [CrossRef]
- Jiang, R.; Tran, D.T.; Li, J.; Chu, D. Ru@RuO2 Core-Shell Nanorods: A Highly Active and Stable Bifunctional Catalyst for Oxygen Evolution and Hydrogen Evolution Reactions. Energy Environ. Mater. 2019, 2, 201–208. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.; Li, C.; Li, W.; Guo, H.; Su, X.; He, P.; Wang, Y.; Xia, Y. Ruthenium oxide coated ordered mesoporous carbon nanofiber arrays: A highly bifunctional oxygen electrocatalyst for rechargeable Zn–air batteries. J. Mater. Chem. A Mater. 2016, 4, 6282–6289. [Google Scholar] [CrossRef]
- Jozwiak, L.; Balcerzak, J.; Tyczkowski, J. Plasma-Deposited Ru-Based Thin Films for Photoelectrochemical Water Splitting. Catalysts 2020, 10, 278. [Google Scholar] [CrossRef] [Green Version]
- Guerrini, E.; Trasatti, S. Electrocatalysis in Water Electrolysis. In Catalysis for Sustainable Energy Production; Wiley: Weinheim, Germany; pp. 235–269. [CrossRef]
- Zhang, J.; Xia, Z.; Dai, L. Carbon-based electrocatalysts for advanced energy conversion and storage. Sci. Adv. 2015, 1, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Dai, H. Recent advances in Zinc-air batteries. Chem. Soc. Rev. 2014, 43, 5257–5275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Kaempgen, M.; Nopphawan, P.; Wee, G.; Mhaisalkar, S.; Srinivasan, M. Silver nanoparticle-decorated carbon nanotubes as bifunctional gas-diffusion electrodes for zinc–air batteries. J. Power Sources 2010, 195, 4350–4355. [Google Scholar] [CrossRef]
- Liang, H.-W.; Wu, Z.-Y.; Chen, L.-F.; Li, C.; Yu, S.-H. Bacterial cellulose derived nitrogen-doped carbon nanofiber aerogel: An efficient metal-free oxygen reduction electrocatalyst for zinc-air battery. Nano Energy 2015, 11, 366–376. [Google Scholar] [CrossRef]
- Li, S.; Cheng, C.; Liang, H.-W.; Feng, X.; Thomas, A. 2D Porous Carbons prepared from Layered Organic-Inorganic Hybrids and their Use as Oxygen-Reduction Electrocatalysts. Adv. Mater. 2017, 29, 1700707. [Google Scholar] [CrossRef]
- Zhang, M.; Chen, J.; Li, H.; Cai, P.; Li, Y.; Wen, Z. Ru-RuO2/CNT hybrids as high-activity pH-universal electrocatalysts for water splitting within 0.73 V in an asymmetric-electrolyte electrolyzer. Nano Energy 2019, 61, 576–583. [Google Scholar] [CrossRef]
- Dalton, F. Historical Origins of the Rotating Ring-Disk Electrode. Electrochem. Soc. Interface 2016, 25, 50–59. [Google Scholar] [CrossRef]










| Compound | Eonset [a] (mV) | E [a],[b] (mV) | nKL [c] | nRRDE [c] | Tafel [d] (mV dec−1) |
|---|---|---|---|---|---|
| RuO2 [RuCl3] | 654 ± 8 | 555 ± 7 | 3.8 | 3.4 | 188 ± 1 |
| RuO2 [Cym] | 697 ± 8 | 462 ± 10 | 2.6 | 2.6 | 82 ± 3 |
| RuO2[PPh3] 500 °C | 677 ± 20 | 522 ± 15 | 2.6 | 3.1 | 155 ± 3 |
| RuO2[PPh3] 700 °C | 714 ± 1 | 514 ± 7 | 2.8 | 3.8 | 106 ± 9 |
| RuO2[PPh3] 900 °C | 753 ± 10 | 602 ± 5 | 3.6 | 3.6 | 106 ± 11 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lorca, S.; Torres, J.; Serrano, J.L.; Pérez, J.; Abad, J.; Santos, F.; Fernández Romero, A.J. Bifunctional P-Containing RuO2 Catalysts Prepared from Surplus Ru Co-Ordination Complexes and Applied to Zn/Air Batteries. Nanomaterials 2023, 13, 115. https://doi.org/10.3390/nano13010115
Lorca S, Torres J, Serrano JL, Pérez J, Abad J, Santos F, Fernández Romero AJ. Bifunctional P-Containing RuO2 Catalysts Prepared from Surplus Ru Co-Ordination Complexes and Applied to Zn/Air Batteries. Nanomaterials. 2023; 13(1):115. https://doi.org/10.3390/nano13010115
Chicago/Turabian StyleLorca, Sebastián, Javier Torres, José L. Serrano, José Pérez, José Abad, Florencio Santos, and Antonio J. Fernández Romero. 2023. "Bifunctional P-Containing RuO2 Catalysts Prepared from Surplus Ru Co-Ordination Complexes and Applied to Zn/Air Batteries" Nanomaterials 13, no. 1: 115. https://doi.org/10.3390/nano13010115
APA StyleLorca, S., Torres, J., Serrano, J. L., Pérez, J., Abad, J., Santos, F., & Fernández Romero, A. J. (2023). Bifunctional P-Containing RuO2 Catalysts Prepared from Surplus Ru Co-Ordination Complexes and Applied to Zn/Air Batteries. Nanomaterials, 13(1), 115. https://doi.org/10.3390/nano13010115