
Metallic Nanostructures Based on DNA Nanoshapes



Abstract
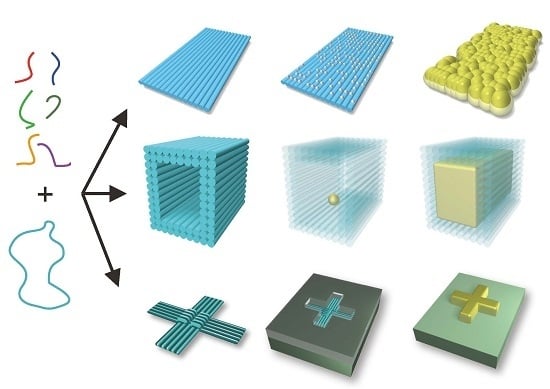
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
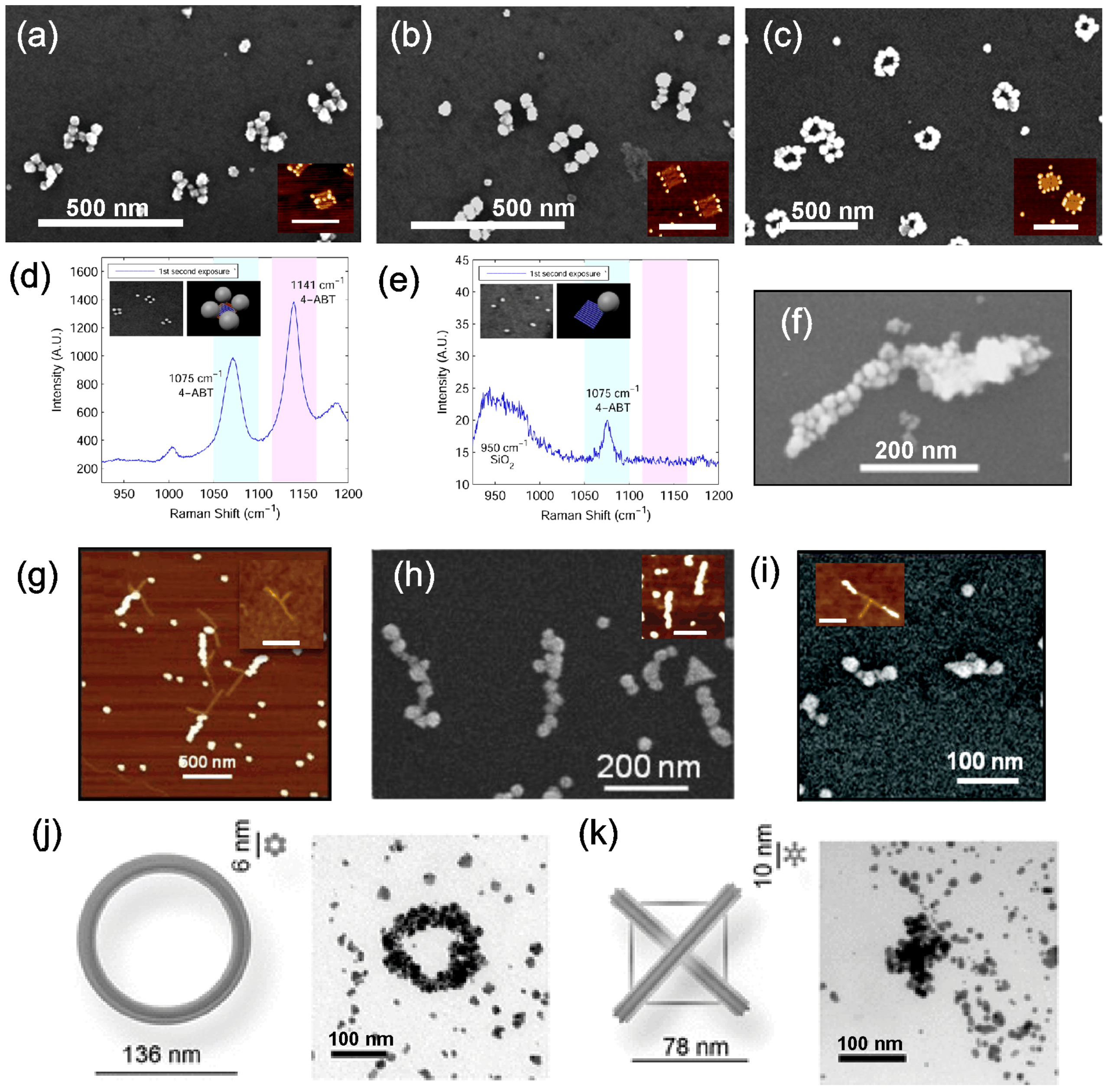{kind=link}
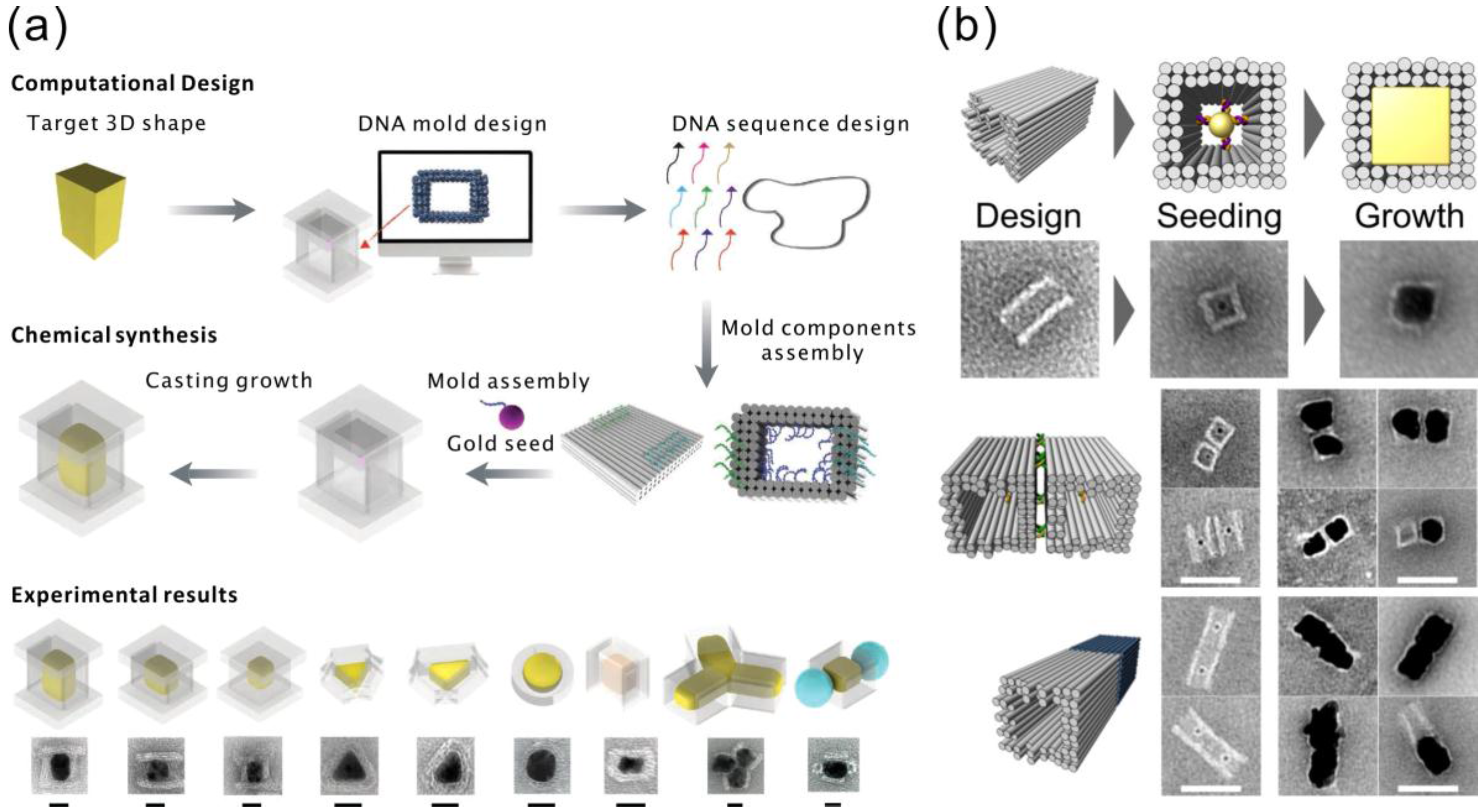{kind=link}
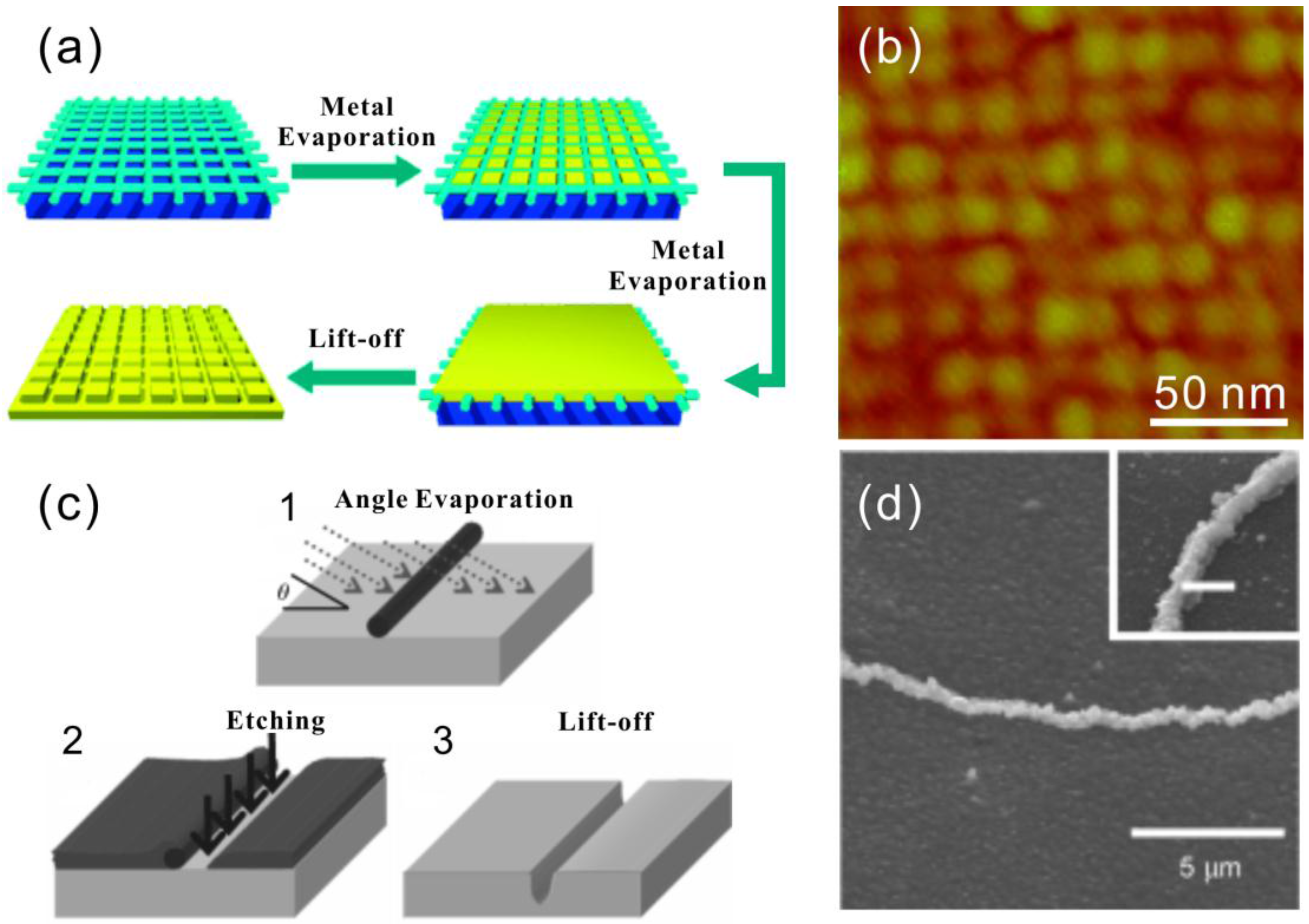{kind=link}
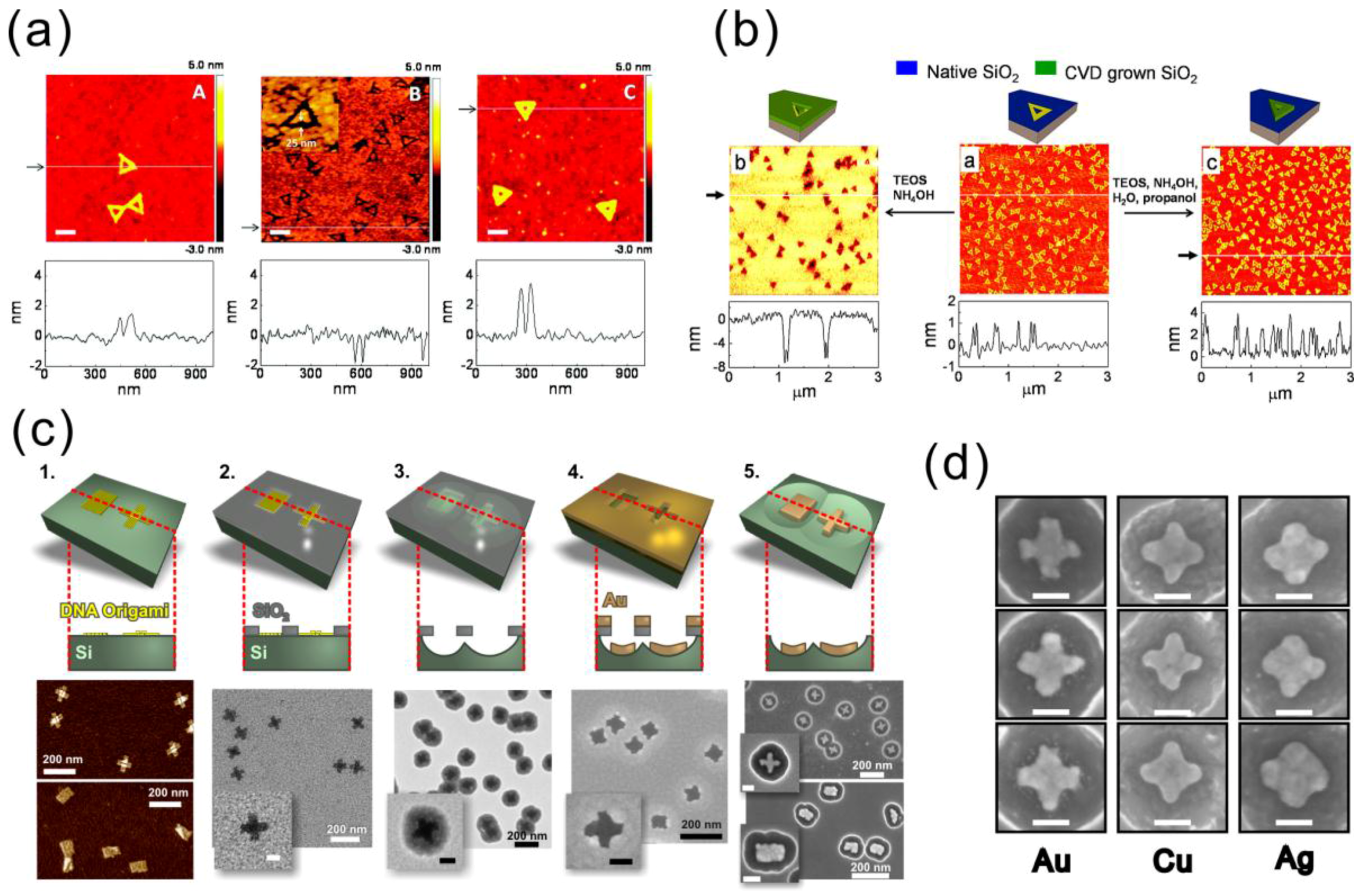{kind=link}
1. Introduction
2. Structural DNA Nanotechnology
2.1. DNA Self-Assembled Nanostructures
2.2. Conjugation of DNA Nanostructures with Metallic NPs
3. Chemical Metallization of DNA Nanostructures
3.1. Metallization of dsDNA and ssDNA
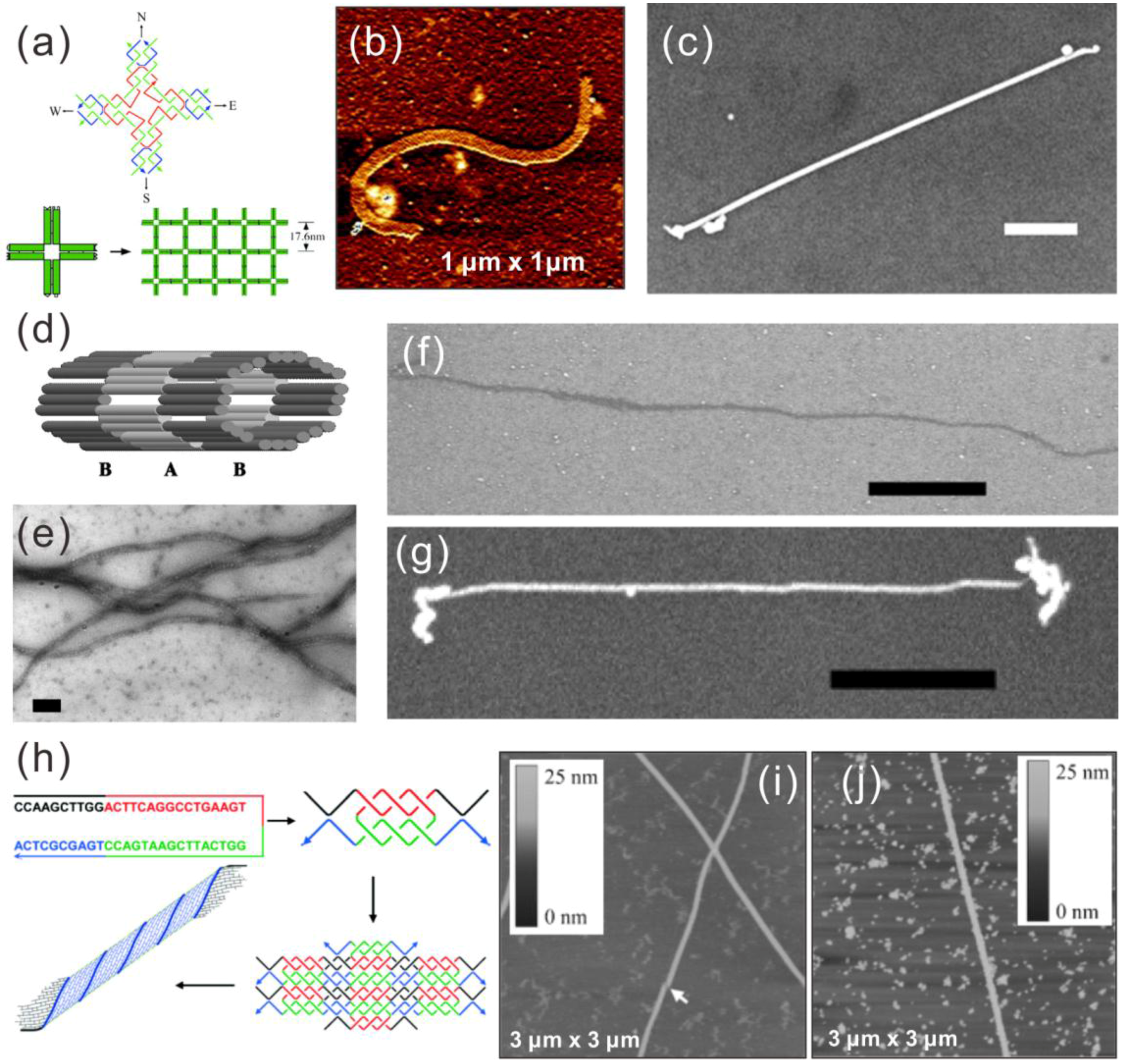
3.2. Tile-Based DNA Nanostructures
3.3. DNA Origami Metallization
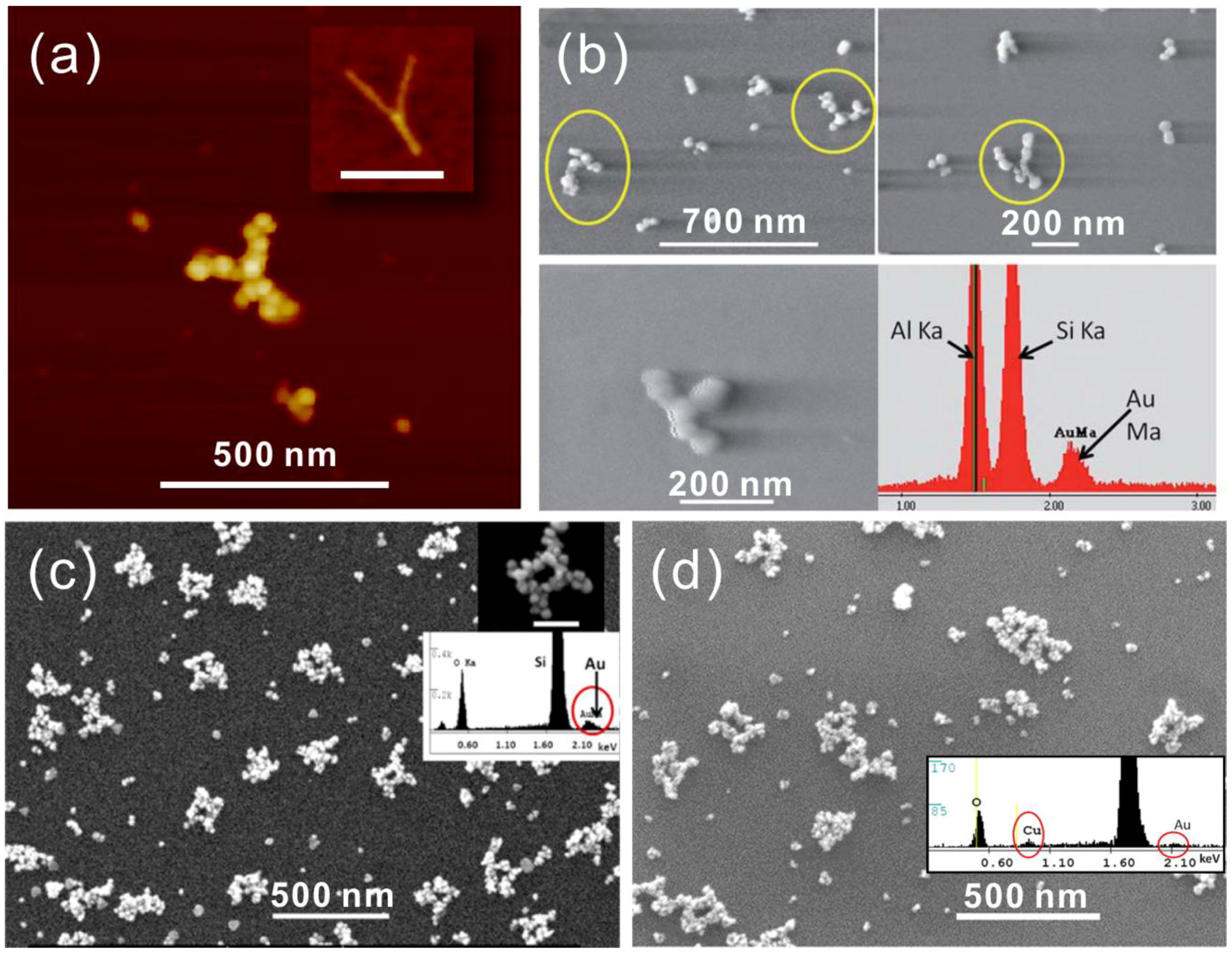
3.4. Metallization Based on Nucleation on Functionalized Nanoparticles
4. Casting of Nanoparticles Using DNA Molds
5. DNA Nanolithography
5.1. dsDNA and DNA Nanogrid as Masks in Lithography
5.2. Silica Mask from DNA Origami for Metal Evaporation
5.3. Patterning of Graphene with Metallized DNA Nanostructures
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| λ-DNA | a linear dsDNA genome from a bacterial virus called lambda phage |
| 2D | two-dimensional |
| 3D | three-dimensional |
| AFM | atomic force microscopy |
| AuNP | gold nanoparticle |
| B-DNA | double-helical DNA in the B-form (geometry attribute) |
| BzMA | benzyl methacrylate |
| cDNA | complementary DNA |
| CVD | chemical vapor deposition |
| DNA | deoxyribonucleic acid |
| DMAB | dimethylaminoborane |
| dsDNA | double-stranded DNA |
| DX | double-crossover |
| EBL | electron beam lithography |
| EDTA | ethylenediaminetetraacetic acid |
| EDX | energy-dispersive X-ray spectroscopy |
| I-V | current-voltage |
| MNP | metal nanoparticle |
| NP | nanoparticle |
| nt | nucleotide |
| PEG | polyethylene glycol |
| PVD | physical vapor deposition |
| RIE | reactive ion etching |
| SEM | scanning electron microscopy |
| SPM | spermine |
| SERS | surface-enhanced Raman spectroscopy |
| ssDNA | single-stranded DNA |
| TEM | transmission electron microscopy |
| TEOS | tetraethyl orthosilicate |
| TOAB | tetraoctylammonium bromide |
| TX | triple-crossover |
| UV | ultraviolet |
References
- Moore, G.E. Cramming more components onto integrated circuits. Proc. IEEE 1965, 86, 82–85. [Google Scholar] [CrossRef]
- Waldrop, M.M. The chips are down for Moore’s law. Nature 2016, 530, 144–147. [Google Scholar] [CrossRef] [PubMed]
- Maier, S.A. Plasmonics: Fundamentals and Applications; Springer US: New York, NY, USA, 2007. [Google Scholar]
- Jahn, M.; Patze, S.; Hidi, I.J.; Knipper, R.; Radu, A.I.; Muhlig, A.; Yuksel, S.; Peksa, V.; Weber, K.; Mayerhofer, T.; et al. Plasmonic nanostructures for surface enhanced spectroscopic methods. Analyst 2016, 141, 756–793. [Google Scholar] [CrossRef] [PubMed]
- Gersten, J.; Nitzan, A. Spectroscopic properties of molecules interacting with small dielectric particles. J. Chem. Phys. 1981, 75, 1139–1152. [Google Scholar] [CrossRef]
- Kinkhabwala, A.; Yu, Z.F.; Fan, S.H.; Avlasevich, Y.; Mullen, K.; Moerner, W.E. Large single-molecule fluorescence enhancements produced by a bowtie nanoantenna. Nat. Photonics 2009, 3, 654–657. [Google Scholar] [CrossRef]
- Lakowicz, J.R.; Geddes, C.D.; Gryczynski, I.; Malicka, J.; Gryczynski, Z.; Aslan, K.; Lukomska, J.; Matveeva, E.; Zhang, J.A.; Badugu, R.; et al. Advances in surface-enhanced fluorescence. J. Fluoresc. 2004, 14, 425–441. [Google Scholar] [CrossRef] [PubMed]
- Fleischmann, M.; Hendra, P.J.; McQuillan, A.J. Raman-spectra of pyridine adsorbed at a silver electrode. Chem. Phys. Lett. 1974, 26, 163–166. [Google Scholar] [CrossRef]
- Albrecht, M.G.; Creighton, J.A. Anomalously intense Raman-spectra of pyridine at a silver electrode. J. Am. Chem. Soc. 1977, 99, 5215–5217. [Google Scholar] [CrossRef]
- Kleinman, S.L.; Frontiera, R.R.; Henry, A.I.; Dieringer, J.A.; Van Duyne, R.P. Creating, characterizing, and controlling chemistry with SERS hot spots. Phys. Chem. Chem. Phys. 2013, 15, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Hartstein, A.; Kirtley, J.R.; Tsang, J.C. Enhancement of the infrared-absorption from molecular monolayers with thin metal overlayers. Phys. Rev. Lett. 1980, 45, 201–204. [Google Scholar] [CrossRef]
- Hatta, A.; Ohshima, T.; Suëtaka, W. Observation of the enhanced infrared-absorption of para-nitrobenzoate on Ag island films with an Atr technique. Appl. Phys. A 1982, 29, 71–75. [Google Scholar] [CrossRef]
- Homola, J. Surface Plasmon Resonance Based Sensors, Springer Series on Chemical Sensors and Biosensors; Springer-Verlag: Berlin/Heidelberg, Germany; New York, NY, USA, 2006. [Google Scholar]
- Willets, K.A.; Van Duyne, R.P. Localized surface plasmon resonance spectroscopy and sensing. Annu. Rev. Phys. Chem. 2007, 58, 267–297. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, A.V.; Han, D.; Shih, W.M.; Yan, H. Challenges and opportunities for structural DNA nanotechnology. Nat. Nanotechnol. 2011, 6, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Linko, V.; Dietz, H. The enabled state of DNA nanotechnology. Curr. Opin. Biotechnol. 2013, 24, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Porath, D.; Cuniberti, G.; Di Felice, R. Charge transport in DNA-based devices. Top. Curr. Chem. 2004, 237, 183–228. [Google Scholar]
- Linko, V.; Paasonen, S.-T.; Kuzyk, A.; Törma, P.; Toppari, J.J. Characterization of the conductance mechanisms of DNA origami by AC impedance spectroscopy. Small 2009, 5, 2382–2386. [Google Scholar] [CrossRef] [PubMed]
- Linko, V.; Leppiniemi, J.; Paasonen, S.-T.; Hytönen, V.P.; Toppari, J.J. Defined-size DNA triple crossover construct for molecular electronics: Modification, positioning and conductance properties. Nanotechnology 2011, 22, 275610. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Linko, V.; Dietz, H.; Toppari, J.J. Dielectrophoretic trapping of multilayer DNA origami nanostructures and DNA origami-induced local destruction of silicon dioxide. Electrophoresis 2015, 36, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Linko, V.; Toppari, J.J. Self-assembled DNA-based structures for nanoelectronics. J. Self-Assem. Mol. Electron. 2013, 1, 101–124. [Google Scholar] [CrossRef]
- Seeman, N.C. Nucleic-acid junctions and lattices. J. Theor. Biol. 1982, 99, 237–247. [Google Scholar] [CrossRef]
- Yan, H.; Park, S.H.; Finkelstein, G.; Reif, J.H.; LaBean, T.H. DNA-templated self-assembly of protein arrays and highly conductive nanowires. Science 2003, 301, 1882–1884. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Chen, Y.; He, Y.; Ribbe, A.E.; Mao, C. Approaching the limit: Can one DNA oligonucleotide assemble into large nanostructures? Angew. Chem. Int. Ed. 2006, 45, 1942–1945. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Birktoft, J.J.; Chen, Y.; Wang, T.; Sha, R.; Constantinou, P.E.; Ginell, S.L.; Mao, C.; Seeman, N.C. From molecular to macroscopic via the rational design of a self-assembled 3D DNA crystal. Nature 2009, 461, 74–77. [Google Scholar] [CrossRef] [PubMed]
- Rothemund, P.W.K. Folding DNA to create nanoscale shapes and patterns. Nature 2006, 440, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Douglas, S.M.; Dietz, H.; Liedl, T.; Högberg, B.; Graf, F.; Shih, W.M. Self-assembly of DNA into nanoscale three-dimensional shapes. Nature 2009, 459, 414–418. [Google Scholar] [CrossRef] [PubMed]
- Dietz, H.; Douglas, S.M.; Shih, W.M. Folding DNA into twisted and curved nanoscale shapes. Science 2009, 325, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Pal, S.; Nangreave, J.; Deng, Z.; Liu, Y.; Yan, H. DNA origami with complex curvatures in three-dimensional space. Science 2011, 332, 342–346. [Google Scholar] [CrossRef] [PubMed]
- Wei, B.; Dai, M.; Yin, P. Complex shapes self-assembled from single-stranded DNA tiles. Nature 2012, 485, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Ke, Y.; Ong, L.L.; Shih, W.M.; Yin, P. Three-dimensional structures self-assembled from DNA bricks. Science 2012, 338, 1177–1183. [Google Scholar] [CrossRef] [PubMed]
- Benson, E.; Mohammed, A.; Gardell, J.; Masich, S.; Czeizler, E.; Orponen, P.; Högberg, B. DNA rendering of polyhedral meshes at the nanoscale. Nature 2015, 523, 441–444. [Google Scholar] [CrossRef] [PubMed]
- Veneziano, R.; Ratanalert, S.; Zhang, K.; Zhang, F.; Yan, H.; Chiu, W.; Bathe, M. Designer nanoscale DNA assemblies programmed from the top down. Science 2016, 352. [Google Scholar] [CrossRef] [PubMed]
- Kuzyk, A.; Schreiber, R.; Fan, Z.; Pardatscher, G.; Roller, E.-M.; Högele, A.; Simmel, F.C.; Govorov, A.O.; Liedl, T. DNA-based self-assembly of chiral plasmonic nanostructures with tailored optical response. Nature 2012, 483, 311–314. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.; Deng, Z.; Yan, H.; Cabrini, S.; Zuckermann, R.N.; Bokor, J. Gold nanoparticle self-similar chain structure organized by DNA origami. J. Am. Chem. Soc. 2010, 132, 3248–3249. [Google Scholar] [CrossRef] [PubMed]
- Subramani, R.; Juul, S.; Rotaru, A.; Andersen, F.F.; Gothelf, K.V.; Mamdouh, W.; Besenbacher, F.; Dong, M.; Knudsen, B.R. A novel secondary DNA binding site in human topoisomerase I unravelled by using a 2D DNA origami platform. ACS Nano 2010, 4, 5969–5977. [Google Scholar] [CrossRef] [PubMed]
- Simmel, F.C. DNA-based assembly lines and nanofactories. Curr. Opin. Biotechnol. 2012, 23, 516–521. [Google Scholar] [CrossRef] [PubMed]
- Linko, V.; Eerikäinen, M.; Kostiainen, M.A. A modular DNA origami-based enzyme cascade nanoreactor. Chem. Commun. 2015, 51, 5351–5354. [Google Scholar] [CrossRef] [PubMed]
- Linko, V.; Nummelin, S.; Aarnos, L.; Tapio, K.; Toppari, J.J.; Kostiainen, M.A. DNA-based enzyme reactors and systems. Nanomaterials 2016, 6, 139–154. [Google Scholar] [CrossRef]
- Maune, H.T.; Han, S.-P.; Barish, R.D.; Bockrath, M.; Goddard, W.A., III; Rothemund, P.W.K.; Winfree, E. Self-assembly of carbon nanotubes into two-dimensional geometries using DNA origami templates. Nat. Nanotechnol. 2010, 5, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Eskelinen, A.-P.; Kuzyk, A.; Kaltiaisenaho, T.K.; Timmermans, M.Y.; Nasibulin, A.G.; Kauppinen, E.I.; Törmä, P. Assembly of single-walled carbon nanotubes on DNA-origami templates through streptavidin-biotin interaction. Small 2011, 7, 746–750. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Ralston, J.; Sedev, R.; Beattie, D.A. Functionalized gold nanoparticles, synthesis, structure and colloid stability. J. Colloid Interface Sci. 2009, 331, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Sperling, R.A.; Parak, W.J. Surface modification, functionalization and bioconjugation of colloidal inorganic nanoparticles. Philos. Trans. R. Soc. A 2010, 368, 1333–1383. [Google Scholar] [CrossRef] [PubMed]
- Turkevich, J.; Stevenson, P.C.; Hillier, J. The formation of colloidal gold. J. Phys. Chem. 1953, 57, 670–673. [Google Scholar] [CrossRef]
- Mirkin, C.A.; Letsinger, R.L.; Mucic, R.C.; Storhoff, J.J. A DNA-based method for rationally assembling nanoparticles into macroscopic materials. Nature 1996, 382, 607–609. [Google Scholar] [CrossRef] [PubMed]
- Alivisatos, A.P.; Johnsson, K.P.; Peng, X.; Wilson, T.E.; Loweth, C.J.; Bruchez, M.P.; Schultz, P.G. Organization of “nanocrystal molecules” using DNA. Nature 1996, 382, 609–611. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Carroll, D.L. Synthesis and characterization of truncated triangular silver nanoplates. Nano Lett. 2002, 2, 1003–1007. [Google Scholar] [CrossRef]
- Pérez-Juste, J.; Pastoriza-Santos, I.; Liz-Marzán, L.M.; Mulvaney, P. Gold nanorods: Synthesis, characterization and applications. Coord. Chem. Rev. 2005, 249, 1870–1901. [Google Scholar] [CrossRef]
- Umar, A.; Akhtar, M.S.; Dar, G.N.; Baskoutas, S. Low-temperature synthesis of α-Fe2O3 hexagonal nanoparticles for environmental remediation and smart sensor application. Talanta 2013, 116, 1060–1066. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.-B.; Zhu, J.-J.; Chen, H.-Y. Photochemical preparation of rectangular PbSe and CdSe nanoparticles. J. Cryst. Growth 2003, 252, 587–592. [Google Scholar] [CrossRef]
- Brust, M.; Walker, M.; Bethell, D.; Schiffrin, D.J.; Whyman, R. Synthesis of thiol-derivatised gold nanoparticles in a two-phase liquid-liquid system. J. Chem. Soc. Chem. Commun. 1994, 801–802. [Google Scholar] [CrossRef]
- Love, J.C.; Estroff, L.A.; Kriebel, J.K.; Nuzzo, R.G.; Whitesides, G.M. Self-assembled monolayers of thiolates on metals as a form of nanotechnology. Chem. Rev. 2005, 105, 1103–1169. [Google Scholar] [CrossRef] [PubMed]
- Durocher, S.; Rezaee, A.; Hamm, C.; Rangan, C.; Mittler, S.; Mutus, B. Disulfine-linked, gold nanoparticle based reagent for detecting small molecular weight thiols. J. Am. Chem. Soc. 2009, 131, 2475–2477. [Google Scholar] [CrossRef] [PubMed]
- Uvdal, K.; Persson, I.; Liedberg, B. Tricyclohexylphosphine adsorbed on gold. Langmuir 1995, 11, 1252–1256. [Google Scholar] [CrossRef]
- Leff, D.V.; Brandt, L.; Heath, J.R. Synthesis and characterization of hydrophobic organically-soluble gold nanocrystals functionalized with primary amines. Langmuir 1996, 12, 4723–4730. [Google Scholar] [CrossRef]
- Weisbecker, C.S.; Merritt, M.V.; Whitesides, G.M. Molecular self-assembly of aliphatic thiols on gold colloids. Langmuir 1996, 12, 3763–3772. [Google Scholar] [CrossRef]
- Niidome, T.; Yamagata, M.; Okomoto, Y.; Akiyama, Y.; Takahashi, H.; Kawano, T.; Katayama, Y.; Niidome, Y. PEG-modified gold nanorods with a stealth character for in vivo applications. J. Control. Release 2006, 114, 343–347. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Deivaraj, T.C.; Too, H.-P.; Lee, J.Y. An alternative phase-transfer method of preparing alkylamine-stabilized platinum nanoparticles. J. Phys. Chem. B 2004, 108, 2181–2185. [Google Scholar] [CrossRef]
- Lee, J.; Kim, S.M.; Lee, I.S. Functionalization of hollow nanoparticles for nanoreactor applications. Nano Today 2014, 9, 631–667. [Google Scholar] [CrossRef]
- Keegan, G.L.; Aherne, D.; Defrancq, E.; Gun´ko, Y.K.; Kelly, J.M. Oligonucleotide functionalization of hollow triangular gold silver alloy nanoboxes. J. Phys. Chem. C 2013, 117, 669–676. [Google Scholar] [CrossRef]
- Liu, S.; Zhihua, Z.; Han, M. Gram-scale synthesis and biofunctionalization of silica-coated silver nanoparticles for fast colorimetric DNA detection. Anal. Chem. 2005, 77, 2595–2600. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Li, H.; Liu, H.; Wu, Z.; Qiang, W.; Xu, D. Fabrication of streptavidin functionalized silver nanoparticle decorated graphene and its application in disposable electrochemical sensor for immunoglobulin E. Electrochem. Commun. 2013, 31, 16–19. [Google Scholar] [CrossRef]
- Schmidt, T.J.; Noeske, M.; Gasteiger, H.A. Electrocatalytic activity of PtRu alloy colloids for CO and CO/H2 electrooxidation: Stripping voltammetry and rotating disk measurements. Langmuir 1997, 13, 2591–2595. [Google Scholar] [CrossRef]
- Götz, M.; Wendt, H. Binary and ternary anode catalyst formulations including the elements W, Sn and Mo for PEMFCs operated on methanol or reformate gas. Electrochim. Acta 1998, 43, 3637–3644. [Google Scholar] [CrossRef]
- Braun, E.; Eichen, Y.; Sivan, U.; Ben-Yoseph, G. DNA-templated assembly and electrode attachment of a conducting silver wire. Nature 1998, 391, 775–778. [Google Scholar] [CrossRef] [PubMed]
- Puchkova, A.O.; Sokolov, P.A.; Kasyanenko, N.A. Metallization of DNA on the surface. J. Struct. Chem. 2011, 52, 1195–1201. [Google Scholar] [CrossRef]
- Wirges, C.T.; Timper, J.; Fischler, M.; Sologubenko, A.S.; Mayer, J.; Simon, U.; Carell, T. Controlled nucleaction of DNA metallization. Angew. Chem. Int. Ed. 2009, 48, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Richter, J.; Seidel, R.; Kirsch, R.; Mertig, M.; Pompe, W.; Plaschke, J.; Schackert, H.K. Nanoscale palladium metallization of DNA. Adv. Mater. 2000, 12, 507–510. [Google Scholar] [CrossRef]
- Nguyne, K.; Monteverde, M.; Filoramo, A.; Goux-Capes, L.; Lyonnais, S.; Jegou, P.; Viel, P.; Goffman, M.; Bourgoin, J.-P. Synthesis of thin and highly conductive DNA-based palladium nanowires. Adv. Mater. 2008, 20, 1099–1104. [Google Scholar] [CrossRef]
- Mertig, M.; Ciacchi, L.C.; Seidel, R.; Pompe, W. DNA as a selective metallization template. Nano Lett. 2002, 2, 841–844. [Google Scholar] [CrossRef]
- Seidel, R.; Ciacchi, L.C.; Weigel, M.; Pompe, W.; Mertig, M. Synthesis of platinum cluster chains on DNA templates: Conditions for a template-controlled cluster growth. J. Phys. Chem. B 2004, 108, 10801–10811. [Google Scholar] [CrossRef]
- Becerril, H.A.; Ludtke, P.; Willardson, B.M.; Woolley, A.T. DNA-templated nickel nanostructures and protein assemblies. Langmuir 2006, 22, 10140–10144. [Google Scholar] [CrossRef] [PubMed]
- Atanasova, P.; Weitz, R.T.; Gerstel, P.; Srot, V.; Kopold, P.; van Aken, P.A.; Burghard, M.; Bill, J. DNA-templated synthesis of ZnO thin layers and nanowires. Nanotechnology 2009, 20, 2844–2850. [Google Scholar] [CrossRef] [PubMed]
- Swami, A.S.; Brun, N.; Langevin, D. Phase transfer of gold metallized DNA. J. Clust. Sci. 2009, 20, 281–290. [Google Scholar] [CrossRef]
- Fischler, M.; Simon, U.; Nir, H.; Eichen, Y.; Burley, G.A.; Gierlich, J.; Gramlich, P.M.E.; Carell, T. Formation of bimetallic Ag-Au nanowires by metallization of artificial DNA duplexes. Small 2007, 3, 1049–1055. [Google Scholar] [CrossRef] [PubMed]
- Monson, C.F.; Woolley, A.T. DNA-templated construction of copper nanowires. Nano Lett. 2003, 3, 359–363. [Google Scholar] [CrossRef]
- Zinchenko, A.A.; Yoshikawa, K.; Baigl, D. DNA-templated silver nanorings. Adv. Mater. 2005, 17, 2820–2823. [Google Scholar] [CrossRef]
- Chen, N.; Zinchenko, A.A.; Yoshikawa, K. Probing biopolymer conformation by metallization with noble metals. Nanotechnology 2006, 17, 5224–5232. [Google Scholar] [CrossRef]
- Pu, S.-Y.; Zinchenko, A.; Qin, L.-L.; Ye, C.-W.; Xu, M.; Murata, S. Photochemical metallization to fabricate DNA-templated gold nanorings. Mater. Lett. 2014, 130, 168–171. [Google Scholar] [CrossRef]
- Keren, K.; Krueger, M.; Gilad, R.; Ben-Yoseph, G.; Sivan, U.; Braun, E. Sequence-specific molecular lithography on single DNA molecules. Science 2002, 297, 72–75. [Google Scholar] [CrossRef] [PubMed]
- Berti, L.; Alessandrini, A.; Facci, P. DNA-templated photoinduced silver deposition. J. Am. Chem. Soc. 2005, 127, 11216–11217. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Sun, B.; Meng, F.; Zhang, M.; Chen, X.; Li, M.; Liu, J. One-step synthesis of UV-induced Pt nanotrees on the surface of DNA network. Mater. Res. Bull. 2009, 44, 1270–1274. [Google Scholar] [CrossRef]
- Erler, C.; Günther, K.; Mertig, M. Photo-induced synthesis of DNA-templated metallic nanowires and their integration into micro-fabricated contact arrays. Appl. Surf. Sci. 2009, 255, 9647–9651. [Google Scholar] [CrossRef]
- Sinha, R.P.; Häder, D.-P. UV-induced DNA damage and repair: A review. Photochem. Photobiol. Sci. 2002, 1, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.J.; Wu, P.L.; Bolze, F.; Leung, H.W.C.; Li, K.F.; Mak, N.K.; Kwong, D.W.J.; Nicoud, J.-F.; Cheah, K.W.; Wong, M.S. Cyanines as new fluorescent probes for DNA detection and two-photon excited bioimaging. Org. Lett. 2010, 12, 2194–2197. [Google Scholar] [CrossRef] [PubMed]
- Henderson, P.T.; Jones, D.; Hampikian, G.; Kan, Y.; Schuster, G.B. Long-distance charge transport in duplex DNA: The phonon-assisted polaron-like hopping mechanism. Proc. Natl. Acad. Sci. USA 1998, 96, 8353–8358. [Google Scholar] [CrossRef]
- Liu, D.; Park, S.H.; Reif, J.H.; LaBean, T.H. DNA nanotubes self-assembled from triple-crossover tiles as templates for conductive nanowires. Proc. Natl. Acad. Sci. USA 2004, 101, 717–722. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Geng, Y.; Pound, E.; Gyawali, S.; Ashton, J.R.; Hickey, J.; Woolley, A.T.; Harb, J.N. Metallization of branched DNA origami for nanoelectronic circuit fabrication. ACS Nano 2011, 5, 2240–2247. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.; Liu, J.; Pound, E.; Gyawali, S.; Harb, J.N.; Woolley, A.T. Rapid metallization of lambda DNA and DNA origami using a Pd seeding method. J. Mater. Chem. 2011, 21, 12126–12131. [Google Scholar] [CrossRef]
- Geng, Y.; Pearson, A.C.; Gates, E.P.; Uprety, B.; Davis, R.C.; Harb, J.N.; Woolley, A.T. Electrically conductive gold- and copper-metallized DNA origami nanostructures. Langmuir 2013, 29, 3482–3490. [Google Scholar] [CrossRef] [PubMed]
- Pearson, A.C.; Liu, J.; Pound, E.; Uprety, B.; Woolley, A.T.; Davis, R.C.; Harb, J.N. DNA origami metallized site specifically to form electrically conductive nanowires. J. Phys. Chem. B 2012, 116, 10551–10560. [Google Scholar] [CrossRef] [PubMed]
- Musick, M.D.; Pena, D.J.; Botsko, S.L.; McEvoy, T.M.; Richardson, J.N.; Natan, M.J. Electrochemical properties of colloidal Au-based surfaces: Multilayer assemblies and seeded colloid films. Langmuir 1999, 15, 844–850. [Google Scholar] [CrossRef]
- Pilo-Pais, M.; Goldberg, S.; Samano, E.; LaBean, T.H.; Finkelstein, G. Connecting the nanodots: Programmable nanofabrication of fused metal shapes on DNA templates. Nano Lett. 2011, 11, 3489–3492. [Google Scholar] [CrossRef] [PubMed]
- Pilo-Pais, M.; Watson, A.; Demers, S.; LaBean, T.H.; Finkelstein, G. Surface-enhanced Raman scattering plasmonic enhancement using DNA origami-based complex metallic nanostructures. Nano Lett. 2014, 14, 2099–2104. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.R.; Natan, M.J. Hydroxylamine seeding of colloidal Au nanoparticles in solution and on surfaces. Langmuir 1998, 14, 726–728. [Google Scholar] [CrossRef]
- Uprety, B.; Gates, E.P.; Geng, Y.; Woolley, A.T.; Harb, J.N. Site-specific metallization of multiple metals on a single DNA origami template. Langmuir 2014, 30, 1134–1141. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.; Kempter, S.; Holler, S.; Schüller, V.; Schiffels, D.; Simmel, S.S.; Nickels, P.C.; Liedl, T. DNA origami-templated growth of arbitrarily shaped metal nanoparticles. Small 2011, 7, 1795–1799. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Boulais, E.; Hakobyan, Y.; Wang, W.L.; Guan, A.; Bathe, M.; Yin, P. Casting inorganic structures with DNA molds. Science 2014, 346. [Google Scholar] [CrossRef] [PubMed]
- Helmi, S.; Ziegler, C.; Kauert, D.J.; Seidel, R. Shape-controlled synthesis of gold nanostructures using DNA origami molds. Nano Lett. 2014, 14, 6693–6698. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Mao, C. Molecular lithography with DNA nanostructures. Angew. Chem. Int. Ed. 2004, 43, 4068–4070. [Google Scholar] [CrossRef] [PubMed]
- Becerril, H.A.; Woolley, A.T. DNA shadow nanolithography. Small 2007, 3, 1534–1538. [Google Scholar] [CrossRef] [PubMed]
- Surwade, S.P.; Zhao, S.C.; Liu, H. Molecular lithography through DNA-mediated etching and masking of SiO2. J. Am. Chem. Soc. 2011, 133, 11868–11871. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Michael, B.; Surwade, S.P.; Ricardo, K.B.; Zhao, S.; Liu, H. Mechanistic study of the nanoscale negative-tone pattern transfer from DNA nanostructures to SiO2. Chem. Mater. 2015, 27, 1692–1698. [Google Scholar] [CrossRef]
- Surwade, S.P.; Zhou, F.; Wei, B.; Sun, W.; Powell, A.; O’Donnell, C.; Yin, P.; Liu, H. Nanoscale growth and patterning of inorganic oxides using DNA nanostructure templates. J. Am. Chem. Soc. 2013, 135, 6778–6781. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Linko, V.; Tapio, K.; Kostiainen, M.A.; Toppari, J.J. Custom-shaped metal nanostructures based on DNA origami silhouettes. Nanoscale 2015, 7, 11267–11272. [Google Scholar] [CrossRef] [PubMed]
- Linko, V.; Shen, B.; Tapio, K.; Toppari, J.J.; Kostiainen, M.A.; Tuukkanen, S. One-step large-scale deposition of salt-free DNA origami nanostructures. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Sun, W.; Ke, Y.; Shih, C.-J.; Paulus, G.L.C.; Wang, Q.H.; Mu, B.; Yin, P.; Strano, M.S. Metallized DNA nanolithography for encoding and transferring spatial information for graphene patterning. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef] [PubMed]







© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, B.; Tapio, K.; Linko, V.; Kostiainen, M.A.; Toppari, J.J. Metallic Nanostructures Based on DNA Nanoshapes. Nanomaterials 2016, 6, 146. https://doi.org/10.3390/nano6080146
Shen B, Tapio K, Linko V, Kostiainen MA, Toppari JJ. Metallic Nanostructures Based on DNA Nanoshapes. Nanomaterials. 2016; 6(8):146. https://doi.org/10.3390/nano6080146
Chicago/Turabian StyleShen, Boxuan, Kosti Tapio, Veikko Linko, Mauri A. Kostiainen, and Jari Jussi Toppari. 2016. "Metallic Nanostructures Based on DNA Nanoshapes" Nanomaterials 6, no. 8: 146. https://doi.org/10.3390/nano6080146
APA StyleShen, B., Tapio, K., Linko, V., Kostiainen, M. A., & Toppari, J. J. (2016). Metallic Nanostructures Based on DNA Nanoshapes. Nanomaterials, 6(8), 146. https://doi.org/10.3390/nano6080146