1. Introduction
Besides nanoparticles composed of titanium oxide, aluminum oxide, carbon and other materials, silica nanoparticles (SiO
2 NPs) are among the most widely produced [
1]. The current and potential applications of engineered amorphous SiO
2 NPs are not restricted to technical fields [
1,
2], but include also their use in biotechnology, such as for instance as transporters for DNA and drug delivery [
3,
4,
5,
6], cancer therapy [
7], enzyme immobilization or as biosensors and biomarkers [
8,
9,
10,
11]. Eventually, SiO
2 NPs find use also in cosmetics, e.g., in toothpaste or skin care products, and also within the food sector. In more detail, they can be found as food additives (E551) like anticaking agents in salt, spices or instant soups, as flavor enhancers or food pigments, as coating material in confectionary products and packaging materials or as health supplements [
12,
13,
14,
15,
16,
17].
Thus, the impact on safety regarding the environment and health due to nanoparticle exposure in general and from SiO
2 NPs in particular, is of great interest, but the scientific knowledge in this respect is relatively limited. Previous studies of SiO
2 NPs mainly focused on inhalational exposure [
1,
2,
18,
19,
20]. Influence on other cell systems, for example, on hepatic [
21] and various epithelial-like [
18,
22] carcinoma cells or primary mouse embryonic cells [
23] have been determined to a minor extent. For a detailed review on recent toxicological studies of synthetic amorphous silica, see Fruijtier-Polloth [
24]. Likewise, only little is known about potential effects on the gastrointestinal (GI) tract after oral uptake of these particles. Therefore, studies on the oral route of exposure are progressively increasing and more data have recently become available.
Several in vitro studies concerning effects of SiO
2 NPs on gastrointestinal cells are published. In a recent study, Yang et al. [
25] considered four different types of SiO
2 NPs as safe up to a concentration of 100 µg/mL for both, human gastric epithelial (GES-1) and colorectal adenocarcinoma (Caco-2) cells after 24 h of incubation. However, longer incubation periods or higher concentrations induced DNA damage and reduced cell viability. In contrast, Tarantini et al. [
26] detected a reduction of cell viability in Caco-2 cells already at a concentration of 32 µg/mL. Furthermore, in another human colon cancer cell line (HT29) 14 nm SiO
2 NPs induced cyto- and genotoxic effects at a concentration of 10 µg/mL [
27].
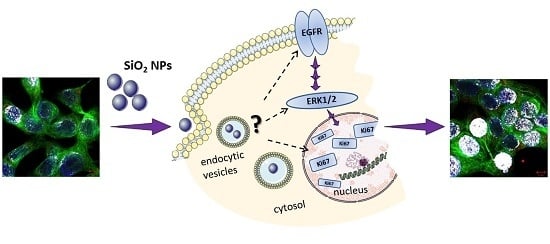
In a previous study, we demonstrated that SiO
2 NPs are taken up by HT29 cells and stimulate cell proliferation when incubated in high fetal bovine serum (FBS)-containing media. In contrast, cytotoxic effects were observed when the cells were incubated under low-serum conditions [
28]. Taken this as a starting point, we investigated the effects of SiO
2 NPs under high-serum conditions in a different carcinoma cell line representative of the stomach (GXF251L) established from a gastric signet ring cell carcinoma [
29] and further addressed the mechanism of the SiO
2 particles with respect to cellular proliferation. Hence, the aim of this study was to determine the effects of SiO
2 NPs with a nominal primary diameter of 12 nm on GXF251L cells mimicking in vitro an oral uptake to some extent and investigating the cytotoxic and proliferation stimulating potential of the nanoparticles by application of a high-content-high-throughput screening assay, as well as, common toxicity/cell growth assays. For purpose of comparison and to figure out if any effects are owed to the nano-scaled size per se, the effects of bigger SiO
2 particles with a nominal primary diameter of 200 nm were also analyzed with regard to their influences on cell viability and cell death. To further assess potential proliferative mechanisms, the effects of the SiO
2 NPs on the nuclear proliferation marker Ki-67 were examined by immunocytochemistry. Previous studies indicate that nanoparticle-induced cell proliferation is associated with interferences with the epidermal growth factor receptor (EGFR)/mitogen-activated protein kinases (MAPK) signaling pathways [
30,
31]. Both signaling cascades, EGFR and MAPK, are well known to be deeply involved in the regulation of cellular processes like differentiation or proliferation [
32,
33] and were determined by the means of SDS-PAGE/Western blot, immunocytochemistry and quantitative real-time polymerase chain reaction (PCR).
3. Discussion
The objective of this in vitro study was to investigate the effects of amorphous SiO
2 NPs with a nominal primary diameter of 12 nm on a human gastric cancer cell line. For this purpose, the gastric cell line GXF251L and its responses to SiO
2 NPs including the influences on viability and possibly related signaling cascades were examined. An extensive description of the physicochemical properties and the cellular uptake of the tested particles has been published previously by Gehrke et al. [
28]. Thereby, the influence of, inter alia, the same particles on HT29 cells cultured in DMEM under high and low serum conditions was examined and was considered as starting point of the paper at hand. In general, a particle characterization under the applied incubation conditions is essential for a toxicological assessment [
34,
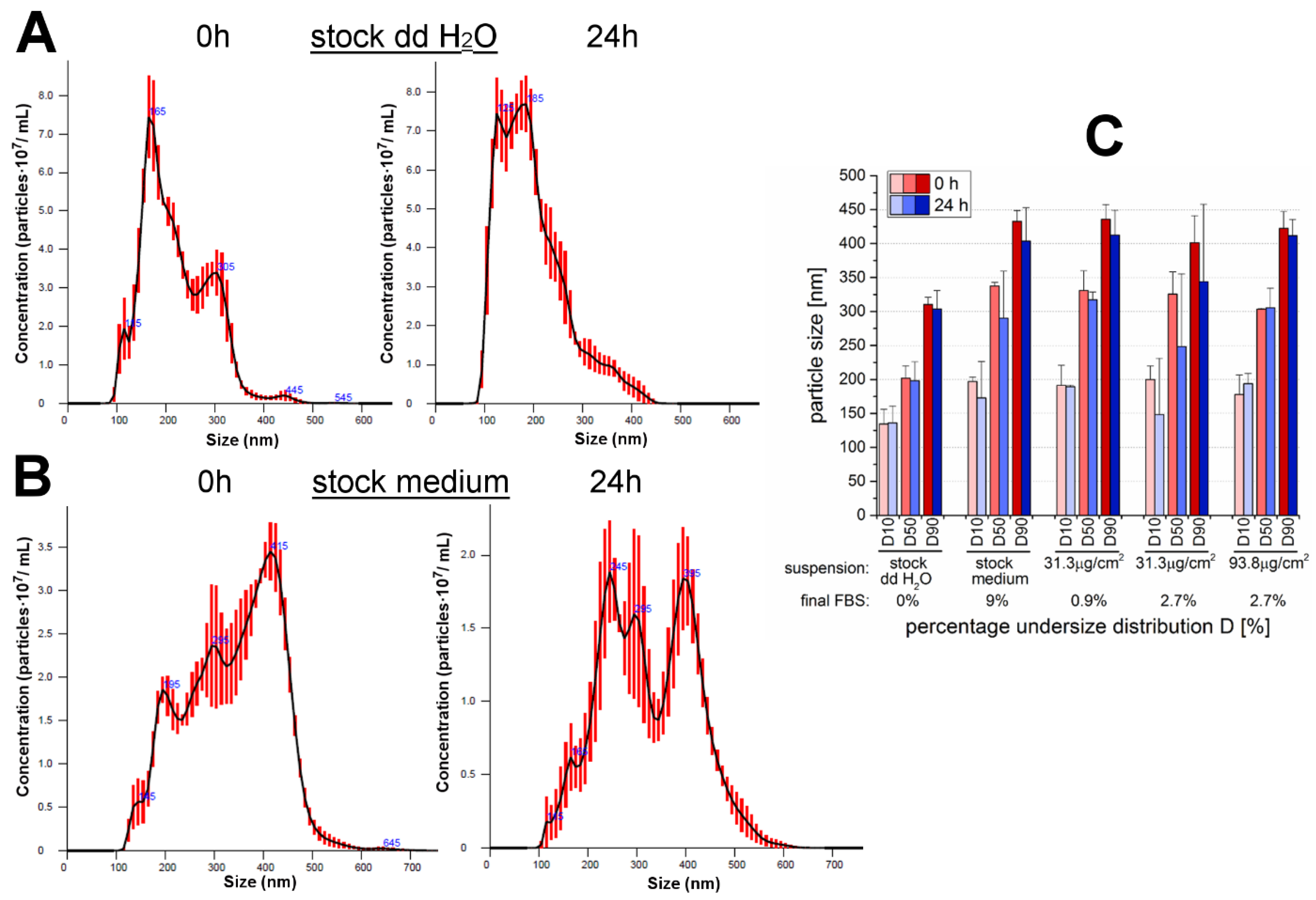
35]. For the present study, the examination of a different cell line (GXF251L) was accompanied by the usage of another cell cultivation medium. For this reason, and because of the availability of an innovative method like NTA, the behavior of the particles of interest was characterized with regard to particles size, PSD and suspension stability (
Table 1 and
Figure 1). In general, the method applied for the determination of particle diameters and PSD is dependent on the particle properties, as well as, the compounds contained in the suspension medium, especially FBS. The examined 12 nm SiO
2 NPs are known as polydisperse particles exhibiting a strong agglomeration and aggregation behavior and a broad PSD not only when suspended in FBS-containing medium but also presumably already due to their manufacturing process (flame synthesis) and their physico-chemical properties [
24,
28,
36,
37]. Hence, the application of methods like dynamic light scattering (DLS), small-angle X-ray scattering etc. was very limited with regard to the given incubation conditions. For example, with DLS the mean particle size is biased towards bigger particle diameters since the light is scattered more intense with increasing particle sizes giving only inappropriate results [
38,
39]. NTA belongs to the state-of-the-art methods for the determination of particle size and PSD of such complex samples [
37,
38,
39]. Moreover, nanoparticle tracking analysis (NTA) tracks and analyses single particles thereby giving a detailed and highly resolved size profile of the nanoparticle population.
For all the cell-based experimental conditions, 9% FBS was included in the cell culture medium in order to maintain physiological conditions and habitual cell growth. The only exceptions were made for the cell-free particle characterization studies due to limitations of the applied particle size determination method, NTA, with regard to high FBS amounts (for more details see
Section 2.1.). Therefore, size analysis was performed with the highest necessary amount of FBS, which was 2.7% for a particle area concentration of 93.8 µg/cm
2 aiming to prevent high background intensities, as well as, strong particle flares and thus, deviating results. Only the medium stock suspension was accepted for analysis with 9% FBS.
With NTA, the particle suspension in water showed the formation of agglomerates and aggregates and the modal values were comparable to the previously performed DLS measurements of the same 12 nm SiO
2 NPs by Gehrke et al. [
28]. While for the latter mentioned DLS measurements no suitable data could be obtained even in low FBS-containing medium, a more explicit statement was possible by means of NTA applied for the present study. Thereby, particles suspended in medium were strongly agglomerated/aggregated even when incubated in low amounts of FBS. This was also proved by the results of the ζ-potential. The values of the stock suspension prepared in double-distilled water were still within the threshold to be claimed as a stable suspension exhibiting only a slight increase of the agglomeration/aggregation status after 24 h of incubation. In contrast, SiO
2 NPs suspended in FBS-containing medium were marked by increased particle repulsion compared to the water sample and thus, may have been subject to a rapid re-agglomeration/-aggregation. This behavior was comparable within the analyzed FBS-medium-suspensions and no significant influences due to the particle concentration, the amount of FBS or the incubation time seemed to be observable. Altogether, it was already confirmed by Gehrke et al. [
28] that the 12 nm SiO
2 NPs were sintered beforehand according to the TEM images. Despite the dispersion process by sonification, the particles were strongly agglomerated/aggregated when suspended in FBS-containing cell culture medium. The results are in accordance with former studies of the same particles in cell culture medium containing FBS and it is assumed that the adsorption of serum proteins is responsible for the increase in size [
25,
28]. Even though studies are controversial with regard to the particle behavior due to the addition of cell culture medium and/or FBS [
28], it can most likely be concluded that the allover suspension composition was responsible for the enhanced agglomeration and aggregation of the SiO
2 particles. Compared with the results of Gehrke et al. [
28] for the DMEM-suspended 200 nm particles the secondary diameters of the 12 nm particles suspended in RPMI 1640 medium with ranges around 300 nm due to formed agglomerates/aggregates are even about 100 nm bigger.
Strictly speaking, the 12 nm SiO
2 NPs cannot be designated as nanoparticles anymore, which is more or less a matter of definition. The common perception is that nanomaterials possess at least one dimension with a maximum range of 100 nm [
34,
40]. Hence, the term nanoparticle only applies to the primary particles with a nominal diameter of 12 nm, whereas the secondary particles exhibited diameters in the range of approximately 100 nm to 500 or 600 nm.
It must be considered that the size measurements were performed at a physiological pH due to method limitations. However, according to Peters et al. [
41] these results seem to resemble the gastric conditions. The working group mimicked the human digestion with the help of an in vitro model and found, depending on the food matrix, that mainly no nano-scaled SiO
2 at all or only small amounts were present at the gastric digestion stage. The formation of large agglomerates was probably due to the low pH and the high concentrations of electrolytes in the gastric fluid whereas in the previous (saliva) and the subsequent (intestine) digestion stages significantly more nano-sized silica particles were found [
41].
For the present study, different incubation times were applied depending on the used assay. Short time effects and especially the relative short residence time of food in the stomach were considered by incubation times of 45 min to 4 h. Furthermore, long term effects, e.g., due to repeated uptake of SiO
2 NPs were taken into account. Especially for the examination of cell growth or proliferation related effects, incubation times of at least 24 h were necessary since the doubling time of the GXF251L cells of about 32 h needed to be considered as well [
42].
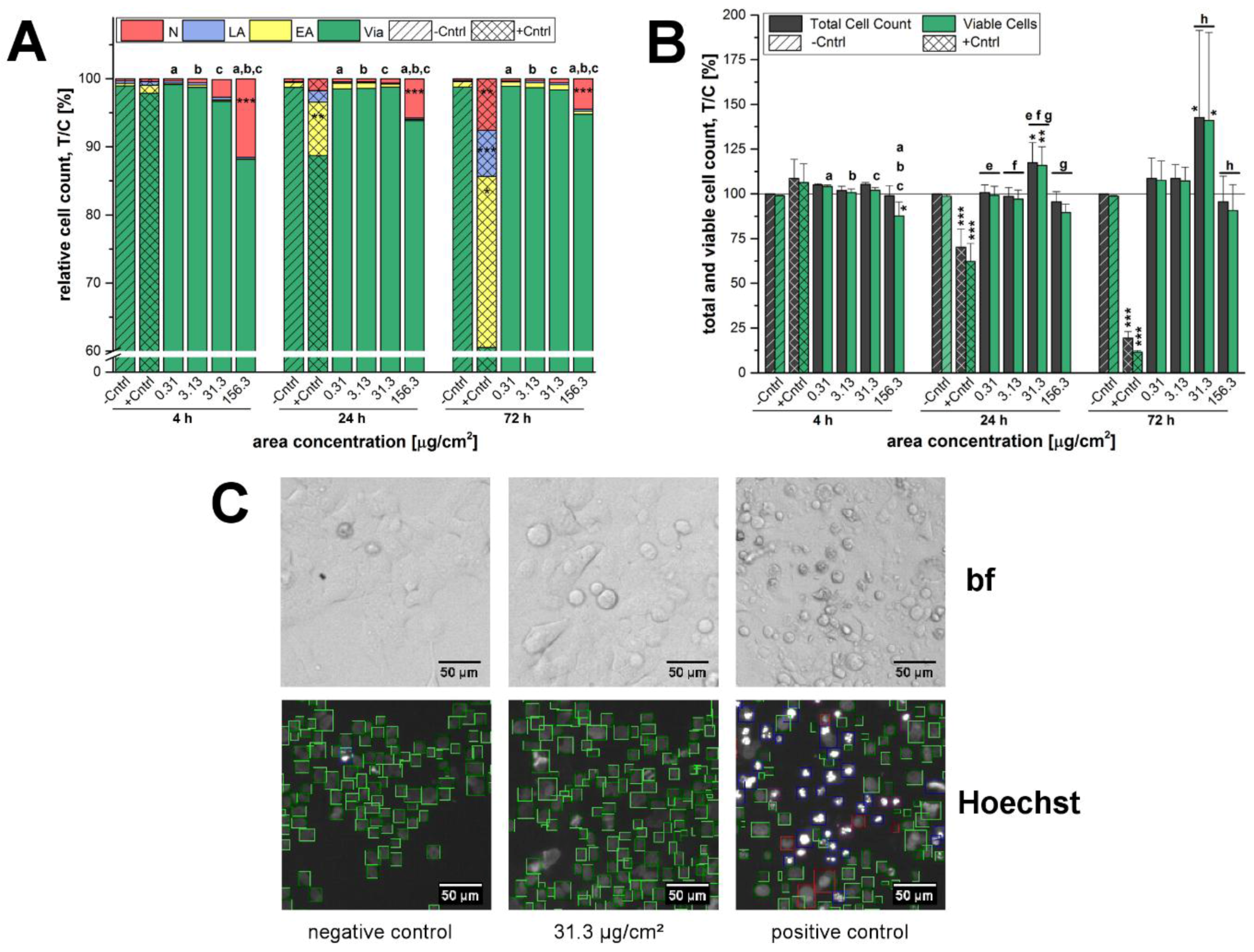
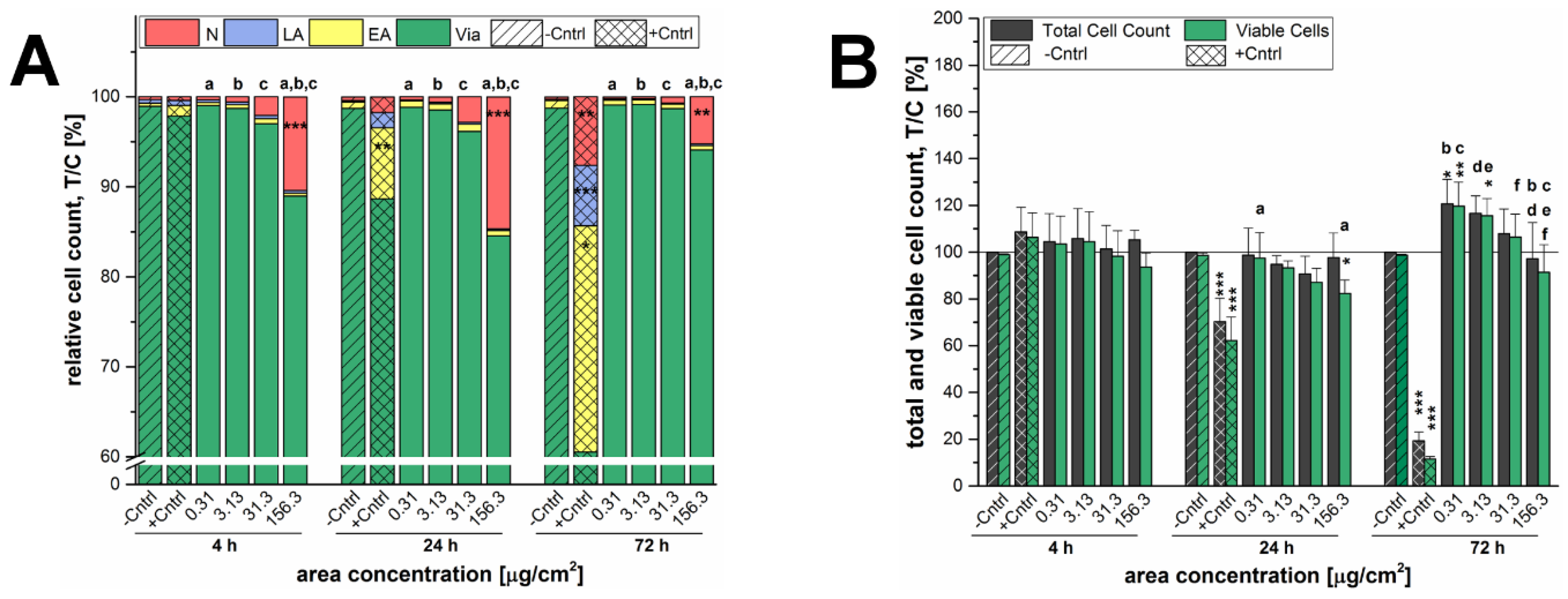
A microscopy-based determination of total cell counts including the distinction between viable cells and different types of cell death was deployed as initial assessment of the cytotoxic potential of the 12 nm SiO
2 NPs. Here, independently of the incubation time the highest applied concentration of SiO
2 NPs exerted necrotic effects whereas cell death due to apoptosis was negligible in general (
Figure 2A). The concentration of 156.3 µg/cm
2 was in fact primarily selected to obtain an overload concentration of the test system and hence to ensure the detection of cellular effects under extreme stress levels in a range even up to physiologically concentrations less likely to be achieved by exposure to nano-silica via food [
25,
43,
44]. As a consequence, necrosis resulted in a decrease of the viable cell number, which was significant only after short term incubation of 4 h (
Figure 2A). This peculiar behavior seems to suggest that the high amount of particles acutely impairs cell viability but at later time points exposed cells are able to handle the administered particles and recover. Comparable results were found for a human keratinocyte cell line exposed to 50 nm silver nanoparticles, both, acutely and chronically [
45]. In contrast, GXF251L cells displayed a distinct proliferation stimulation at a concentration of 31.3 µg/cm
2 after 24 h and 72 h of incubation. Interestingly, similar results were found for the SiO
2 particles with a bigger nominal primary diameter of 200 nm (
Figure 3). Necrotic GXF251L cells were also only found for the highest examined concentration resulting in reduced cell viability and this effect was diminished after 72 h of incubation. As well, cell proliferation was stimulated but not until 72 h of incubation and only for the lowest examined concentration in a decreasing manner with increasing particle concentrations. Consequently, the effects causing cell death, as well as, cell proliferation are present for both SiO
2 particle preparations independently of the nominal particle diameter.
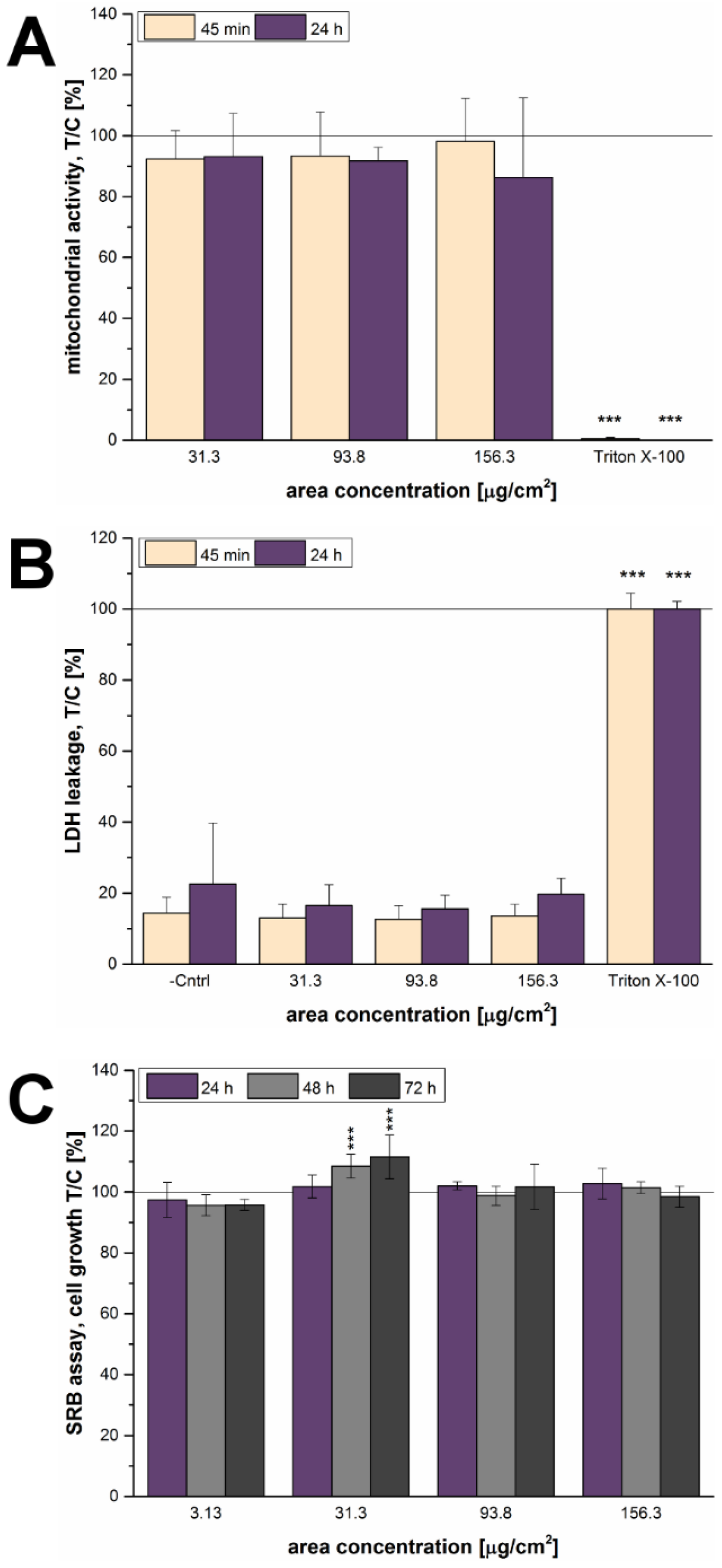
In order to explore the effects of the 12 nm SiO
2 NPs in detail, their cytotoxic potential was determined as a next step. The particles did not exert any effects on the mitochondrial activity (WST-1 assay) or membrane integrity (LDH assay) of GXF251L cells, neither after 45 min nor after 24 h of incubation (
Figure 4A,B). Interestingly, the SRB assay showed a stimulation of growth at a concentration of 31.3 µg/cm
2 after 48 h and 72 h of incubation (
Figure 4C), which is in accordance with the increase of cell number detected with the microscopy-based assay. Though, a growth stimulus after 24 h was not detectable in the latter assay, which might be due to the different endpoints analyzed by the two assays and the assumption that the microscopy-based assay might be more sensitive than the SRB assay. A similar behavior regarding a growth stimulus was already described for the same particles in HT29 colon cells at a concentration of 31.3 µg/cm
2 and also at 93.8 µg/cm
2 [
28]. In both cases, differences between the results of the WST-1 and SRB assay suggest that the behavior of NPs cannot be easily categorized and described by one single assay [
46,
47]. In another study, 20 nm SiO
2 NPs also exerted slight growth stimulating effects on the gastric cancer cell line MGC80-3 after 24 h detected by MTT assay [
48]. However, longer incubation times, as well as, particle concentrations of more than 300 µg/mL lead to significant cytotoxic effects towards MGC80-3 cells.
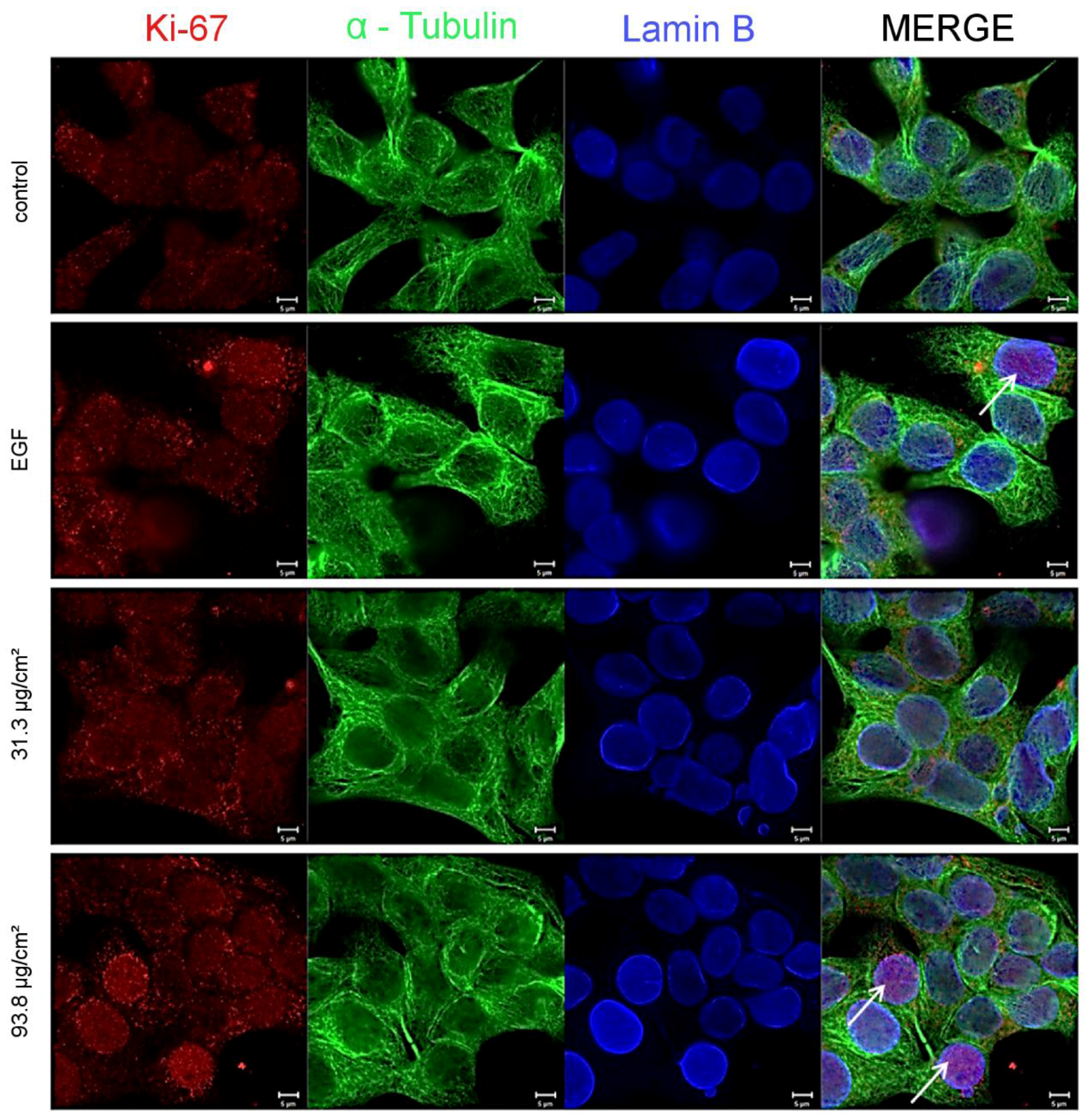
As a consequence for the present study, a mechanistic approach was carried out with respect to the underlying mechanism of the proliferating effects of the SiO
2 NPs. Therefore, the cellular proliferation marker, Ki-67 was evaluated by immunocytochemistry. Ki-67 detects a nuclear antigen which is only found in proliferating cells and is absent in resting cells [
49]. A concentration-dependent increase of the proliferation marker could also be demonstrated at the SiO
2 NPs concentrations of 31.3 µg/cm
2, as well as, 93.8 µg/cm
2 after 24 h of incubation (
Figure 5 and
Figure 6).
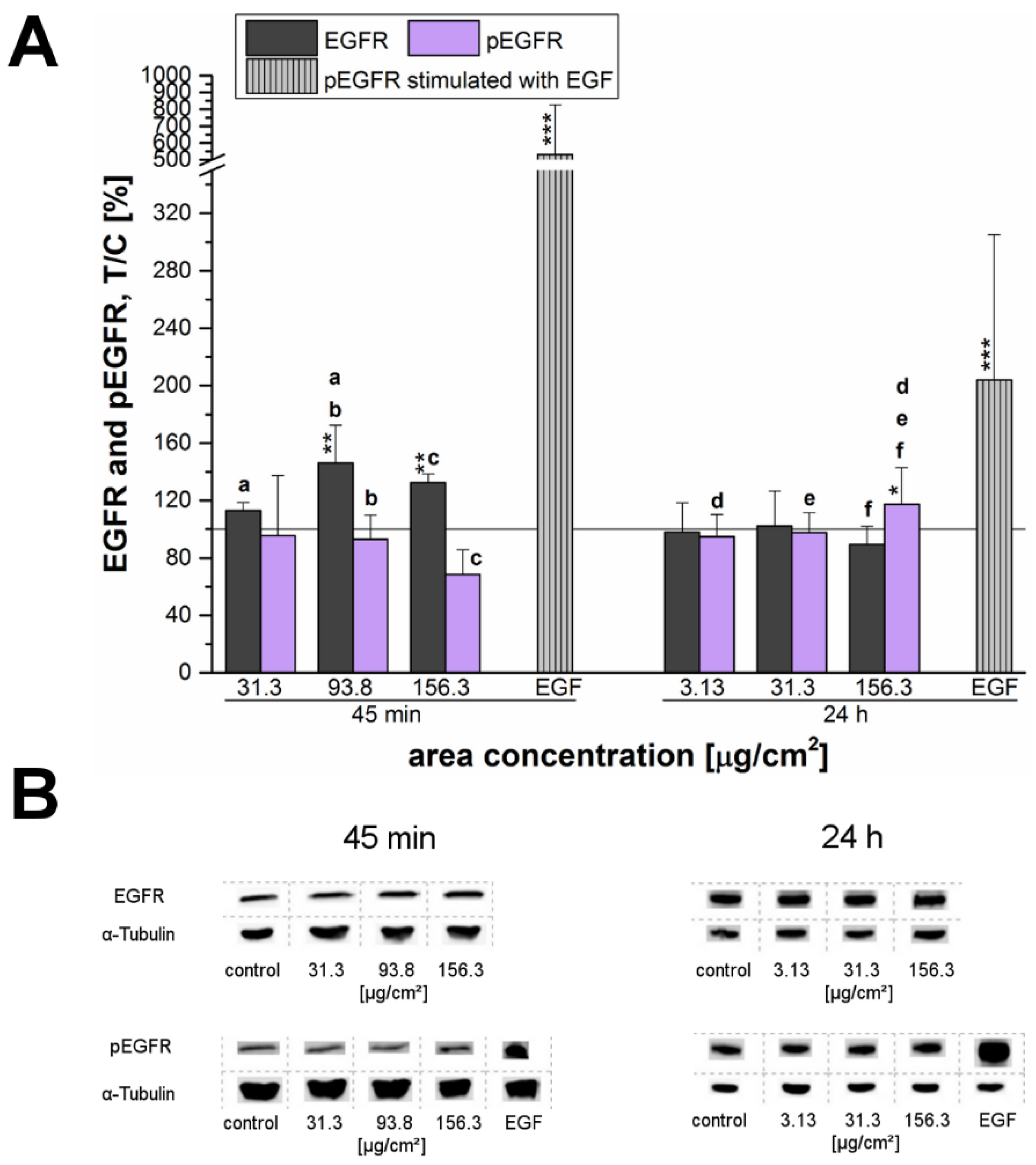
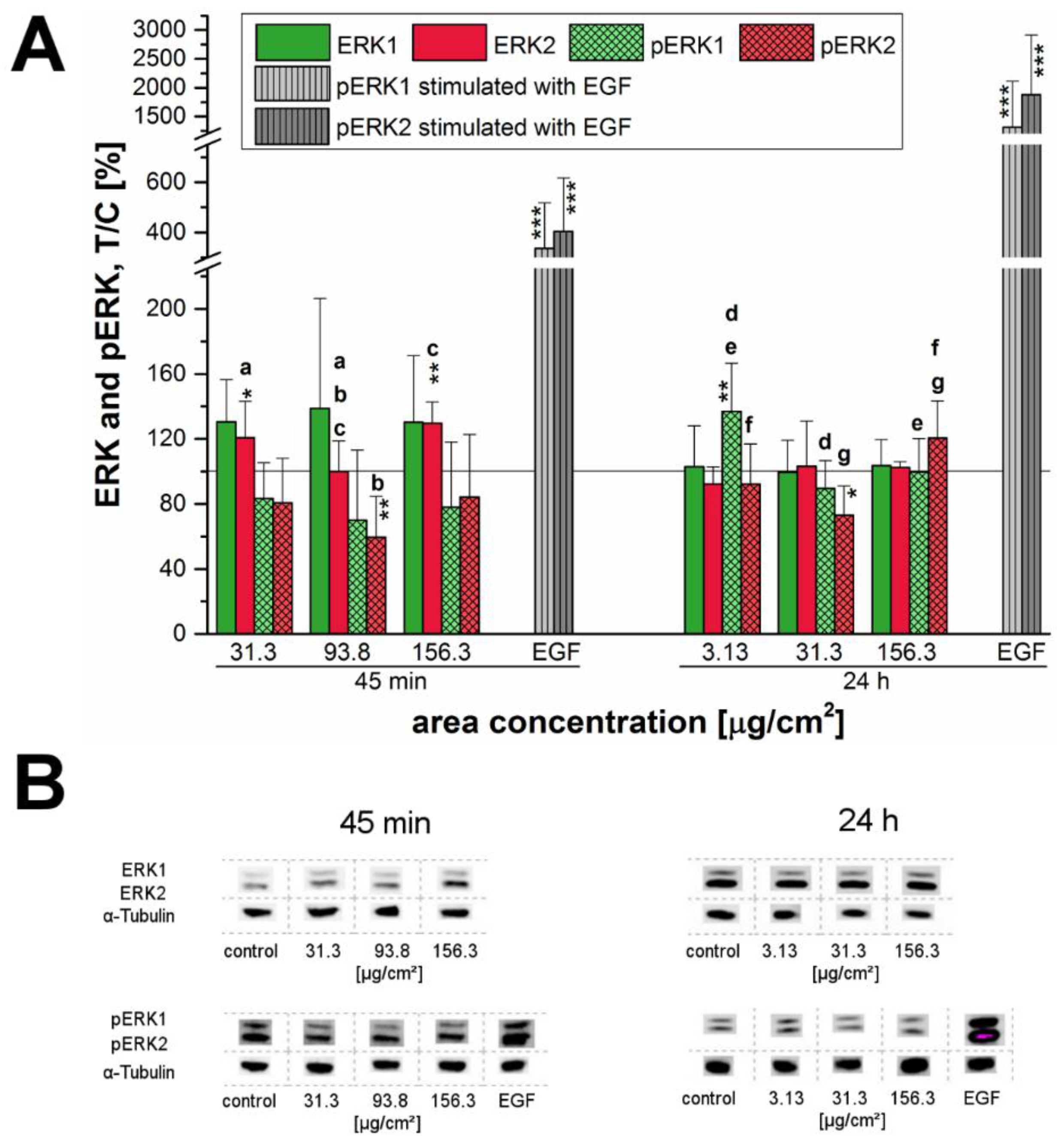
Furthermore, the phosphorylation of the ERK1/2 and its upstream regulator, the EGFR, were examined after exposure of cells to the 12 nm SiO
2 NPs (
Figure 7 and
Figure 8). The EGFR (also known as ErbB1 and HER1 in humans) is a membrane spanning cell surface receptor [
50] and plays an important role in cell adhesion, motility, signal transduction, cell division, differentiation and is mediating cellular proliferation, as well as, cell-to-cell communication in mammalian cells [
32,
51,
52]. Previously, it was demonstrated that the induction of nanoparticle-dependent cell proliferation is associated with the activation of the EGFR signaling cascade with a subsequent proliferation-specific ERK1/2 activation, whereas an EGFR-downstream activation of the c-Jun NH
2-terminal kinases1/2 (JNK1/2) is specific for apoptosis [
30]. Furthermore, it was found that the MAPK/ERK1/2 signaling pathway is a key component of pathways controlling cell-cycle processes, differentiation and proliferation, as well as, cell death [
28,
31,
33,
53]. Kim et al. [
54] as well observed a brief activation of the ERK1/2 pathway followed by an increase in cell proliferation in human adipose tissue-derived stem cells (hADSCs) after incubation with SiO
2 NPs (diameters in the range of 50–120 nm).
In the present study, a connection between cell proliferation and activation of the EGFR and ERK1/2 signaling pathways was assumed and therefore investigated in more detail. After short-term incubation of 45 min, the endogenous EGFR levels increased especially at high incubation concentrations. This behavior seemed to suggest that EGFR-trafficking was obstructed by the high amount of the applied particles, thus interfering with EGFR homeostasis. However, this hypothesized influence due to a particle overload only seemed to be a matter of short duration—longer incubation of 24 h resulted in a normalization of the endogenous levels comparable to those of the negative control (
Figure 7).
After 24 h, the endogenous levels of both, EGFR and ERK1/2 remained constant and only for the EGFR a slight but significant activation was observed at the highest examined concentration (
Figure 7A). Interestingly, phosphorylation of ERK1 significantly increased at a low concentration of just 3.13 µg/cm
2. However, ERK2 did not seem to be affected here—in contrast, its phosphorylation seemed to be declining. Thus, after 24 h of incubation a consistent activation signal for EGFR and downstream for ERK1/2 could only be confirmed for the overload concentration. Obviously, the tested silica particles seem to influence the EGFR/MAPK signaling pathway in a time-dependent manner with regard to the endogenous receptor levels as well as the receptor activation. Comfort et al. [
45] associated an observable increased Ki-67 expression, as well as, an activation of the EGFR/MAPK signaling cascade in human keratinocytes to augmented stress and altered functionality of human keratinocytes. However, the results could be assigned to chronic administration of very low silver nanoparticles-concentrations in contrast to an acute exposure of 24 h.
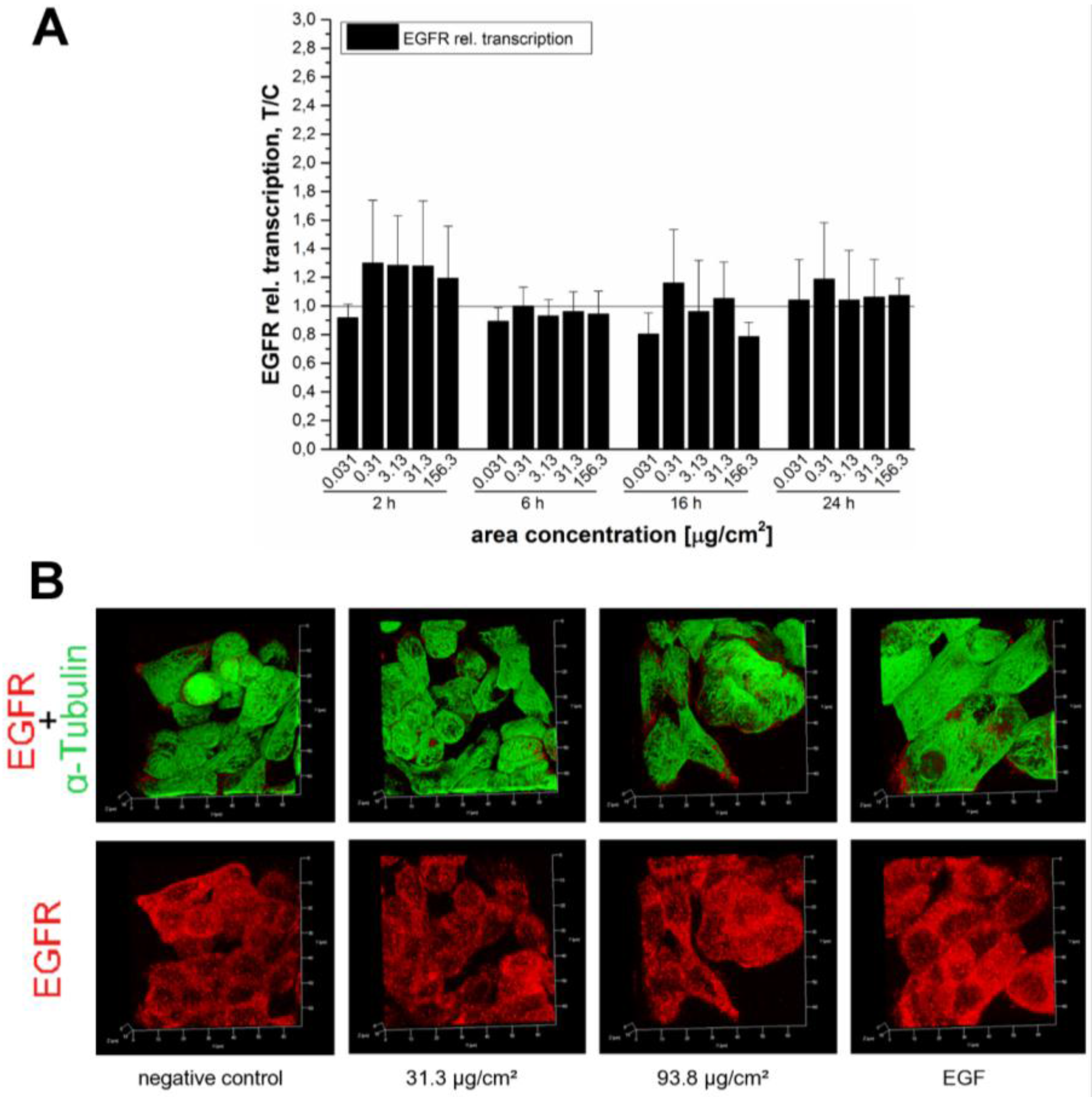
In order to verify whether the endogenous EGF receptor enhancement after 45 min was due to an increased transcription rate or the result of impaired receptor trafficking due to particle overload, the mRNA expression of EGFR was examined. The relative transcriptions rates were measured after several incubation periods of 2 h, 6 h, 16 h and 24 h to capture the kinetics of receptor expression (
Figure 9A). In spite of this extensive analysis, we were not able to detect any noteworthy enhancement of EGF receptor mRNA levels. Hence, influences on EGFR trafficking seemed more likely. Immunocytochemistry revealed an enhanced EGFR signal at a SiO
2 NP area concentration of 93.8 µg/cm
2 (
Figure 9B), which is in accordance with the results of the Western blot (
Figure 7). Stimulation of the EGFR pathway induces receptor aggregation on the cell surface to enhance the chances of dimer formation and subsequently auto-phosphorylation [
55,
56]. The results of the present study suggest that the examined SiO
2 NPs may induce a concentration-dependent receptor recruitment. Since no effect was detectable at the level of transcription, the effect of the NPs on the cellular growth might be due to a more “direct” effect mediated by the interaction of the silica particles with the cell membrane. The 12 nm SiO
2 NPs are taken up by HT29 cells [
28] and it is reasonable to assume that they are also internalized by GXF251L cells. In this respect, even though the exact uptake mechanism is still unclear, endocytosis seems to be most likely. In fact, SiO
2 NPs were previously found to be internalized in a clathrin- or caveolin-dependent manner [
17,
57] or by passive cellular uptake due to an adhesive interaction of 14 nm SiO
2 NPs with lipid membranes [
27]. Activation of EGFR can presumably occur by different cellular mechanisms but its signaling capacity is strongly dependent on sorting and targeting for lysosomal degradation and its recycling to the plasma membrane [
58,
59]. Furthermore, substantial amounts of EGFRs and ligands can accumulate intracellularly due to the fact that the rate of receptor internalization is much faster than its degradation rate [
60]. In this respect, it is possible to speculate that the uptake mechanisms of the NPs through cell membranes may lead to an impairment of the EGFR trafficking and homeostasis, causing, as a final effect, an alteration of the cellular proliferation and growth stimuli.
In spite of that, downstream responses seem to depend on particular cellular sites. That silica nanoparticles increase cell proliferation dependent on ERK1/2 activation is a valid hypothesis. However, the kinetics and dose-response in the various assays monitoring effects on growth and signaling varied. Complex cellular interactions are responsible for different signaling outcomes also in the case of ERK1/2 possibly resulting in both, anti- but also pro-apoptotic effects [
61]. Here, further investigations are in progress to clarify this aspect in our cell system.
Altogether, even if it is difficult to predict the relevance of the SiO
2 NP concentrations for daily dietary uptake due to different study results and estimations, we consider our results important toward a better comprehension of their biological effects on the GI level. The “Scientific Panel on Dietetic Products, Nutrition and Allergies” of the “European Food Safety Authority” estimated a typical intake of silicon of 20 to 50 mg/day in a 60 kg adult (0.3–0.8 mg/kg bodyweight/day) [
43] which equals around 43 to 107 mg SiO
2 per day (0.7–1.8 mg/kg bodyweight/day). Dekkers et al. [
44] estimated a comparable intake value according to analyzes of silica additives in food products. Including these values, Yang et al. [
25] deduced an daily exposure dose of the intestine of around 0.01–0.05 µg/cm
2. The interior gastric surface area of adults with normal body weight is assumed to be 383 cm
2 up to 800 cm
2 in total [
62,
63]. Consequently, the gastric exposure dose would be around 54–279 µg/cm
2. Further, an excess uptake of products containing (nano-) silica particles must be considered and additionally, the conjecture that probably not all food products which may contain E551 were identified in the respective study [
44]. Therefore, Dekkers et al. [
44] estimated an elevated uptake of 9.4 mg/kg bodyweight which would result in an enormous gastric exposure of 705–1473 µg/cm
2 calculated for a 60 kg adult, distinctly exceeding the applied concentrations in the present study. Yet, current investigations suggest a daily intake of 5 mg/kg bodyweight as harmless throughout lifetime [
64]. However, these amounts of intake are assumed to be safe [
43,
65] but it must be considered the given data refer to silicon and silica in general without taking into account nano-sized types of SiO
2. Analyses of products containing E551 indicated a nano-silica content up to 43% [
44]. Nonetheless risk assessment is difficult, thus enforcing, in our opinion the need for characterization of the nano-sized material.
All in all, the presence, behavior and bio-distribution, as well as the excretion of nano-sized materials have been extensively studied in the last decade. Thereby, oral uptake and with it the different gastrointestinal digestion stages received attention only in the recent years. Different test systems were applied to assess safety or hazard of silica-containing food, like in vitro [
25,
28] and in vivo [
66,
67,
68,
69] toxicity screenings or simulation of the gastrointestinal status and barrier [
41,
70]. However, it is not yet clear how amorphous SiO
2 NPs influence health after oral uptake and if they can indeed per se be considered as safe in food products.
In conclusion, the examined food-grade silica particles with a nominal primary diameter of 12 nm are strongly agglomerated and aggregated in the applied suspension. They exert slight cytotoxic effects, as well as, growth and proliferation stimulation in gastric cells, which however, displayed differences in comparison to the effect on other cell lines [
25,
28]. Toxic effects were limited, but detectable in a time- and concentration-dependent manner. The stimulation of proliferation in the gastric carcinoma GXF251L cell line could not be attributed to the classical activation of the EGFR and/or ERK 1/2 signaling pathways, usually reflected by enhanced phosphorylation. However, depending on incubation time and applied concentration, both signalling elements were clearly influenced by the administered particles. Further, influences on receptor trafficking and homeostasis are assumed, whereby impact on transcription could be ruled out. However, particle characterization and the comparison with bigger particles indicate that the observed effects cannot necessarily be attributed to particles below 100 nm and thus might not be regarded as a “nano-specific” effect
per se. However, it has to be pointed out, that the examined silica particles enhanced proliferation of the studied gastric carcinoma cell line, suggesting a potential risk for gastric cancer patients and underlining the necessity of further studies to clarify whether non-transformed gastric cells might be targeted as well.
4. Materials and Methods
4.1. Cell Culture
Experiments were performed using the established gastric cell line GXF251L (kindly provided by H. H. Fiebig, Institute for Experimental Oncology, Oncotest GmbH, Freiburg, Germany). GXF251L cells are a human tumor xenograft cell line derived from a signet ring cell carcinoma and were cultivated in Roswell Park Memorial Institute (RPMI) Medium 1640 containing l-glutamine. The media for GXF251L cells was supplemented with 25 mM HEPES (pH 7.2), 50 µg/mL gentamycin sulfate and 9% (v/v) heat-inactivated fetal bovine serum (FBS). To ensure homogeneous response and growth, cells were kept in culture for maximum 25 passages in humidified incubators with 5% CO2 at 37 °C. Media and supplements were purchased from Gibco® Life Technologies™, Karlsruhe, Germany.
Cells were routinely tested for mycoplasm contamination and found to be negative. For all experiments only a cell density of around 70% to 80% confluence was selected for particle application. Exceptions were made for the microscopy-based determination of cell counts, apoptosis and necrosis and for the sulforhodamine B assay for which a confluence of around 40% to 50% was chosen to guarantee exponential cell growth over an extended incubation time of 24 h to 72 h.
4.2. Particles and Incubation Conditions
The commercially available, engineered amorphous silica nanoparticles Aerosil® 200 F (hydrophilic, pyrogenic), especially designed as supplement for food and feed, were a kind gift from Evonik Industries (Essen, Germany). According to the manufacturer, the particle’s specific surface area is indicated as 200 ± 25 m2/g (BET method) and their nominal primary diameter as 12 nm. For the purpose of comparison, bigger SiO2 particles (Ångström-Sphere™) with a nominal primary diameter of 200 nm were included for some assays. According to the manufacturer Fibre Optic Centre Inc. (New Bedford, MA, USA), their specific surface area, also determined by the BET method, is given as 4 ± 2 m2/g and they are claimed to be applicable in food industry as well.
Previous studies conducted by Gehrke et al. [
28] with the same 12 nm SiO
2 particles revealed a broad particle size distribution and diameters of 16 to 40 nm of individual particles examined by transmission electron microscopy (TEM). TEM images and dynamic light scattering (DLS) analysis showed the presence of agglomerates and/or aggregates with a Sauter mean diameter (SMD) of more than 500 nm in FBS-containing Dulbecco’s Modified Eagle Media (DMEM). The suspension of the 200 nm SiO
2 particles resulted in an instant formation of micrometer-sized agglomerates and aggregates which sedimented quickly. TEM analysis further revealed individual particles of sizes around 200 nm in the suspended fraction which was also confirmed by DLS measurements and was in accordance with the manufacturer’s information. Interestingly, neither the suspension medium (water or DMEM) nor the addition of low or high FBS-amounts seemed to influence the diameter of the 200 nm particles substantially. For both particles suspended in high-FBS containing DMEM, ζ-potentials of around—10 mV revealed quite unstable suspensions with a strong agglomeration/aggregation tendency. Further information regarding the agglomeration behavior as well as a detailed particle characterization in DMEM and in water have been published by Gehrke et al. [
28]. In order to exclude specific influences of the particle behavior due to a differently applied cell culture medium, a detailed characterization of the 12 nm NPs suspended in FBS-containing RPMI 1640 medium was performed for the present study.
The particles were applied to cells according to a specific internal standard operating protocol (SOP) to guarantee equal treatment and incubation conditions between the different test systems. A stock suspension of 10 mg/mL was prepared by weighing 20 to 30 mg of the particles and addition of an appropriate volume of complete media, including a standard concentration of 9% FBS to avoid effects related to cytotoxicity or stress. All suspensions and dilutions were prepared in the respective cell culture media (RPMI 1640). The stock suspension was carefully vortexed and sonicated for 30 strokes with a Branson Sonifier
® S-450A (230 V ± 10 V, 50/60 Hz and 400 Watts with the following settings: duty cycle: 50%; output control: 4; including microtip 102C). Afterwards, the stock suspension was diluted 1:10 (
v/
v) gaining a particle suspension with a concentration of 1 mg/mL. Subsequently, the incubation suspensions in the range of 0.03 µg/cm
2 up to 156.3 µg/cm
2 (equivalent to 0.1 µg/mL up to 500 µg/mL) were prepared. Area and volume concentration conversions are based on a 96-well plate with a surface area of 0.32 cm
2 and an application volume of 100 µL and scaled upon need (for more detailed information refer to Gehrke et al. [
28]). Therefore, the applied volume varied depending on the area of the dish. Treatment of control cells was the same except for incubating the cells with the respective culture media without particles.
4.3. Nanoparticle Tracking Analysis
For analysis of particle size and particle size distribution (PSD) a state-of-the-art method, namely nanoparticle tracking analysis (NTA) was performed with the NanoSight NS500 (Malvern Instruments Ltd, Malvern, UK). Thereby, a laser beam was passed through a chamber containing the sample. The present particles moving under Brownian motion scattered the light and single particles could be tracked by means of a microscope equipped with a camera. Background and camera levels were set manually in dependency of the particle intensity and finally, the hydrodynamic diameters and PSDs were analyzed by the software (NTA 3.1, Malvern Instruments Ltd, Malvern, UK) using the Stokes-Einstein equation. In comparison to DLS, a bias of particle intensity is avoided and the resolution of particle sizes is improved [
38].
Nevertheless, preliminary experiments revealed a limitation of NTA. The measurement of the incubation suspensions as described in
Section 2.1 resulted in high background levels, strong particle flares outshining smaller particles and subsequently, in vague, deviating values—an issue owed to the high amounts of FBS (9%). In consequence, the FBS-amount was adjusted to the highest necessary volume, which was 2.7% for the analyzed suspension with an area concentration of 93.8 µg/cm
2. The 12 nm SiO
2 NPs were pre-suspended in 9%-FBS containing RPMI 1640 medium (including 1% HEPES) and also double-distilled water with a concentration of 10 mg/mL as described beforehand. The suspensions were diluted to 1 mg/mL with 9%-FBS containing medium or double-distilled water, respectively and further diluted with medium to the final incubation concentrations of 31.3 and 93.8 µg/cm
2. For the latter dilution, FBS was added separately, resulting in various final amounts of 0.9% and 2.7% in order to discern possible FBS-related effects. The suspensions were analyzed shortly after preparation (0 h) and after 24 h of incubation at 37 °C. For NTA measurements, the suspensions were diluted with double-distilled water in a manner that single particles could be discerned and were analyzed after maintaining the temperature of the sample chamber to 25 °C with individually set background and camera levels. In order to avoid drift of the samples and to ensure reproducible results, the chamber was pre-loaded with the dilution medium (RPMI 1640 in water) prior to and in-between each analysis until not more than 5 particles were recorded. Five different frames, 60 s each, were recorded and merged by the software. The mean diameter ± SD of at least two independent experiments was calculated and the PSD was considered individually.
4.4. ζ-Potential
The dispersion of particles may probably result in the formation of agglomerates and/or aggregates. The re-agglomeration behavior and thus, the stability of the particle suspensions was analyzed also by means of ζ-potential measurements (Malvern Zetasizer Nano-ZS, Malvern Instruments Ltd, Malvern, UK). The method is based on laser Doppler electrophoresis and particle movement under Brownian motion due to an applied electric field. Thereby, the moving particles scatter the light of a laser beam passing through the suspension resulting in fluctuations of the light intensity. Since the frequency shift is proportional to particle velocity, particle motion can be calculated by the software (Zetasizer software v7.11, Malvern Instruments Ltd, Malvern, UK) from the ratio of velocity to electric field strength. The 12 nm SiO
2 NP stock suspensions (1 mg/mL) were prepared in double-distilled water and RPMI 1640 medium (containing 1% HEPES and 9% FBS). The incubation suspensions with area concentrations of 31.3 and 93.8 µg/cm
2 were pre-suspended in medium and examined with regard to different final FBS amounts (see also
Section 2.1). The samples were analyzed without further dilution inside of a disposable capillary cell with electrodes shortly after sample preparation (0 h) and after 24 h of incubation at 37 °C. The ζ-potential was recorded in triplicates after sample equilibration to 25 °C. The mean of the triplicates and subsequently, the mean ± SD of at least two independent experiments was calculated.
4.5. Microscopy-Based Determination of Cell Counts, Apoptosis and Necrosis
In order to obtain a direct evaluation of the cytotoxic potential of the 12 nm, as well as, 200 nm SiO
2 particles, a high-content-high-throughput screening assay covering multiple endpoints at the single cell level was chosen. The microscopic determination of total and viable cell counts, as well as, the distinction of different types of cell death was performed according to the method published in Donauer et al. [
71] with slight modifications. The number of seeded cells for each treatment was chosen depending on the final particle incubation time as described above in
Section 2.2 cell culture to ensure homogeneous and comparable cell density during the treatment. For this experimental set-up, incubation times of 4 h, 24 h and 72 h were chosen. Cells were allowed to adhere for 23 h achieving a confluence of approximately 70% (4 h of incubation) and 50% (24 h and 72 h of incubation), respectively, at the beginning of the incubation period. The pure incubation media served as a negative control and staurosporine (800 nM) was included as a positive control. Thereafter, cells were stained with 0.3 µg/mL Hoechst33342 and 0.25 µg/mL propidium iodide (PI) and images were acquired with the help of the Olympus IX-81-ZDC microscope and the ScanR Acquisition software. Finally, the data were analyzed with the ScanR Analysis software (OLYMPUS, Hamburg, Germany).
Every concentration was applied as quadruplicate at least and every experiment was performed three times using cells from different passages and/or cell culturing flasks. Per well four images were taken for evaluation resulting in a total of 48 replicates per treatment. The effects on cellular growth were determined as percentage of total cell count (treated-over-control ratio × 100%) and percentage of viable, apoptotic and necrotic cells in relation to total cell count (treated-over-control ratio × 100%), respectively.
4.6. Cytotoxicity
The influence of the 12 nm SiO2 NPs on cell proliferation and cytotoxicity was determined by four different bulk assays measuring various endpoints like mitochondrial activity, membrane integrity and protein content/cell number. This approach was chosen in order to verify the data and to reduce the risk of experimental artifacts.
4.6.1. Water Soluble Tetrazolium (WST-1) Assay
In order to determine the effects on cell toxicity and viability, the commercially available WST-1 test kit was used (Roche Applied Science, Mannheim, Germany). The test principle is based on the ability of cells to reduce the tetrazolium salt 4-[3-(4-iodophenyl)-2-(4-nitrophenyl)-2H-5-tetrazolio]-1,3-benzene disulfonate to a water soluble formazan salt, which can be measured in a plate reader at λ = 450 nm. Cells were seeded into a 96-well plate and allowed to grow for 48 h (cell confluence of around 70%). Thereafter, cells were incubated with the respective SiO2 NPs prepared according to the SOP for 45 min or 24 h. Triton X-100 was included in the test system as a positive control. Then, cells were washed twice with 100 µL PBS and incubated with 110 µL of a 1:10 WST-1 reagent/medium dilution (v/v). Subsequently, the plates were measured including a reference wavelength of λ = 650 nm. Each SiO2 NPs preparation was tested three times with cells from at least three different passages and every concentration was measured six times resulting in eighteen replicates for each experimental point. The effect on the mitochondrial activity was expressed as percentage of viability (treated-over-control ratio × 100%).
4.6.2. Lactate Dehydrogenase (LDH) Assay
In parallel to the WST-1 assay, the potential for cytotoxicity of SiO2 NPs was also determined by measuring the lactate dehydrogenase release in the cell medium. To this aim the commercial LDH kit (Roche Applied Science, Mannheim, Germany) was used. In the case of cell lysis, released LDH catalyzes in a coupled enzymatic reaction the reduction of the tetrazolium salt 2-[4-iodophenyl]-3-[4-nitrophenyl]-5-phenyltetrazolium chloride (INT) to a red colored, water soluble formazan salt, which enables the correlation of the spectrophotometric signal with the cell status. After particle incubation, 50 µL of the supernatant and 50 µL of PBS were carefully transferred into a clean 96-well plate for the LDH assay. Then 100 µL of the freshly prepared reaction mixture (according to manufacturer’s handbook) were added to every well. The plates were incubated under agitation for 5 min at room temperature and measured immediately with a plate reader at λ = 490 nm. A reference wavelength (λ = 650 nm) was recorded and subtracted. For each particle preparation, tests were carried out at least three times using cells from at least three different cell preparations. Each silica concentration was measured six times resulting in eighteen replicates. The effects on the LDH release were determined as percentage of LDH leakage (treated-over-assay control ratio × 100%).
4.6.3. Sulforhodamine B (SRB) Assay
To determine the influence on cellular growth, the SRB assay was performed according to Skehan et al. [
72] with slight modifications. Therefore, cells were seeded into 96-well plates and allowed to grow for 48 h prior to treatment up to 40% of confluence. Cells were incubated with SiO
2 particle suspensions for 24 h, 48 h or 72 h, respectively, in complete medium. Incubation was stopped by addition of 10 µL of trichloroacetic acid (50%
v/
v solution). After 1 h at 4 °C, plates were washed four times with water. The dried plates were stained with a 0.4% (
w/
v) solution of SRB. Again, plates were washed twice with water and acetic acid solution (1%
v/
v). The dried protein-bound dye was dissolved with 100 µL Tris-buffer (10 mM, pH 10.5) and quantified photometrically (λ = 570 nm). The effects on cellular growth were determined as percentage of survival (treated-over-control ratio × 100%).
4.7. Western Blot Analysis
Phosphorylation of the ERK 1/2 as well as the phosphorylation of the EGFR were analyzed by Western blot according to the method prior published by Gehrke et al. [
28]. In brief, GXF251L cells were seeded into Petri dishes (Ø 10 cm) using 10 mL of the respective growth media supplemented with 9% FBS and allowed to grow for 96 h. Cells were starved in low-serum medium for additional 24 h to arrest cell growth and to equalize cell cycle phase. Thereafter, cells were treated with the 12 nm SiO
2 NP suspensions for 45 min or 24 h. After the incubation period, cells were stored on ice and cell lysis was performed using a commercially available Cell Lysis Buffer (10× stock; New England BioLabs GmbH, Frankfurt, Germany) including 1 mM PMSF. Protein content was adjusted by means of Bradford assay and subsequently, proteins were separated by SDS-PAGE and transferred onto a nitrocellulose membrane by Western blot. After blocking in 5% (
w/
v) milk powder diluted with Tris-buffered saline containing 0.1% Tween-20, membranes were incubated using rabbit antibodies against human EGFR (polyclonal, detection of total EGFR protein), pEGFR (polyclonal, detection of EGFR only when phosphorylated at Tyr1068), p44/42 MAPK (ERK1/2) (monoclonal, detection of total p44/42 MAP kinase protein) and phospho-p44/42 MAPK (pERK1/2) (polyclonal, detection of p44/42 MAP kinase when individually or dually phosphorylated at Thr202 and Tyr204 of ERK1/Thr185 and Tyr187 of ERK2), all purchased from Cell Signaling Technology (provided by New England BioLabs GmbH, Frankfurt, Germany). α-tubulin (mouse monoclonal, raised against amino acids 149–448 of α-tubulin of human origin; Santa Cruz Biotechnology, Inc., Heidelberg, Germany) was included as a loading control and detected on the same membrane after cutting it in two pieces. Anti-rabbit as well as anti-mouse IgG horseradish peroxidase conjugates (Santa Cruz Biotechnology, Inc., Heidelberg, Germany) were used as secondary antibodies. The respective chemoluminescent signals (LumiGlo Reagent & Peroxide: Cell Signaling Technology provided by New England BioLabs GmbH, Frankfurt, Germany) were analyzed using the LAS 4000 system with the Multi Gauge Image Analyzer software version 3.2 for quantification (Fujifilm, Tokyo, Japan). Arbitrary light units were plotted as treated-over-control ratios (T/C × 100%).
4.8. Immunocytochemistry
Immunocytochemical (IC) experiments were performed for the localization of Ki-67 and the EGF receptor. In the first case, GXF251L cells were incubated with the respective concentrations of 12 nm SiO2 NPs for 24 h and in the latter for 45 min following the same protocol of seeding and incubation already described for the Western blot experiments. At the end of the incubation time cells were washed with PBS and fixed with 3.7% formaldehyde (15 min, room temperature). After fixation, cells were washed, permeabilized with 0.2% Triton X-100 and blocked with 1% BSA (Ki-67) or 10% normal goat serum (NGS; EGFR) (1 h, room temperature). For the IC analysis, Lamin B goat polyclonal antibody (sc-6216), Ki-67 rabbit polyclonal antibody (sc-15402) and α-tubulin mouse monoclonal antibody (sc-5286) (all purchased from Santa Cruz Biotechnology, Inc., Heidelberg, Germany) were used. In addition, a rabbit polyclonal antibody against human EGFR was used (New England BioLabs GmbH, Frankfurt, Germany). Fluorescent secondary antibodies raised against relevant species were then used for the detection according to manufacturer details (Alexa Fluor 647 Anti-Goat, Alexa Fluor 568 Anti-Rabbit and Alexa Fluor 488 Anti-Mouse antibodies, Life Technologies, Vienna, Austria). After incubation with the secondary antibodies cells were washed and post-fixed with 3.7% of formaldehyde and free aldehyde groups were then quenched with PBS glycine (100 mM). Slides were mounted and sealed with Roti-Mount FluorCare (Roth, Graz, Austria) and imaged after 48 h with a Confocal LSM Zeiss 710 meta equipped with a Plan Apochromat 63X/1.4 oil objective, zoom 2. For image analysis and quantification, the software Zeiss ZEN was used. Images were acquired from three different cell preparations keeping the acquisition parameters constant within the experimental session (Laser power, gain, background correction). Image quantification was performed in the central section of the z-stack of the nucleus and for every experimental condition more than 60 nuclei were quantified and signal intensities were evaluated as treated-over-control ratios (T/C × 100%).
4.9. RNA Extraction and qRT-PCR
For RNA extraction, GXF251L cells were seeded into Petri dishes (Ø 6 cm) and allowed to grow to a confluence of around 70%–80%. Thereafter, cells were treated with the respective 12 nm SiO2 NPs suspensions for 2 h, 6 h, 16 h and 24 h, prepared according to the SOP and harvested subsequently. Total RNA was extracted with the QuantiTect RNeasy Mini Kit (Qiagen GmbH, Hilden, Germany) and RNA content was determined with the Nanodrop 2000c (Peqlab). The QuantiTect Reverse Transcription Kit (Qiagen GmbH, Hilden, Germany) along with the Peltier Thermal Cycler (BioRad) were used for reverse transcription of the extracted RNA into cDNA. Subsequently, transcripts were amplified and quantified by the StepOne Plus SYBR-Green based quantitative real-time PCR (Life Technologies, Applied Biosystems, Vienna, Austria) by means of the QuantiTect SYBR Green PCR Kit and QuantiTect Primer Assay (Qiagen GmbH, Hilden, Germany). Thereby, the EGFR (Hs_EGFR_1_SG) gene was analyzed and β-actin (Hs_ACTB_1_SG), as well as, GAPDH (Hs_GAPDH_2_SG) were analyzed as housekeeping genes. No-template controls and melting curves were included to exclude potential contaminations and non-specific amplifications. The respective relative transcription rates were normalized to the housekeeping genes and then calculated using the ΔΔCT-method.
4.10. Statistics
Unless stated otherwise, all results are given as mean value of at least three different independent experiments ± standard deviation (SD) and are depicted as treated-over-control T/C [%]. Data were analyzed using the OriginPro 9.1.0G software (OriginLab Corporation, Northampton, MA, USA). Tests for normal distribution were performed by Shapiro-Wilk test (p ≥ 0.05). For statistical analysis of differences of the mean between the several tested groups One-way ANOVA and subsequently “Fisher’s Least Significant Differences (LSD)” post-hoc test were performed. “Student’s t-test for two independent samples” was performed to compare significances between only two groups. Symbols are used in figures to indicate a comparison to the respective negative control with statistical levels of p ≤ 0.05 for *, p ≤ 0.01 for ** and p ≤ 0.001 for ***. Bold printed letters in a figure indicate a significant difference of these data and were considered statistically significant with a significance level of at least p ≤ 0.05.


 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}