
Synthesis of Polyhydroxybutyrate Particles with Micro-to-Nanosized Structures and Application as Protective Coating for Packaging Papers


Abstract
:
1. Introduction
2. Results and Discussion
2.1. Synthesis and Characterization of PHB Microparticles and Submicron Particles
2.1.1. Morphological Analysis by Scanning Electron Microscopy
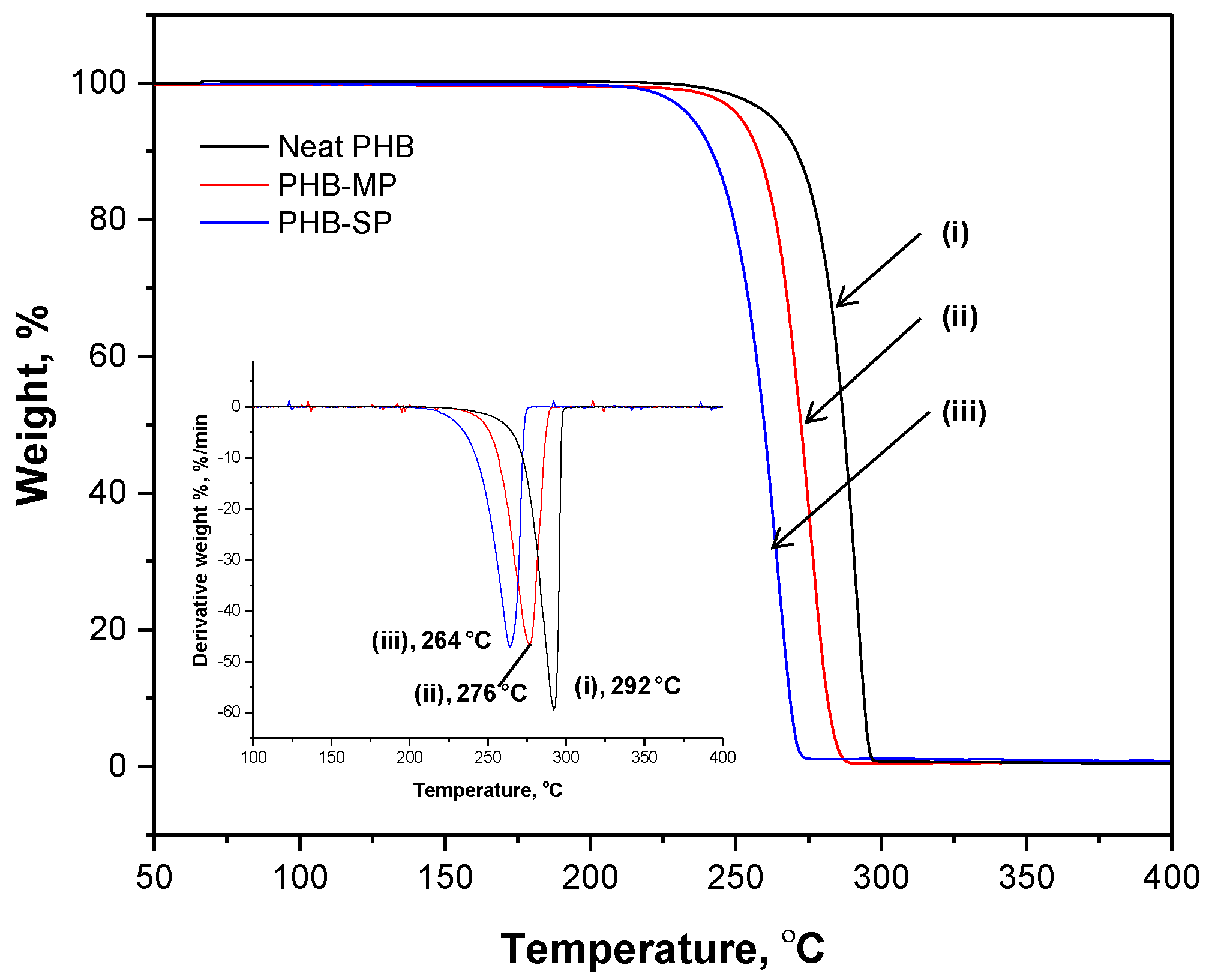
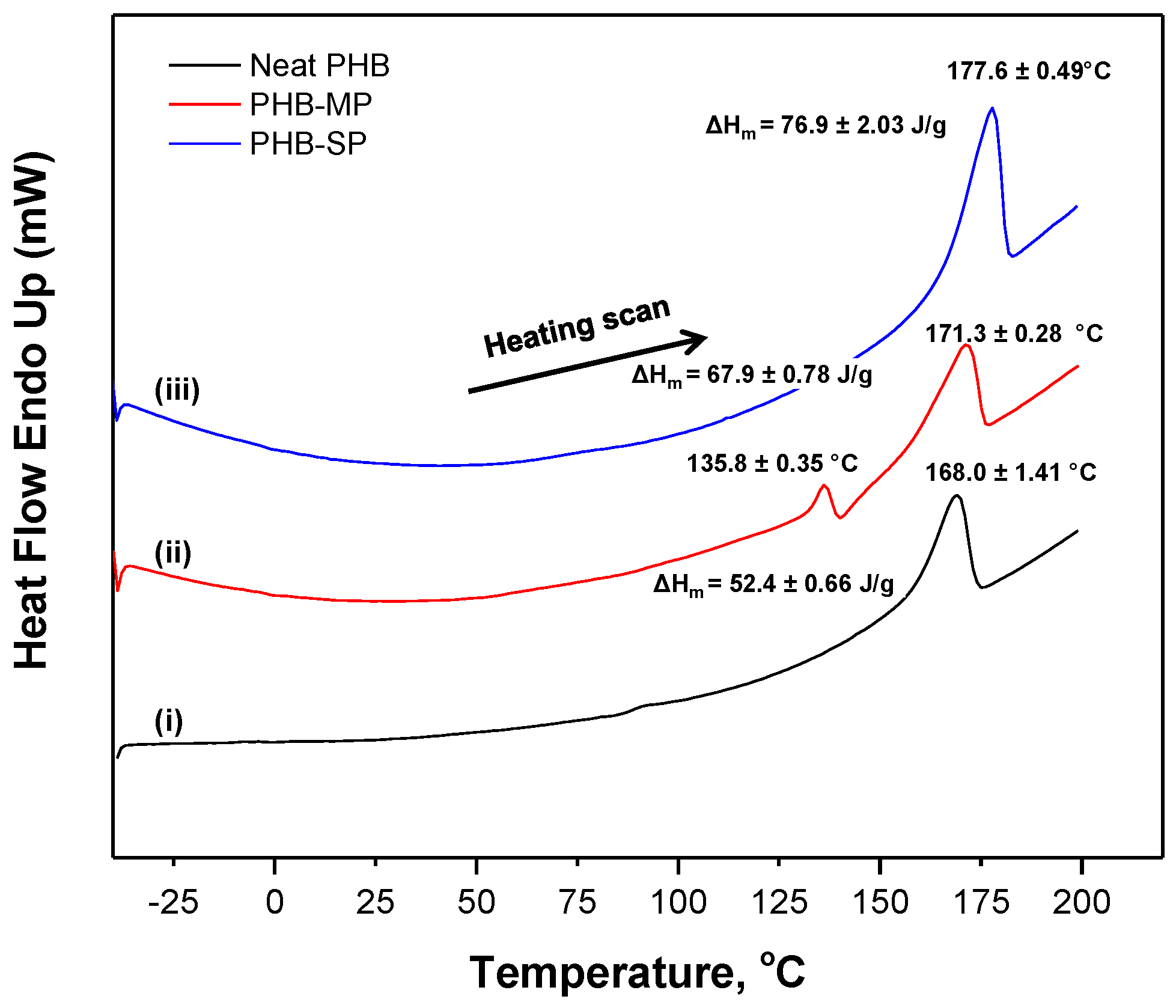
2.1.2. Thermal Analysis
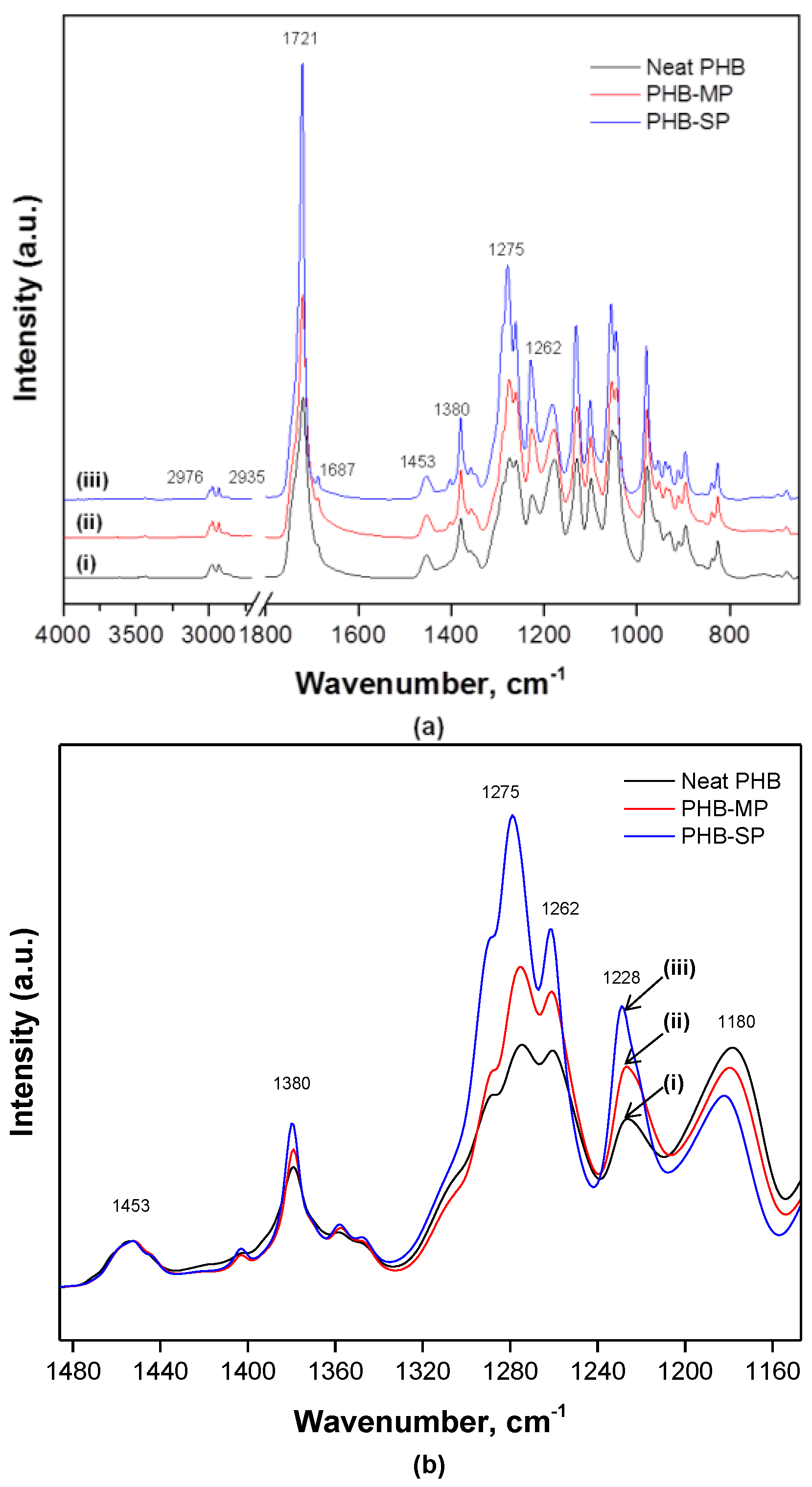
2.1.3. Chemical Analysis by Fourier-Transform Infrared Spectroscopy
2.2. First Approach: Application of PHB-MP as Paper Coating
2.2.1. Morphological Analysis (SEM) and Water Contact Angle Measurements
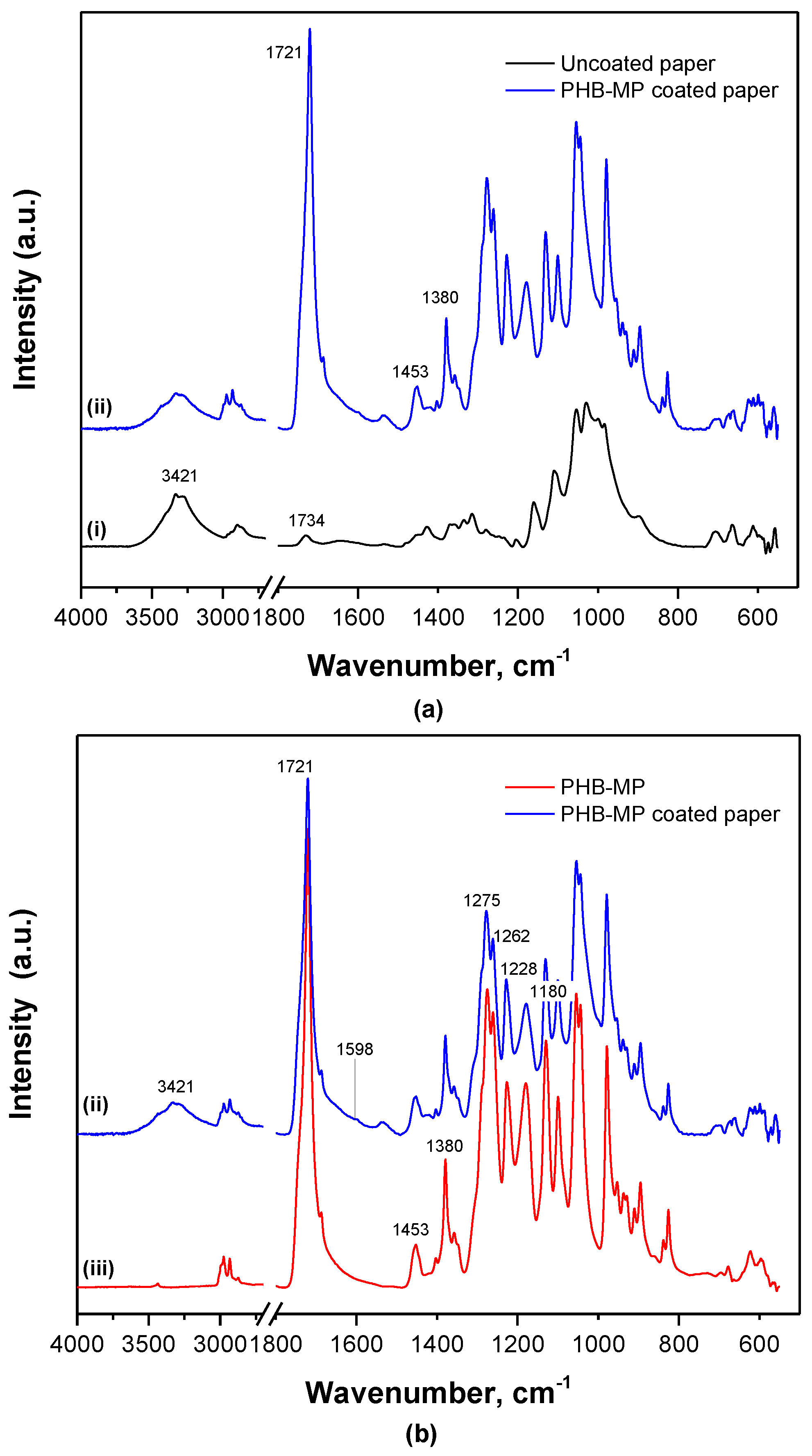
2.2.2. Interactions between PHB-MP and the Paper Substrate
2.3. Second Approach: Application of PHB-SP as Paper Coating
2.3.1. Morphological Analysis (SEM) and Water Contact Angle Measurements
2.3.2. Mechanisms of Interactions between PHB-SP/NFC and the Paper Substrate
2.4. Coating Weight and Coating Thickness
3. Experimental Section
3.1. Materials
3.2. Synthesis of PHB Microparticles and Submicron Particles and Deposition as Paper Coating
3.3. Characterization of PHB Microparticles and Submicron Particles and Coated Papers
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| PHB | Polyhydroxybutyrate |
| NFC | Nanofibrillated cellulose fibers |
| PHB-MP | Polyhydroxybutyrate microparticles |
| PHB-SP | Polyhydroxybutyrate submicron particles |
| DMF | Dimethylformamide |
| THF | Tetrahydrofuran |
| PVA | Polyvinyl alcohol |
| TGA | Thermogravimetric analysis |
| DSC | Differential scanning calorimetry |
| SEM | Scanning electron microscopy |
| FTIR | Fourier transforms infrared spectroscopy |
| AFM | Atomic force microscopy |
| Xc, % | Degree of crystallinity |
References
- Koch, K.; Barthlott, W. Superhydrophobic and superhydrophilic plant surfaces: An inspiration for biomimetic materials. Philos. Trans. A 2009, 367, 1487–1509. [Google Scholar] [CrossRef] [PubMed]
- Genzer, J.; Marmur, A. Biological and Synthetic Self-Cleaning Surfaces. MRS Bull. 2008, 33, 742–746. [Google Scholar] [CrossRef]
- Nosonovsky, M.; Bhushan, B. Multiscale friction mechanisms and hierarchical surfaces in nano- and bio-tribology. Mater. Sci. Eng. R 2007, 58, 162–193. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.M.; Joshi, P.; Datta, S.; Zhao, J.G.; France, P. Plasma assisted hydrophobic coatings on porous materials: Influence of plasma parameters. J. Phys. D 2002, 35, 1927–1933. [Google Scholar] [CrossRef]
- Smook, G.A. Handbook for Pulp and Paper Technologists, 2nd ed.; Angus Wilde Publications: Vancouver, BC, Canada, 1992; pp. 223–224. [Google Scholar]
- Rhim, J.W.; Lee, J.-H.; Hong, S.-I. Increase in water resistance of paperboard by coating with poly(lactide). Packag. Technol. Sci. 2007, 20, 393–402. [Google Scholar] [CrossRef]
- Cyras, V.P.; Commisso, M.S.; Mauri, A.N.; Vázquez, A. Biodegradable double-layer films based on biological resources: Polyhydroxybutyrate and cellulose. J. Appl. Polym. Sci. 2007, 106, 749–756. [Google Scholar] [CrossRef]
- Rastogi, V.K.; Samyn, P. Bio-based coatings for paper applications. Coatings 2015, 5, 887–930. [Google Scholar] [CrossRef]
- Hablot, E.; Bordes, P.; Pollet, E.; Avérous, L. Thermal and thermo-mechanical degradation of poly(3-hydroxybutyrate)-based multiphase systems. Polym. Degrad. Stab. 2008, 93, 413–421. [Google Scholar] [CrossRef]
- Manangan, T.; Shawaphun, S. PHB and PLA Coated Bagasse Paper for Biodegradable Food Packaging. J. KMUTNB 2010, 20, 215–223. [Google Scholar]
- Ma, M.; Hill, R.M. Superhydrophobic surfaces. Curr. Opin. Colloid Interface Sci. 2006, 11, 193–202. [Google Scholar] [CrossRef]
- Callies, M.; Chen, Y.; Marty, F.; Pépin, A.; Quéré, D. Microfabricated textured surfaces for super-hydrophobicity investigations. Microelectron. Eng. 2005, 78–79, 100–105. [Google Scholar] [CrossRef]
- Hikita, M.; Tanaka, K.; Nakamura, T.; Kajiyama, T.; Takahara, A. Super-liquid-repellent surfaces prepared by colloidal silica nanoparticles covered with fluoroalkyl groups. Langmuir 2005, 21, 7299–7302. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.; Wang, Z.; Zhang, X. Combining a layer-by-layer assembling technique with electrochemical deposition of gold aggregates to mimic the legs of water striders. Adv. Mater. 2005, 17, 1005–1009. [Google Scholar] [CrossRef]
- Ogihara, H.; Xie, J.; Saji, T. Factors determining wettability of superhydrophobic paper prepared by spraying nanoparticle suspensions. Colloids Surf. A 2013, 434, 35–41. [Google Scholar] [CrossRef]
- Li, S.; Wei, Y.; Huang, J. Facile Fabrication of Superhydrophobic Cellulose Materials by a Nanocoating Approach. Chem. Lett. 2010, 39, 20–21. [Google Scholar] [CrossRef]
- Samyn, P.; Schoukens, G.; Van den Abbeele, H.; Vonck, L.; Stanssens, D. Application of polymer nanoparticle coating for tuning the hydrophobicity of cellulosic substrates. J. Coat. Technol. Res. 2011, 8, 363–373. [Google Scholar] [CrossRef]
- Stanssens, D.; Van den Abbeele, H.; Vonck, L.; Schoukens, G.; Deconinck, M.; Samyn, P. Creating water-repellent and super-hydrophobic cellulose substrates by deposition of organic nanoparticles. Mater. Lett. 2011, 65, 1781–1784. [Google Scholar] [CrossRef]
- Mertaniemi, H.; Laukkanen, A.; Teirfolk, J.-E.; Ikkala, O.; Ras, R.H.A. Functionalized porous microparticles of nanofibrillated cellulose for biomimetic hierarchically structured superhydrophobic surfaces. RSC Adv. 2012, 2, 2882. [Google Scholar] [CrossRef]
- Geissler, A.; Loyal, F.; Biesalski, M.; Zhang, K. Thermo-responsive superhydrophobic paper using nanostructured cellulose stearoyl ester. Cellulose 2014, 21, 357–366. [Google Scholar] [CrossRef]
- Obeso, C.G.; Sousa, M.P.; Song, W.; Rodriguez-Pérez, M.A.; Bhushan, B.; Mano, J.F. Modification of paper using polyhydroxybutyrate to obtain biomimetic superhydrophobic substrates. Colloids Surf. A 2013, 416, 51–55. [Google Scholar] [CrossRef]
- Koller, M. Poly(hydroxyalkanoates) for Food Packaging: Application and Attempts towards Implementation. Appl. Food Biotechnol. 2014, 1, 3–15. [Google Scholar]
- Fukuzumi, H.; Saito, T.; Iwata, T.; Kumamoto, Y.; Isogai, A. Transparent and high gas barrier films of cellulose nanofibers prepared by TEMPO-mediated oxidation. Biomacromolecules 2009, 10, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Hult, E.-L.; Iotti, M.; Lenes, M. Efficient approach to high barrier packaging using microfibrillar cellulose and shellac. Cellulose 2010, 17, 575–586. [Google Scholar] [CrossRef]
- Horowitz, D.M.; Sanders, J.K.M. Amorphous, biomimetic granules of polyhydroxybutyrate: Preparation, characterization, and biological implications. J. Am. Chem. Soc. 1994, 116, 2695–2702. [Google Scholar] [CrossRef]
- Botana, A.; Mollo, M.; Eisenberg, P.; Torres Sanchez, R.M. Effect of modified montmorillonite on biodegradable PHB nanocomposites. Appl. Clay Sci. 2010, 47, 263–270. [Google Scholar] [CrossRef]
- Shan, G.-F.; Gong, X.; Chen, W.-P.; Chen, L.; Zhu, M.-F. Effect of multi-walled carbon nanotubes on crystallization behavior of poly(3-hydroxybutyrate-co-3-hydroxyvalerate). Colloid Polym. Sci. 2011, 289, 1005–1014. [Google Scholar] [CrossRef]
- Wróbel-Kwiatkowska, M.; Zuk, M.; Szopa, J.; Dymińska, L.; Maczka, M.; Hanuza, J. Poly-3-hydroxy butyric acid interaction with the transgenic flax fibers: FT-IR and Raman spectra of the composite extracted from a GM flax. Spectrochim. Acta Mol. Biomol. Spectrosc. 2009, 73, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Chan, R.T.H.; Garvey, C.J.; Marçal, H.; Russell, R.A.; Holden, P.J.; Foster, L.; John, R. Manipulation of Polyhydroxybutyrate Properties through Blending with Ethyl-Cellulose for a Composite Biomaterial. Int. J. Polym. Sci. 2011, 2011, 651549. [Google Scholar] [CrossRef]
- Xu, J.; Guo, B.-H.; Yang, R.; Wu, Q.; Chen, G.-Q.; Zhang, Z.-M. In situ FTIR study on melting and crystallization of polyhydroxyalkanoates. Polymer 2002, 43, 6893–6899. [Google Scholar] [CrossRef]
- Peppas, N.A. Tear propagation resistance of semicrystalline polymeric networks. Polymer 1977, 18, 403–407. [Google Scholar] [CrossRef]
- Bugnicourt, E. Polyhydroxyalkanoate (PHA): Review of synthesis, characteristics, processing and potential applications in packaging. EXPRESS Polym. Lett. 2014, 8, 791–808. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Yan, R.; Chen, H.; Lee, D.H.; Zheng, C. Characteristics of hemicellulose, cellulose and lignin pyrolysis. Fuel 2007, 86, 1781–1788. [Google Scholar] [CrossRef]
- Alemdar, A.; Sain, M. Isolation and characterization of nanofibers from agricultural residues—Wheat straw and soy hulls. Bioresour. Technol. 2008, 99, 1664–1671. [Google Scholar] [CrossRef] [PubMed]
- Arbatan, T.; Zhang, L.; Fang, X.-Y.; Shen, W. Cellulose nanofibers as binder for fabrication of superhydrophobic paper. Chem. Eng. J. 2012, 210, 74–79. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Coating Type | PHB Coating Weight, mg | Wax Coating Weight, mg | Total Coating Weight, mg | Net Coat Weight, g/m2 | Net Coating Thickness, µm | Contact Angle before Wax Coating, ° | Contact Angle after Wax Coating, ° |
|---|---|---|---|---|---|---|---|
| No coating (filter paper) | - | 2.5 ± 0.1 | 2.5 ± 0.1 | 4.2 ± 0.2 | 5 ± 0.5 | <40 * | 106 ± 2 |
| Neat PHB | 21.7 ± 1.2 | 2.2 ± 0.1 | 23.9 ± 1.2 | 39.8 ± 2.0 | 65 ± 1 | 70 ± 1 | 112 ± 1 |
| PHB-MP 1 | 6.1 ± 0.3 | 2.8 ± 0.2 | 8.8 ± 0.4 | 14.7 ± 0.6 | 25 ± 2 | 105 ± 4 | 129 ± 3 |
| PHB-MP 2 | 14.2 ± 0.4 | 3.2 ± 0.3 | 17.2 ± 0.5 | 28.7 ± 0.8 | 40 ± 2 | 122 ± 2 | 144 ± 2 |
| PHB-SP/NFC, 0% | 1.0 ± 0.1 | 2.8 ± 0.1 | 3.8 ± 0.1 | 6.3 ± 0.2 | 15 ± 1 | <40 * | 112 ± 2 |
| PHB-SP/NFC, 1% | 3.5 ± 0.3 | 3.0 ± 0.1 | 6.5 ± 0.3 | 10.3 ± 0.5 | 20 ± 2 | <40 * | 125 ± 3 |
| PHB-SP/NFC, 2% | 5.5 ± 0.2 | 4.1 ± 0.4 | 9.5 ± 0.4 | 15.8 ± 0.7 | 25 ± 3 | <40 * | 133 ± 4 |
| PHB-SP/NFC, 5% | 8.5 ± 0.5 | 5.2 ± 0.3 | 13.5 ± 0.6 | 22.5 ± 1.0 | 35 ± 1 | <40 * | 142 ± 2 |
| PHB-SP/NFC, 7% | 10.0 ± 0.6 | 5.5 ± 0.4 | 15.5 ± 0.7 | 25.8 ± 1.2 | 50 ± 1 | <40 * | 152 ± 1 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rastogi, V.K.; Samyn, P. Synthesis of Polyhydroxybutyrate Particles with Micro-to-Nanosized Structures and Application as Protective Coating for Packaging Papers. Nanomaterials 2017, 7, 5. https://doi.org/10.3390/nano7010005
Rastogi VK, Samyn P. Synthesis of Polyhydroxybutyrate Particles with Micro-to-Nanosized Structures and Application as Protective Coating for Packaging Papers. Nanomaterials. 2017; 7(1):5. https://doi.org/10.3390/nano7010005
Chicago/Turabian StyleRastogi, Vibhore Kumar, and Pieter Samyn. 2017. "Synthesis of Polyhydroxybutyrate Particles with Micro-to-Nanosized Structures and Application as Protective Coating for Packaging Papers" Nanomaterials 7, no. 1: 5. https://doi.org/10.3390/nano7010005
APA StyleRastogi, V. K., & Samyn, P. (2017). Synthesis of Polyhydroxybutyrate Particles with Micro-to-Nanosized Structures and Application as Protective Coating for Packaging Papers. Nanomaterials, 7(1), 5. https://doi.org/10.3390/nano7010005