1. Introduction
Hepatocellular carcinoma (HCC) is the most common primary malignant tumor of the liver and one of the most common causes of cancer-related deaths worldwide. Some patients can have curative responses to the treatment of surgery or chemotherapy, whereas those patients with advanced HCC have to be treated with palliative intent [
1]. Alternatively, gene therapy has been tested to treat advanced tumors by overcoming the limitations of current therapeutic paradigms. Among them, RNA interference (RNAi), a post-transcriptional gene silencing mechanism, has been emerging as a promising therapeutic strategy [
2]. The survivin gene is the smallest member of the inhibitor of apoptosis (IAP) family and plays an important role in oncogenesis, due to its interaction with multiple signaling networks and action in preservation of endothelial cell viability [
3]. It has been demonstrated that overexpression of survivin indicates a poor prognosis in up to 70% of HCC patients [
4]. In addition, small interfering RNA (siRNA) against survivin has been shown to be able of inducing apoptosis of HCC cells [
3]. The success of survivin-targeted RNAi treatment depends mainly on the efficient delivery of specific siRNA to HCC cells, since siRNAs can be rapidly degraded by extracellular RNase and cleared by renal filtration [
5]. Therefore, a robust siRNA delivery system is highly desirable for gene therapy of HCC. Moreover, a gene delivery system with image agents, i.e., superparamagnetic iron oxide nanoparticles (SPIO), being loaded would endow the nanoprobe a theranostic nature, which enables noninvasive detection of cancer and simultaneous monitoring of the target therapy.
Amylose is a natural biopolymer with many advantages, such as abundance, cheapness, biocompatibility, and non-toxicity [
6,
7]. Its natural linear polysaccharide is composed of glucose residues through α (1 → 4) glycosidic linkages which determines the efficient degradable performance by various enzymes [
8]. Amylose can be modified by substituting the hydroxides with quaternary ammonium salt to increase its solubility and enzymatic digestibility in order to meet the requirements of gene delivery. As a nanocarrier backbone, amylose has been constructed into a variety of vehicles for controllable drug and gene delivery, such as flufenamic acid, testosterone, caffeine [
9], curcumin, indomethacin [
10], image labels [
11], therapeutic DNA [
12], and RNA [
13]. Further modification with a targeted moiety can significantly increase the active and specific accumulation of the nanovehicle in tumor cells of interest. Folate receptors are universally expressed on the surface of tumor cells more frequently than on normal cells [
14]. A variety of folate-decorated nanoparticles have been used as promising nanocarriers for solid tumor detection and targeting treatment [
15]. Previously, folic acid-conjugated SPIOs were shown to possess good diagnostic sensitivity with magnetic resonance imaging (MRI) in folate-receptor-positive cancer cells [
16,
17].
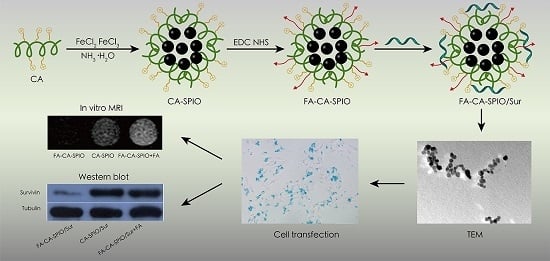
In this study, we synthesized a new class of amylose nanoparticles, where the cationic amylose was used as the backbone, folate as the targeting ligand, and SPIO as the image label to deliver survivin-siRNA to HCC cells. The aim of this study was to investigate the potential of folate-functionalized, SPIO-loaded cationic amylose nanoparticles as a new biocompatible polysaccharide-based multifunctional nanoparticle to transfer siRNA against survivin and meet the requirement of simultaneous MRI detection of HCC.
2. Materials and Methods
2.1. Materials
Amylose (>98%) from potato was purchased from Fluka Co., Ltd. (Buchs, Switzerland). Molar mass distribution of the obtained purified amylose, as detected by gel permeation chromatography (GPC; Waters Corporation, Milford, MA, USA) was: Mn = 83.0 kDa, MW = 107.6 kDa, and a polydispersity index (PDI) = 1.30. siRNA transfection media was purchased from GenePharm Co., Ltd. (Shanghai, China). Glycidyltrimethylammonium chloride (GTA, MW = 151.63 Da) was purchased from Sigma-Aldrich Co. Ltd. (St. Louis, MO, USA). 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC·HCl, MW = 191.70 Da), N-Hydroxysuccinimide (NHS, MW = 115.09 Da), and folic acid (FA, MW = 441.4 Da) were obtained from Aladdin Reagent Database Inc. (Shanghai, China). 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) was obtained from Promega Co., Ltd. (Madison, WI, USA). The fluorescent staining agent 4′,6-diamidion-2-phenylindole (DAPI) was purchased from Roche Co., Ltd. (Mannheim, Germany). All other chemicals and reagents were of analytical grade. Survivin siRNA (sense: 5′-CACCGCAUCUCUACAUUCATT-3′; antisense: 5′-UGAAUGUAGAGAUGCGGUGTT-3′) and carboxyfluorescein(FAM)-conjugated scramble RNA (sense: 5′-UUCUCCGAACGUGUCACGUTT-3′; antisense: 5′-ACGUGACACGUUCGGAGAATT-3′) used as negative control were synthesized by GenePharm Co., Ltd. (Shanghai, China).
2.2. Synthesis of Magnetic Amylose Nanoparticles
Amylose (0.5 g) was dissolved in 25 mL distilled water, and adjusted to pH 12–14 using 4 mol/L NaOH. The solution was mixed and dispersed by ultrasound. Then, 2 g 2,3-epoxypropyl trimethyl ammonium chloride (Sigma-Aldrich Co. Ltd., St. Louis, MO, USA) was slowly added into the solution, and the mixture was stirred at 80 °C for 24 h. After adjusting the pH to 6–7, the mixture was dialyzed for three days (MWCO = 8000–14,000) and lyophilized to obtain the cationic amylase (CA). One-pot synthesis of magnetic nanoparticles was used to obtain SPIO-loaded CA. In brief, 0.2 g CA was dissolved in 20 mL distilled water by continuous stirring. FeCl
3·6H
2O (0.2 g) and 0.10 g FeCl
2·4H
2O dissolved in 5 mL distilled water was added, stirred, and purged with nitrogen for 30 min. Then, 2.5 mL 25% NH
3·H
2O was added under vigorous stirring and the reaction was allowed to proceed for 1 h at 80 °C. The mixture was cooled to room temperature and dialyzed against distilled water for two days (MWCO = 14,000). After centrifugation, the upper aqueous dispersion was collected and lyophilized to obtain SPIO-loaded CA (CA-SPIO). To obtain folate-functionalized CA-SPIO, 20 mg folic acid was dissolved in 4 mL dimethyl sulphoxide (DMSO) by continuous stirring for 1 h, after which 40 mg EDC·HCl and 20 mg NHS were added, the mixture was stirred and allowed to react completely for 4 h away from light to obtain the folic acid ester in DMSO. Then the solution of CA-SPIO (0.1 g CA) was added in a 25 mL flask and acetic acid was added to adjust the pH to 5. After that, the folic acid ester in DMSO was added at room temperature away from light for 48 h. After reaction, the mixture was dialyzed for three days (MWCO = 8000–14,000) and lyophilized to obtain folate-conjugated CA-SPIO (FA-CA-SPIO) (
Figure 1).
2.3. Synthesis of siRNA-Complexed Magnetic Amylose Nanoparticles
FA-CA-SPIO and CA-SPIO were dissolved in deionized water and stored at 4 °C. Survivin-siRNA was diluted and stored according to the manufacturer’s instructions. To prepare siRNA-complexed magnetic amylose nanoparticles, survivin siRNA and a designated amount of FA-CA-SPIO or CA-SPIO were dissolved separately. The two solutions were mixed by vigorous pipetting, and then the mixture was kept at room temperature for 30 min to allow for the formation of the complexes, survivin-siRNA-complexed FA-CA-SPIO (FA-CA-SPIO/Sur), or survivin-siRNA-complexed CA-SPIO (CA-SPIO/Sur). To prepare negative control RNA-complexed magnetic amylose nanoparticles, FAM-conjugated scramble RNA was added in place of survivin-siRNA in the same manner to obtain FAM-conjugated scramble RNA-complexed CA-SPIO (CA-SPIO/FAM-RNA) or FA-CA-SPIO (FA-CA-SPIO/FAM-RNA). To assess the siRNA condensation ability, gel electrophoresis was performed on a Bio-Rad Sub-Cell® GT electrophoresis cell (Bio-Rad Laboratories, Inc.; Hercules, CA, USA). For the test, nanocomplexes (containing 0.64 μg survivin siRNA) were induced at different w/w ratios (nanoparticle/siRNA) ranging from 4 to 35, loaded onto 3% agarose gels with EtBr (0.1 µg/mL) and run with Trisacetate-EDTA running buffer at 100 V for 30 min. Visualization and image capture were performed using a UV-transilluminator under a Kodak EDAS 290 digital imaging suite (Fisher Scientific; Pittsburgh, PA, USA).
2.4. Characterization of Nanoparticles
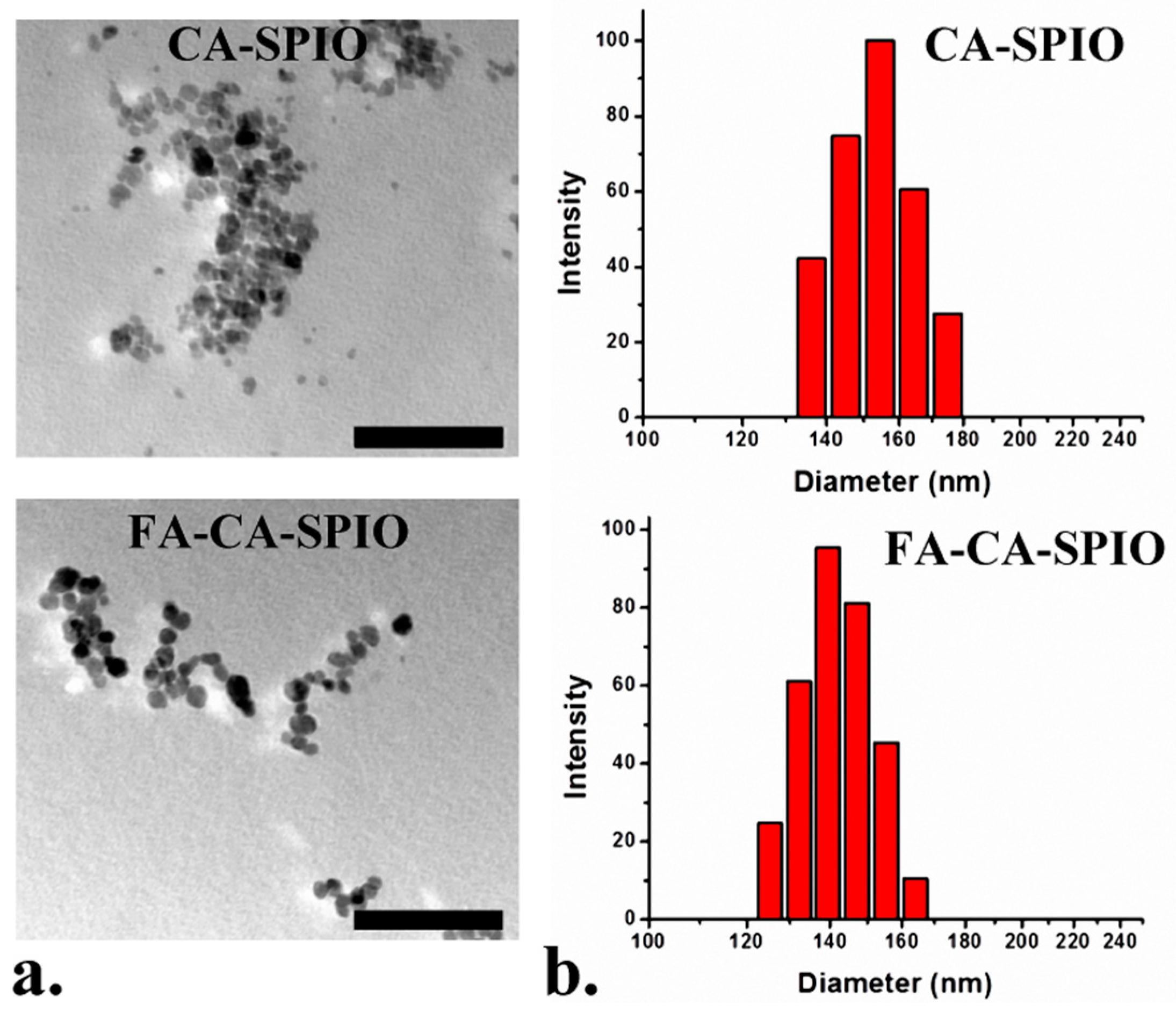
The Fourier transform infrared (FT-IR) spectra was performed on a Nicolet/Nexus 670 FT-IR spectrophotometer (Thermo Nicolet; Madison, WI, USA) at a resolution of 4 cm−1 and frequencies ranging from 450 to 4000 cm−1 using KBr pellets. 1H NMR analysis was carried out on an AVANCE III 400 MHz NMR Spectrometer (BrukerBiospin; Fällanden, Switzerland) at room temperature using DMSO-d6 or D2O as the solvent. The thermogravimetric analyses (TGA) were carried out by using a Pyris 1 thermogravimetric analyzer (Perkin Elmer; Waltham, MA, USA). For TGA, the samples (about 3 mg) were heated from room temperature to 700 °C at a heating rate of 20 °C/min in N2. The X-ray diffraction (XRD) measurements were performed on a SmartLab X-ray diffractometer (Rigaku, Tokyo, Japan) using Cu Kα radiation. The magnetic measurements were carried out with a superconducting quantum interference device (SQUID), model MPMS XL-7 magnetometer (Quantum Design, San Diego, CA, USA). The transmission electron microscopy (TEM) images were obtained on a JEM-2010HR 200kV transmission electron microscope (Jeol Ltd., Tokyo, Japan). For TEM, the sample suspension was prepared by drying a drop (5 μL) of the sample solution on a copper grid coated with amorphous carbon and then blotted with a filter paper after 1 h. After the samples were stained with 10 μL of uranyl acetate solution (2 wt % in water), blotted with a piece of filter paper after 1 min, the grid was finally air dried. The measurements of particle sizes and zeta potentials were performed on a ZetaPALS instrument (Brooken Haven; Long Island, NY, USA). The iron content was detected by using a polarized Zeman atomic absorption spectrometer (AAS; Hitachi Z-200; Tokyo, Japan). For AAS, the samples were dissolved in 1 M HCl solution for thorough release and dissolution of SPIO, then analyzed at the specific Fe absorption wavelength (248.3 nm) based on a pre-established calibration curve. All tests were performed in triplicate.
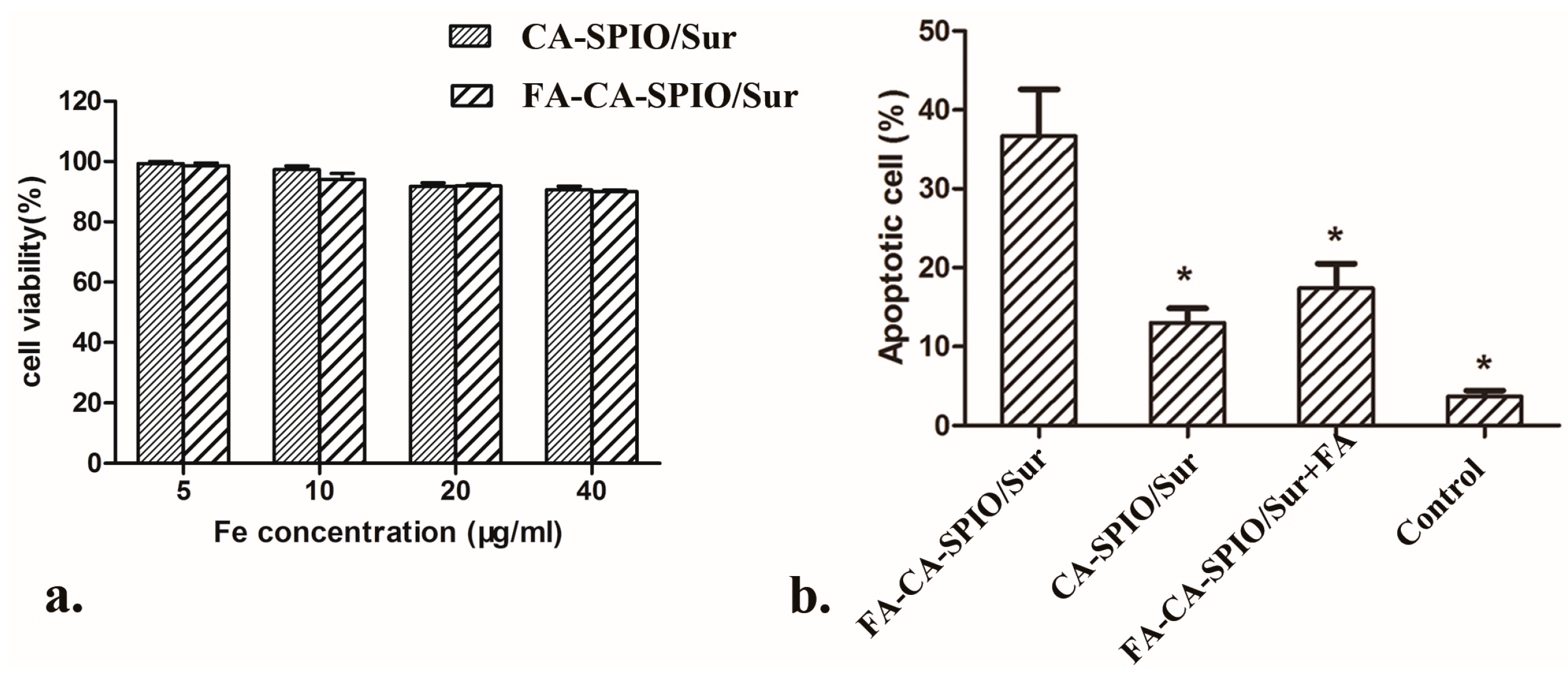
2.5. Cytotoxicity and Apoptosis
For cytotoxic analysis, the influence of FA-CA-SPIO/FAM-RNA and CA-SPIO/FAM-RNA on the viability of HepG2 cells was measured using an MTS assay. HepG2 cells were purchased from the Experimental Animal Center of Sun Yat-Sen University. Briefly, 5 × 103 cells/well were seeded in 96-well plates with 100 μL of complete medium. The nanocomplexes were prepared at various iron concentrations of 0, 5, 10, 20, and 40 μg/mL and at the w/w ratio of 12. Each nanocomplex concentration was replicated in five wells. Cells in five wells were not transfected and served as controls. FA-CA-SPIO/FAM-RNA or CA-SPIO/FAM-RNA with different concentrations of iron were added and incubated with cells for 30 min. Then, the culture medium was replaced by 100 μL of fresh RPMI-1640 medium. After 48 h of incubation at 37 °C, 10 uL of MTS solution (Promega, Madison, USA) was added to each well. After 3 h of incubation, the absorbance at 492 nm of each well was recorded on a microplate reader (MRXII; Dynex Technologies, Chantilly, VA, USA). For analysis of apoptosis induced by FA-CA-SPIO/Sur or CA-SPIO/Sur, cell apoptosis was measured by flow cytometry. Briefly, 2 × 105 HepG2 cells were seeded in six-well plates in culture medium free of folic acid or supplemented with 1 mmol/L of folic acid. Folic acid was added 30 min before the start of nanoparticle treatment to block folate receptors. After incubation with 24 μL FA-CA-SPIO/Sur or CA-SPIO/Sur for 30 min, cells were washed with phosphate-buffered solution (PBS) three times. Cell apoptosis was assessed by the Annexin V/propidium iodide (PI) double staining method and analyzed using flow cytometry (FACSCalibur, BD Biosciences, San Jose, CA, USA). All experiments were performed in triplicate.
2.6. Cellular Uptake Assay
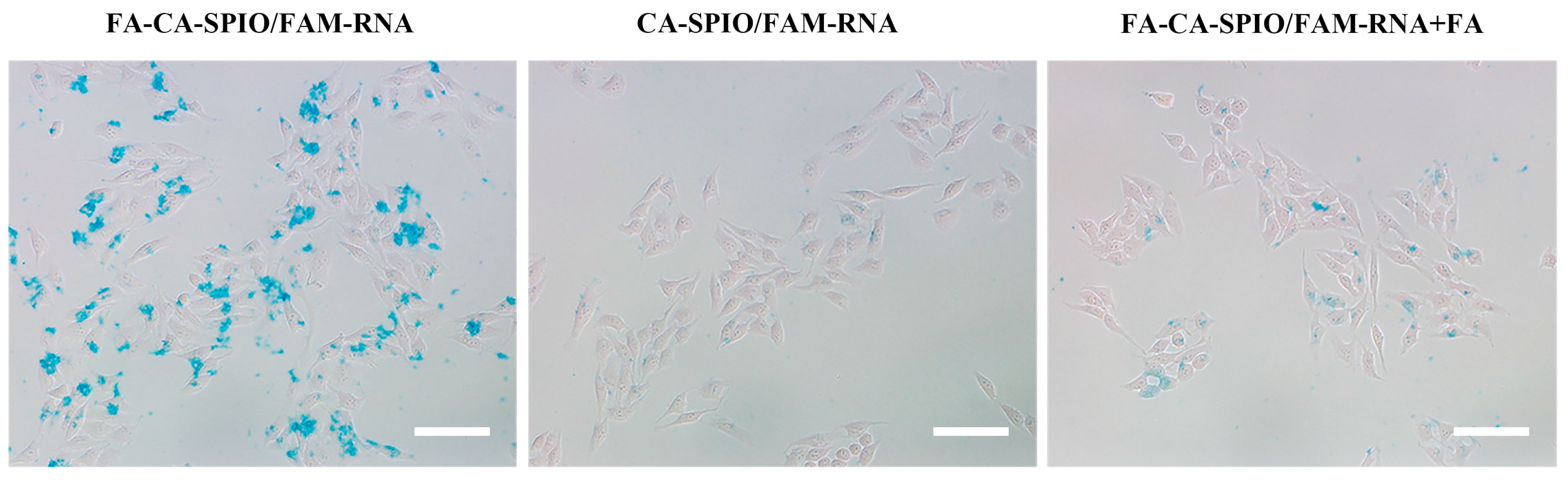
The cellular uptake of nanocomplexes was assessed by Prussian blue staining, fluorescence microscopy, and flow cytometry. HepG2 cells were seeded into 24-well plates at a density of 3 × 104 cells/well and incubated in folic acid-free RPMI-1640 medium or medium supplemented with 1 mmol/L of folic acid for 24 h at 37 °C. Folic acid was added 30 min before the start of nanoparticle treatment to block folate receptors. For Prussian blue staining, 6 μL of FA-CA-SPIO/FAM-RNA or CA-SPIO/FAM-RNA was added to transfect cells at 37 °C for 30 min. After that, the cells were washed with PBS three times and fixed by 1 mL 4% paraformaldehyde solution for 20 min. Then, 1 mL of a 1:1 mixture of 2% potassium ferrocyanate trigydrate and 4% hydrochloric acid (in PBS) was added. The cells were incubated in the dark for 30 min at 37 °C, then washed and counterstained with nuclear fast red for 5 min. Prussian blue staining was examined with an optical microscope (Olympus BX51, Tokyo, Japan). For fluorescence microscopy and flow cytometry assays, HepG2 cells were seeded into the six-well plate at a density of 2 × 105 cells/well and incubated in culture medium free of folic acid or supplemented with 1 mmol/L of folic acid for 24 h. For fluorescence microscopy anlysis, 24 μL of FA-CA-SPIO/FAM-RNA or CA-SPIO/FAM-RNA was added to transfect cells at 37 °C for 30 min, then the DNA-staining agent DAPI (1 mg/mL) was added. Cells were washed three times with PBS, examined by fluorescence microscope (Nikon, Tokyo, Japan). For flow cytometry analysis, 2 mL of PBS containing 24 μL of FA-CA-SPIO/FAM-RNA or CA-SPIO/FAM-RNA was added and incubated at 37 °C for 30 min. Then cells were washed three times with PBS, and analyzed using a flow cytometer (Bio-Rad, Hercules, CA, USA). The experiments were performed in triplicate. The same number of untreated cells were used as controls.
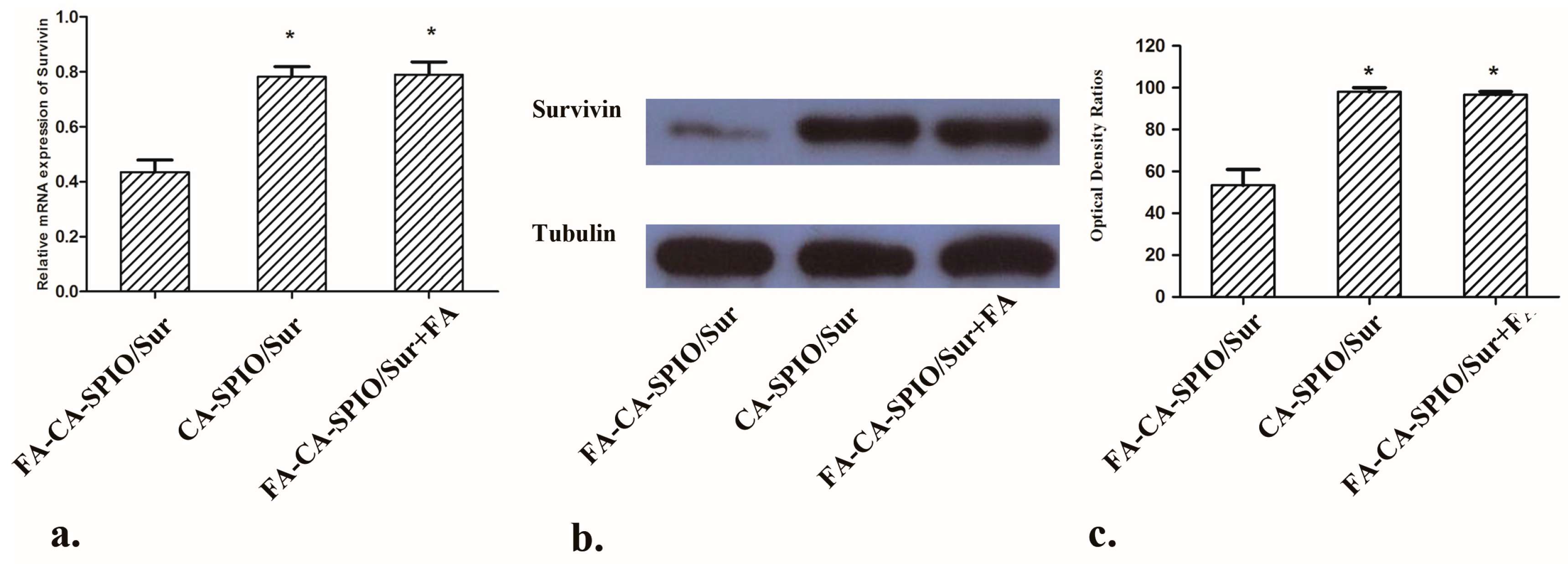
2.7. Gene Silencing Effect
To test the gene silencing effect of nanocomplexes, HepG2 cells were seeded in six-well plates at a density of 2 × 105 cells/well. FA-CA-SPIO/Sur, CA-SPIO/Sur and FA-CA-SPIO/Sur plus folic acid were added and incubated at 37 °C for 30 min. Gene silencing of survivin was measured at the mRNA level using the ABI 7500 real-time PCR system. In brief, total RNA was extracted using the RNAiso Plus reagent (TAKARA, Shiga, Japan) according to the manufacturer’s instruction. qPCR was performed according to the protocols from SYBR Premix Ex Taq™ kit (TAKARA; Takara Bio, Seta, Japan). The values of sample copies were obtained after quantitative amplification and normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) using the 2−ΔΔCt method. Water was used as negative and quality controls, and each sample was measured in triplicate.
The expression of survivin protein was detected by Western blot analysis. In brief, HepG2 cells were seeded in six-well plates at a density of 2 × 105 cells/well and were incubated with FA-CA-SPIO/Sur, CA-SPIO/Sur, and FA-CA-SPIO/Sur plus folic acid for 30 min. Then the culture medium was replaced by fresh medium and cells were incubated for 48 h. Total protein was extracted using the ProteoJETTM Mammalian Cell Lysis Reagent (Fermentas, Burlington, ON, Canada) with phenylmethanesulfonyl fluoride (PMSF). The protein content was determined using the bicinchoninic acid protein assay kit (Invitrogen, Carlsbad, CA, USA). Ten micrograms of protein were separated on a 10% sodium dodecyl sulfate (SDS) polyacrylamide gel and transferred to polyvinylidene difluoride (PVDF) membranes. The membranes were blocked with 5% fat-free milk in Tris-buffered saline Tween-20 (TBST) for 1 h at room temperature and were subsequently incubated with rabbit anti-human survivin antibody (1:1000 dilution in PBS/Tween; Cell Signaling Technology, Danvers, MA, USA) or rabbit anti-human tubulin antibody (1:10,000; Cell Signaling Technology, Danvers, MA, USA) overnight at 4 °C. Membranes were washed twice with TBST and incubated with HRP-conjugated goat anti-rabbit antibody (Cell Signaling Technology, Danvers, MA, USA) diluted 1:5000 in TBST buffer for 1 h at room temperature. An enhanced chemiluminescence detection reagent (Millipore; Billerica, MA, USA) was then used, and the protein bands were visualized after exposure to X-ray film. Non-transfected cells without transfection were used as controls. The detection of the bands was performed using a VersaDoc model 5000 imaging system quantifying the intensity with Quantity One computer software (Bio-Rad, Hercules, CA, USA). The densitometry of the bands was normalized to the corresponding tubulin band. The experiment was conducted in triplicate.
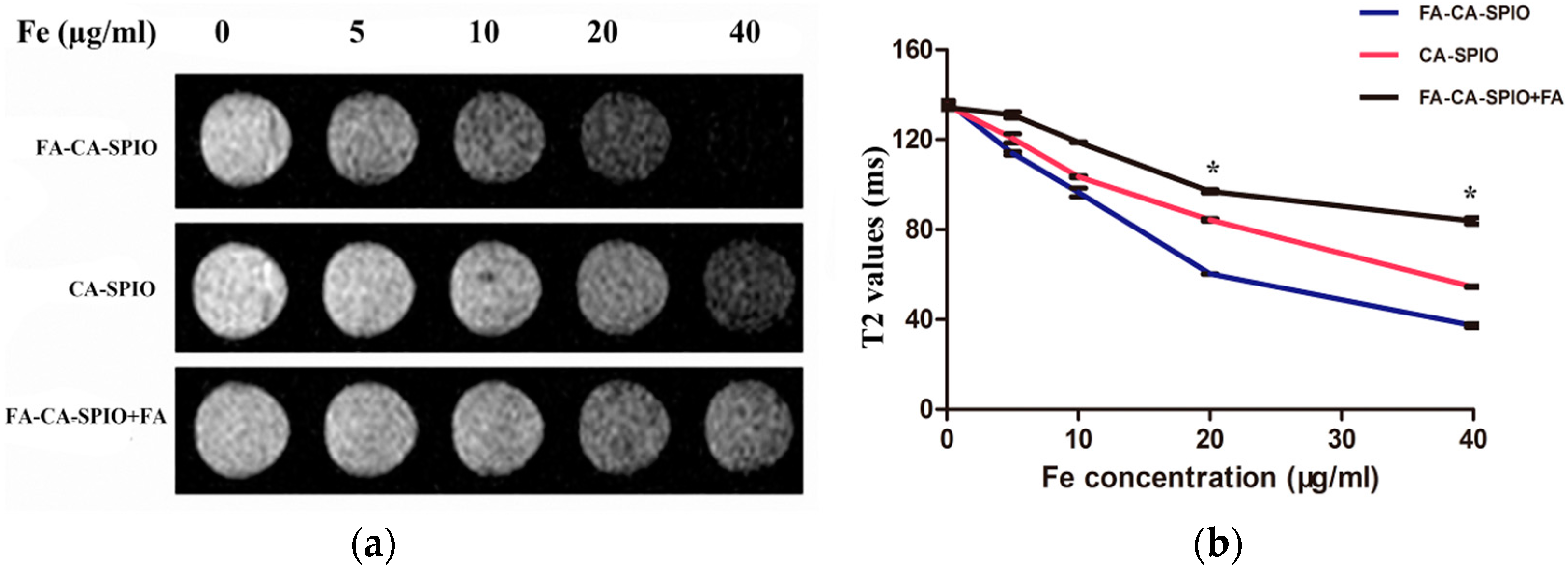
2.8. Magnetic Resonance Imaging
HepG2 cells (1 × 104) were incubated with FA-CA-SPIO or CA-SPIO at various iron concentrations (5, 10, 20, 40 μg/mL) for 30 min, the cells were rinsed with PBS three times, trypsinized, centrifuged, and resuspended in PBS containing 0.5% agarose in 96-well plates for magnetic resonance imaging (MRI). MRI was carried out on a 1.5 T scanner (Intera; Philips Medical Systems, Best, the Netherlands) at room temperature. The sequences included fast field echo T2*-weighted imaging and T2-mapping. T2*-weighted imaging using a fast spin echo sequence with a repetition time (TR)/echo time(TE) = 300/11.5 ms; flip angle = 20°; number of acquisitions (NSA) = 4; acquisition matrix = 268 × 96, field of view (FOV) = 80 mm × 80 mm; and slice thickness/gap = 2/0 mm. T2-mapping was performed by using single-slice multi-echo spin echo sequence for transverse relaxation time (T2 value) calculations with the acquisition parameters: TR/TE = 2600/20–80 ms; four stepped echoes; NSA = 1; acquisition matrix = 308 × 180; FOV = 80 mm × 80 mm; and slice thickness was 2 mm. The experiment was carried out in triplicate.
2.9. Statistical Analysis
The data was expressed as the mean ± standard deviation and the average of multiple repeats is shown. The cell viability and apoptosis were analyzed using the Student’s t-test. The T2 values were compared using analysis of variance, followed by post-hoc pairwise comparisons using the least significant difference t-test. p values less than 0.05 was considered to be statistically significant. All statistical tests were performed using the Statistical Package for the Social Sciences software version 17.0 (SPSS Inc., Chicago, IL, USA).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






