Challenges for the Self-Assembly of Poly(Ethylene Glycol)–Poly(Lactic Acid) (PEG-PLA) into Polymersomes: Beyond the Theoretical Paradigms

 ,
,  and
and
Abstract
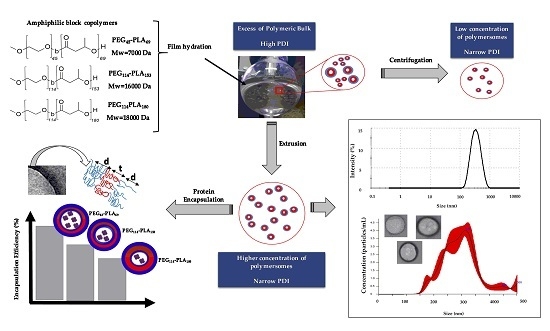
:
1. Introduction
2. Materials and Methods
2.1. Selection of Amphiphilic Copolymer
2.2. Film Hydration
2.3. Centrifugation and Extrusion
2.4. Dynamic Light Scattering (DLS)
2.5. Nanoparticle Tracking Analysis (NTA)
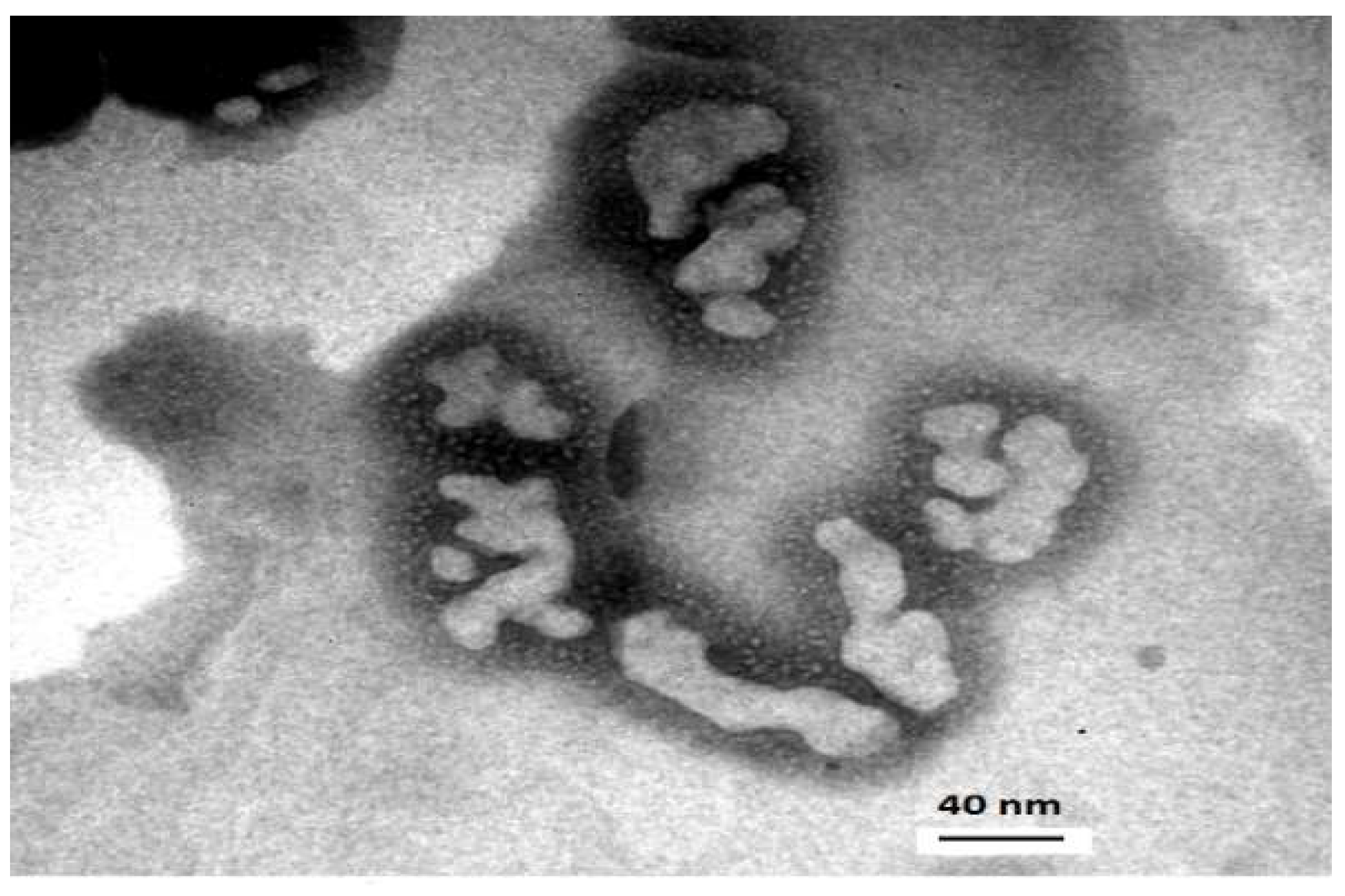
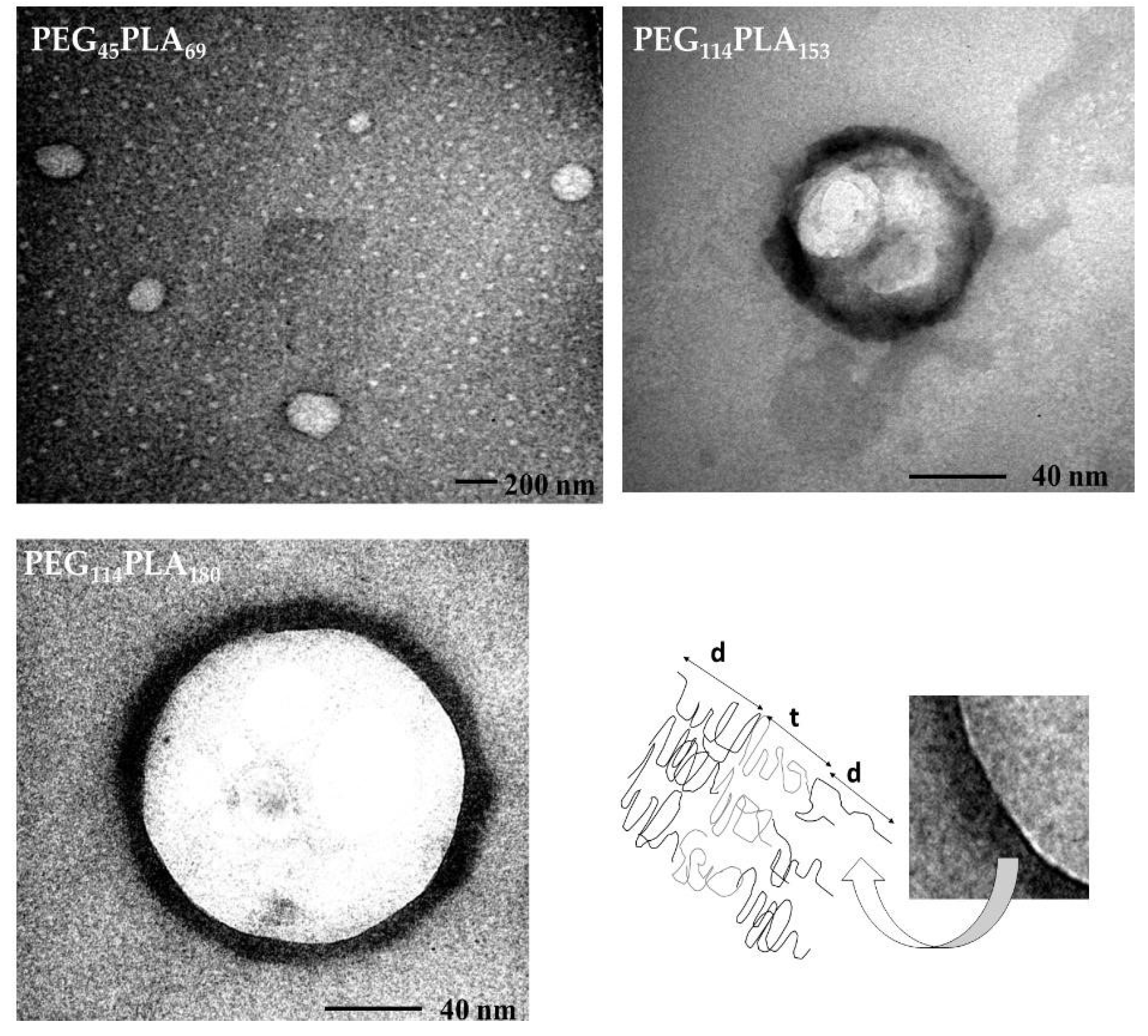
2.6. Transmission Electron Microscopy
2.7. Encapsulation of Model Globular Protein BSA and Therapeutic Protein l-Asparaginase
2.8. Statistical Analysis
3. Results and Discussion
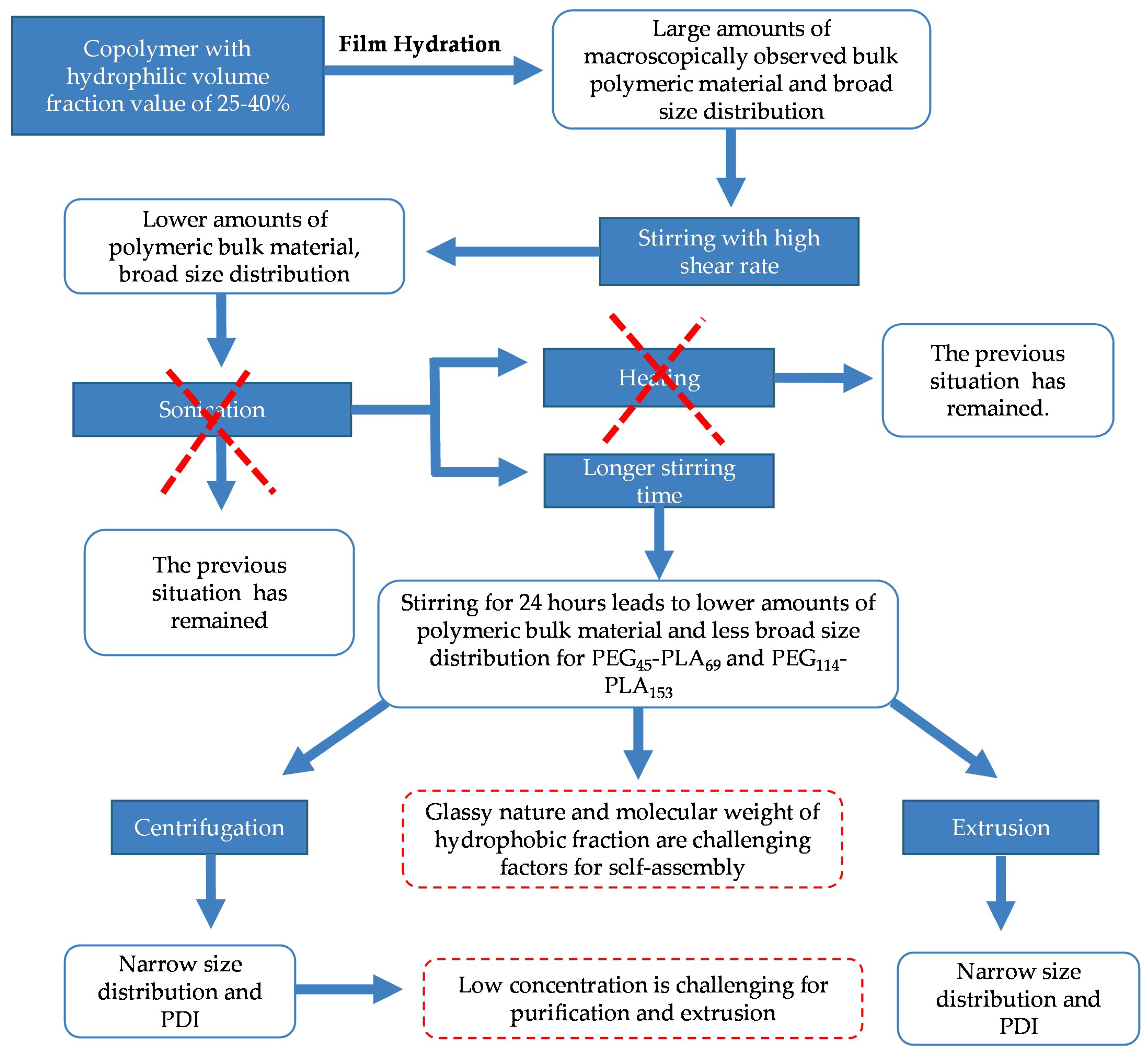
3.1. Self-Assembly by Film Hydration under Orbital Agitation Versus Magnetic Stirring
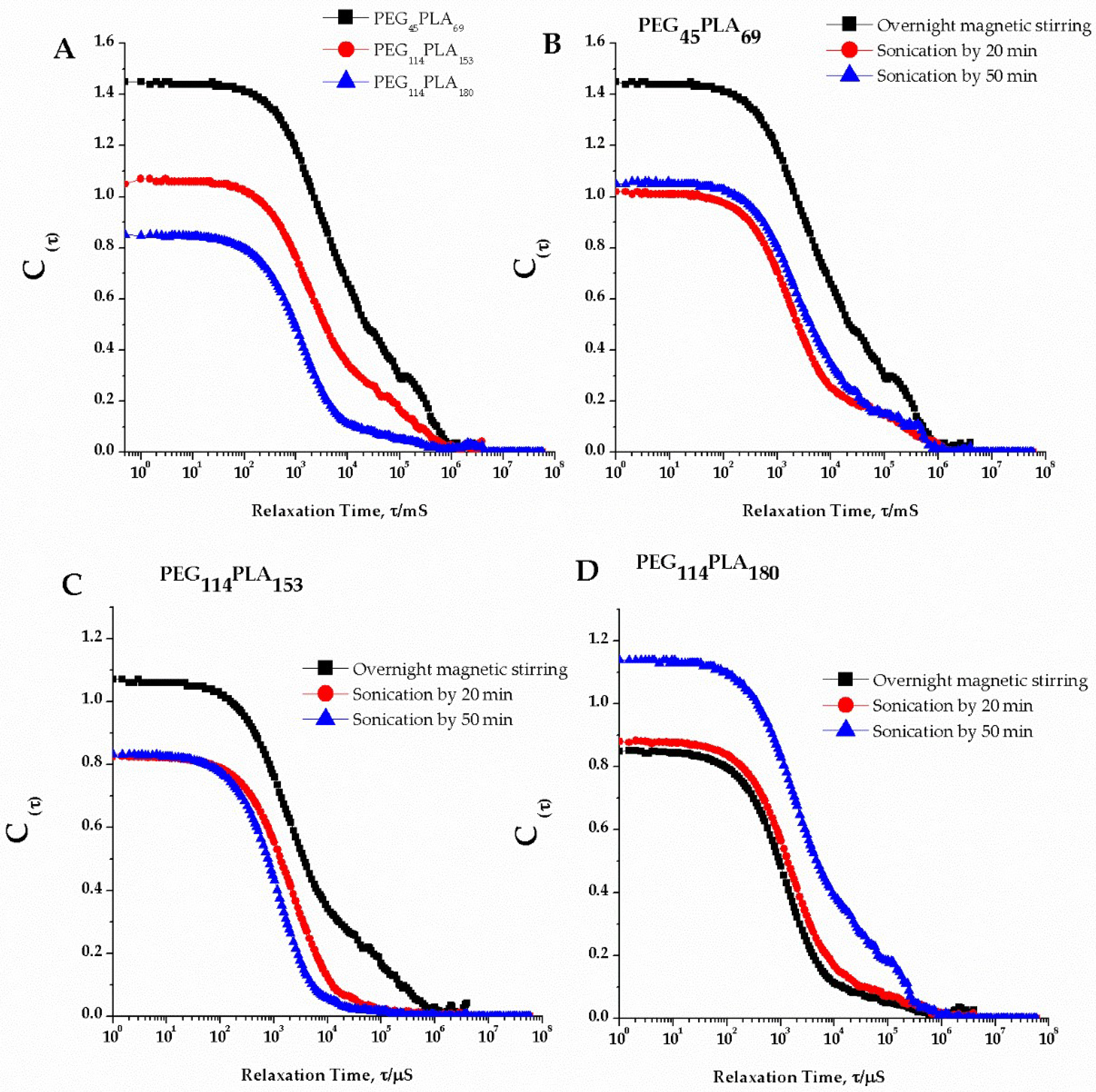
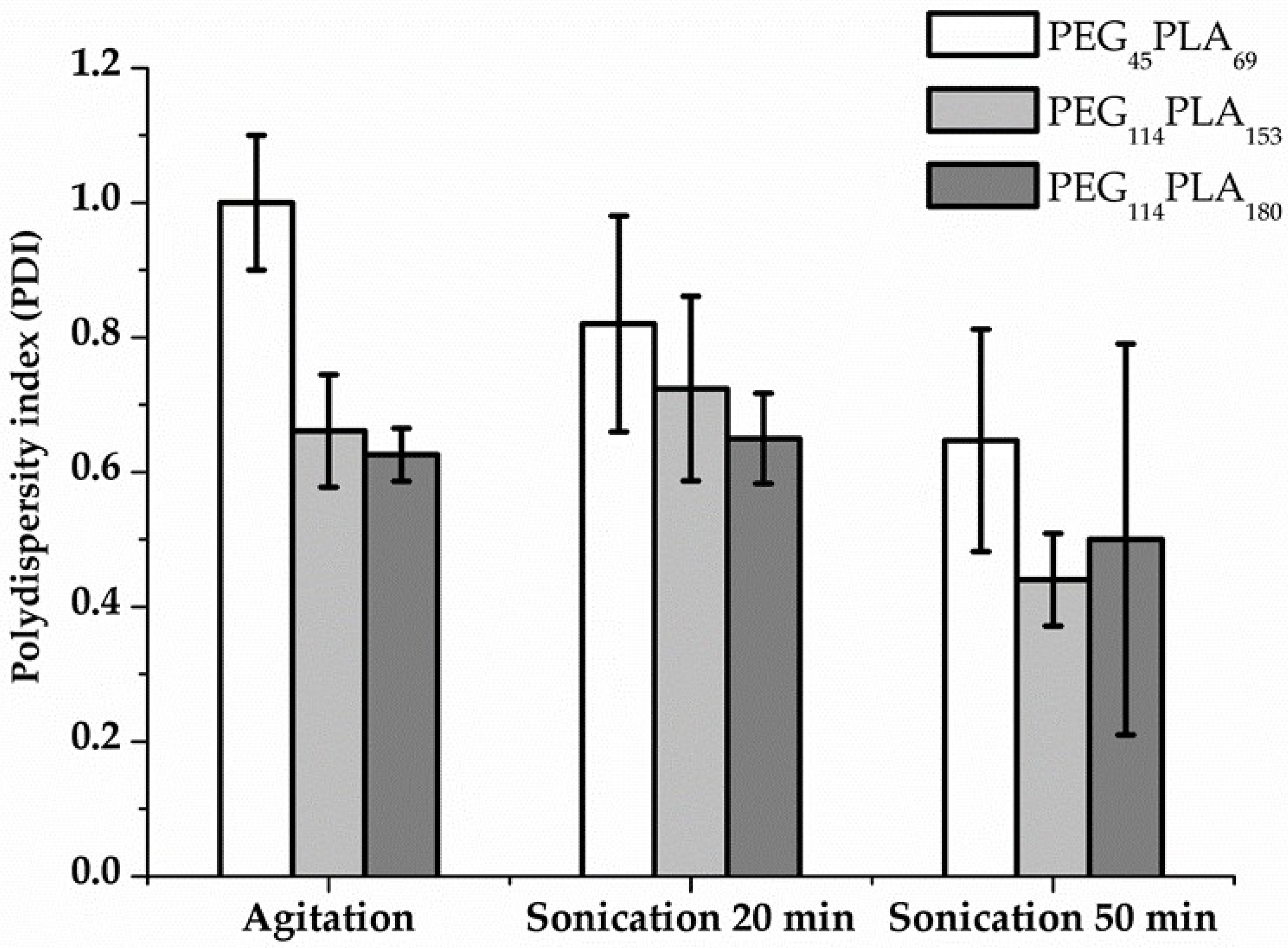
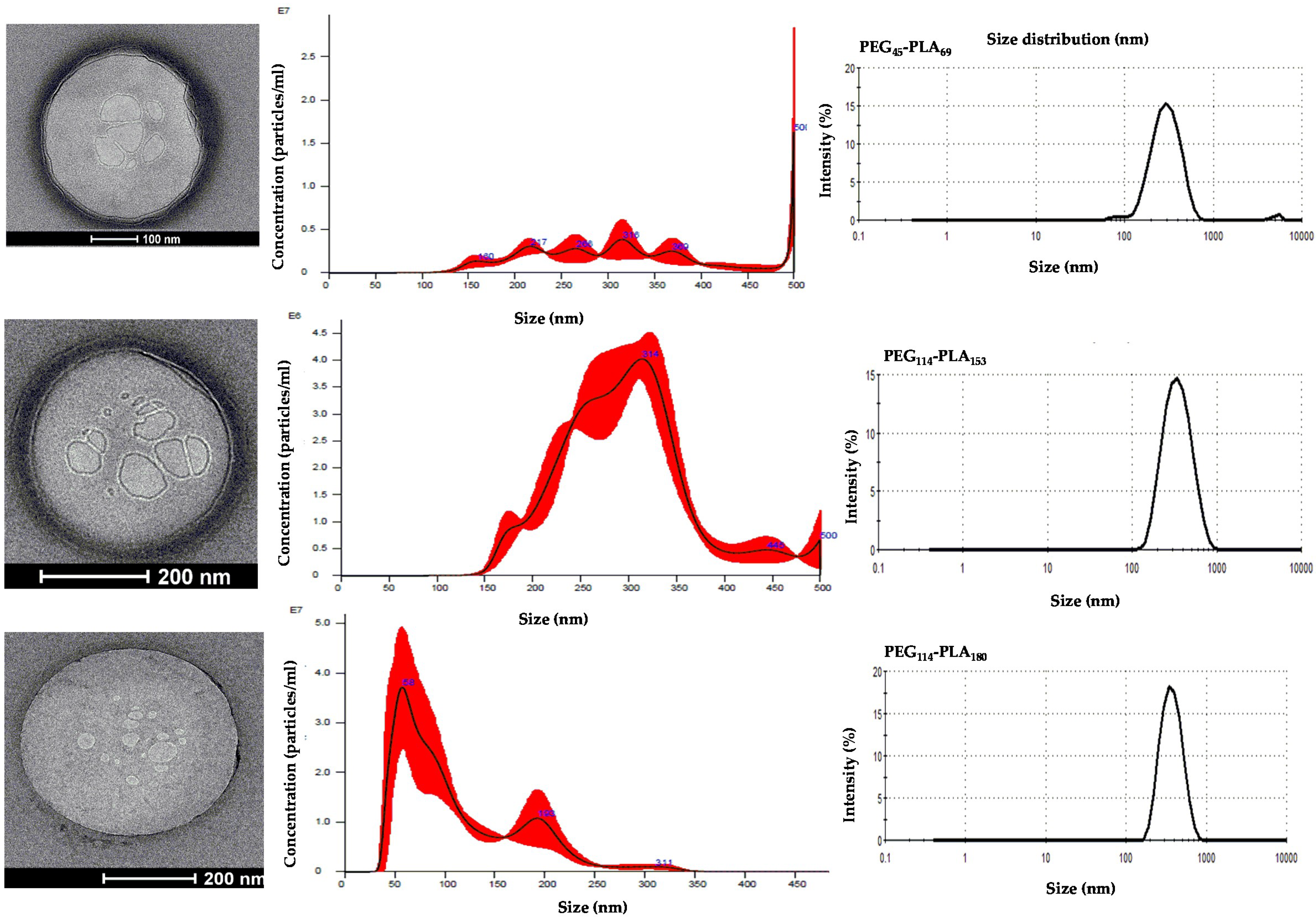
3.2. Self-Assembly by Film Hydration under Magnetic Stirring and Sonication
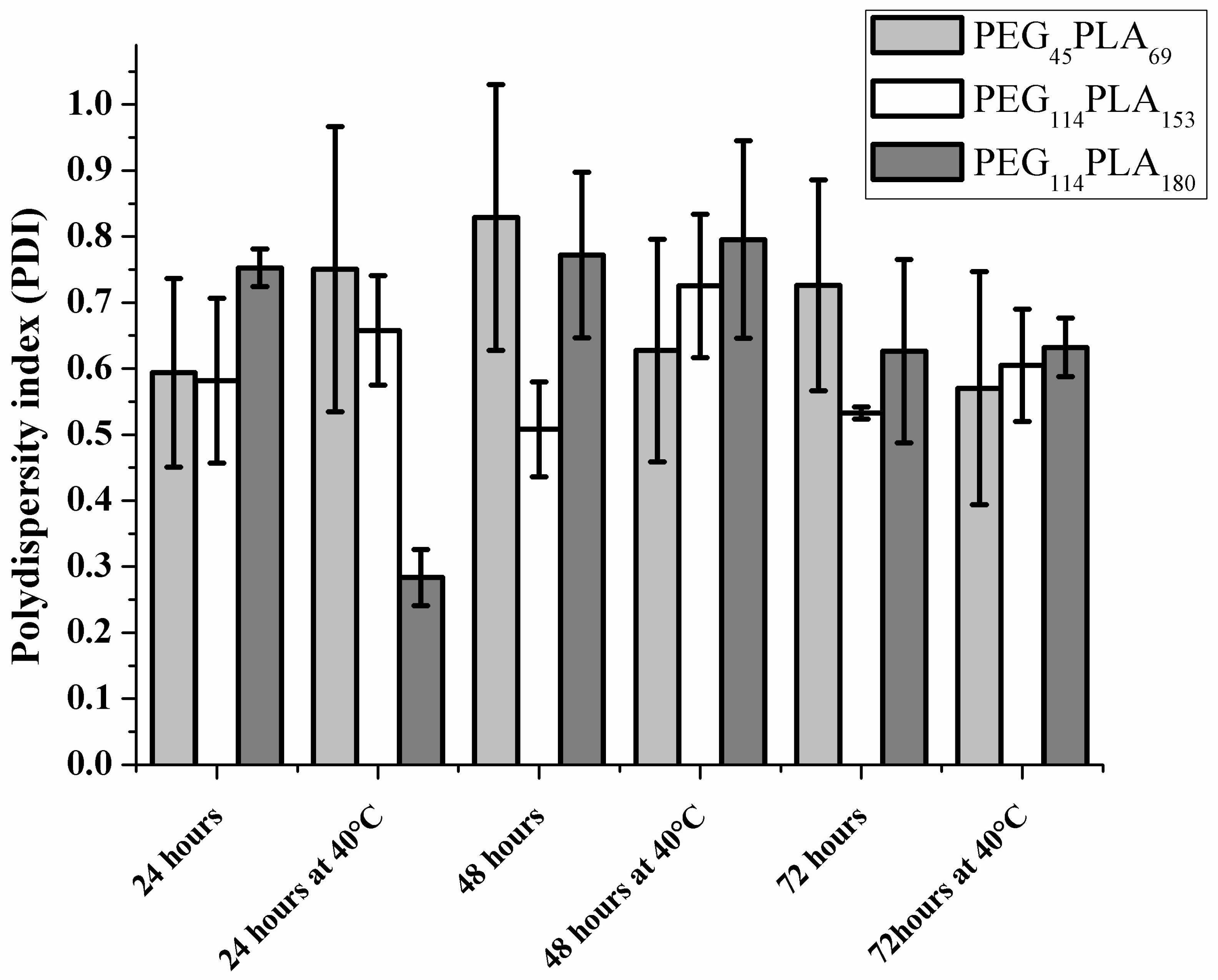
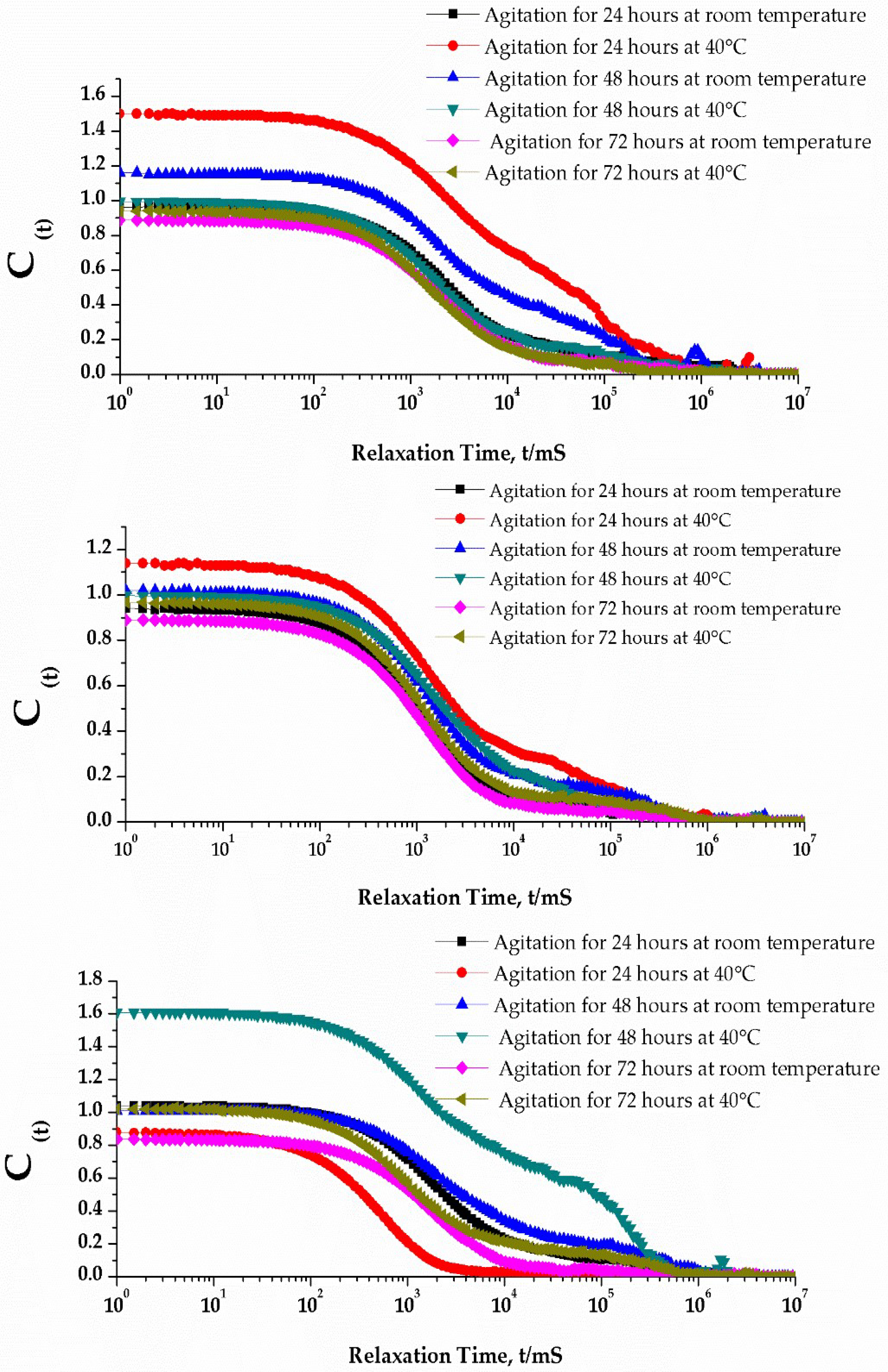
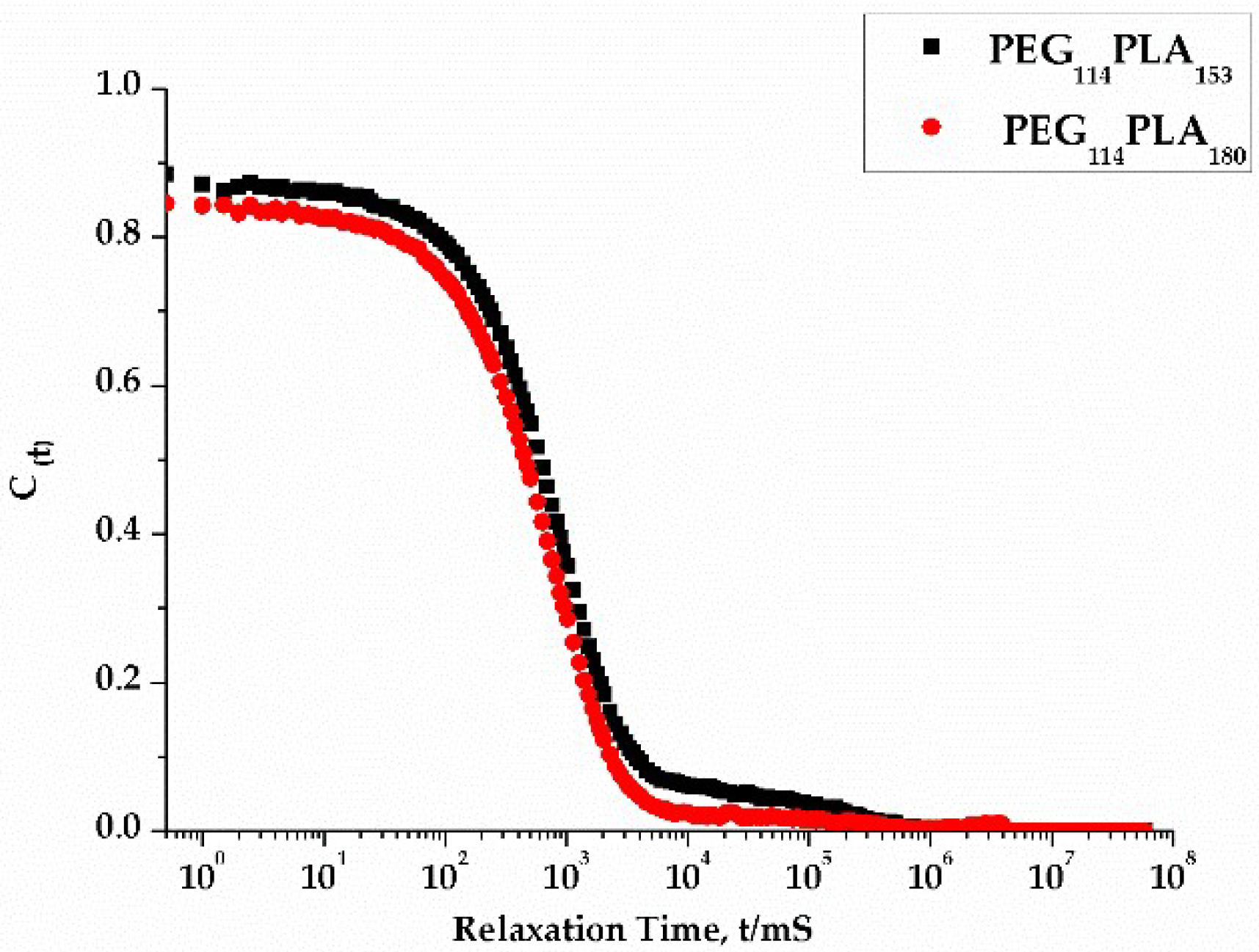
3.3. Effect of Centrifugation and Extrusion Post-Film Hydration
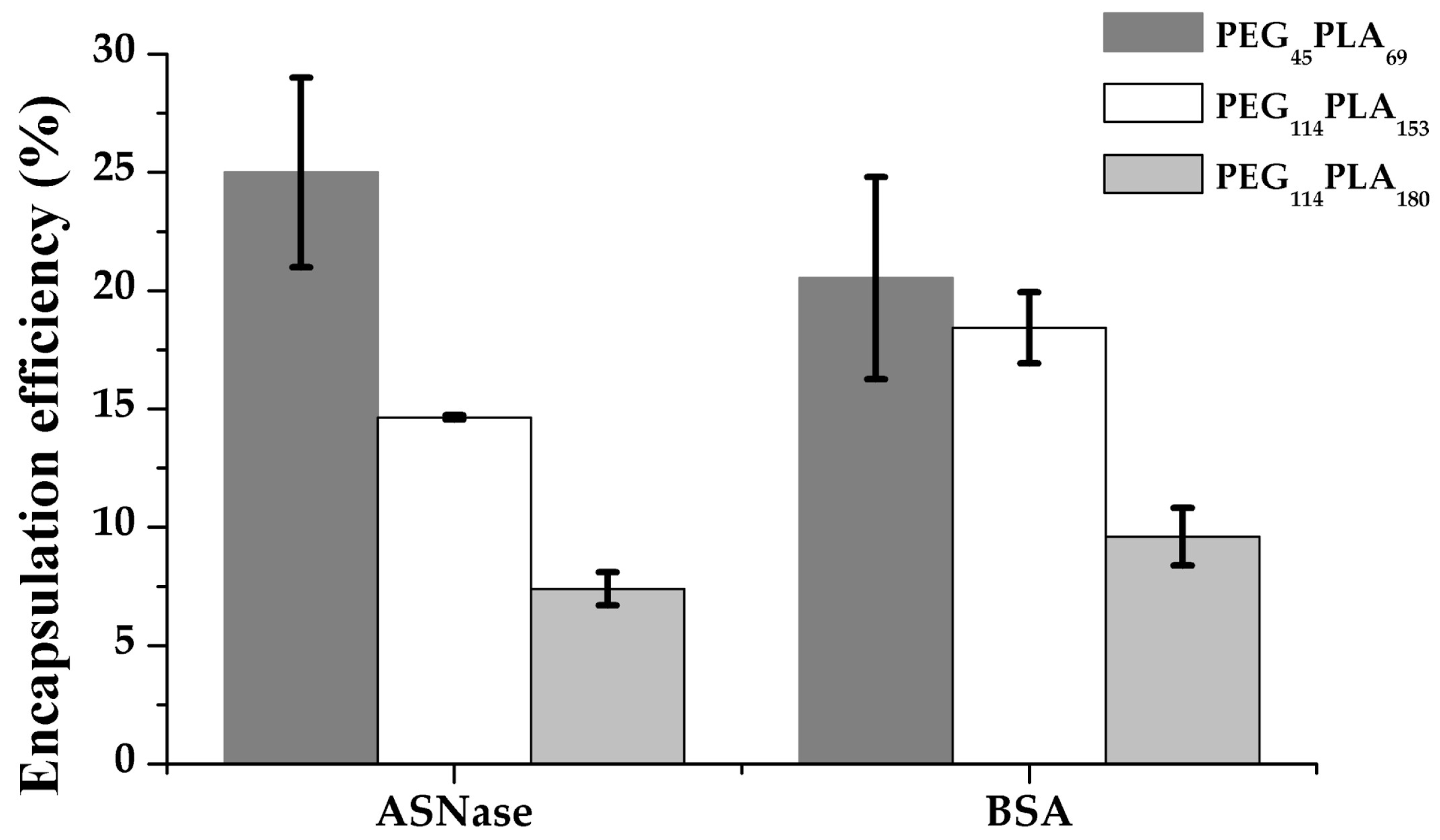
3.4. Protein Encapsulation into PEG-PLA Polymersomes
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Messager, L.; Gaitzsch, J.; Chierico, L.; Battaglia, G. Novel aspects of encapsulation and delivery using polymersomes. Curr. Opin. Pharmacol. 2014, 18, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Pachioni-Vasconcelos, J.D.A.; Lopes, A.M.; Apolinário, A.C.; Valenzuela-Oses, J.K.; Costa, J.S.R.; Nascimento, L.D.O.; Pessoa, A.; Barbosa, L.R.S.; Rangel-Yagui, C.D.O. Nanostructures for protein drug delivery. Biomater. Sci. 2016, 4, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Loos, K. Editorial: Self-assembly. Polymer 2016, 107, 341–342. [Google Scholar] [CrossRef]
- Lopresti, C.; Massignani, M.; Fernyhough, C.; Blanazs, A.; Ryan, A.J.; Madsen, J.; Warren, N.J.; Armes, S.P.; Lewis, A.L.; Chirasatitsin, S.; et al. Controlling polymersome surface topology at the nanoscale by membrane confined polymer/polymer phase separation. ACS Nano 2011, 5, 1775–1784. [Google Scholar] [CrossRef] [PubMed]
- Lipowsky, R. The conformation of membranes. Nature 1991, 349, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Howse, J.R.; Jones, R.A.L.; Battaglia, G.; Ducker, R.E.; Leggett, G.J.; Ryan, A.J. Templated formation of giant polymer vesicles with controlled size distributions. Nat. Mater. 2009, 8, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Pegoraro, C.; Cecchin, D.; Madsen, J.; Warren, N.; Armes, S.P.; MacNeil, S.; Lewis, A.; Battaglia, G. Translocation of flexible polymersomes across pores at the nanoscale. Biomater. Sci. 2014, 2, 680–692. [Google Scholar] [CrossRef] [PubMed]
- Discher, D.E.; Ahmed, F. Polymersomes. Annu. Rev. Biomed. Eng. 2006, 8, 323–341. [Google Scholar] [CrossRef] [PubMed]
- Dionzou, M.; Morère, A.; Roux, C.; Lonetti, B.; Marty, J.-D.; Mingotaud, C.; Joseph, P.; Goudounèche, D.; Payré, B.; Léonetti, M.; et al. Comparison of methods for the fabrication and the characterization of polymer self-assemblies: What are the important parameters? Soft Matter 2016, 12, 2166–2176. [Google Scholar] [CrossRef] [PubMed]
- Fetsch, C.; Gaitzsch, J.; Messager, L.; Battaglia, G.; Luxenhofer, R. Self-Assembly of Amphiphilic Block Copolypeptoids—Micelles, Worms and Polymersomes. Sci. Rep. 2016, 6, 33491. [Google Scholar] [CrossRef] [PubMed]
- Bartenstein, J.E.; Robertson, J.; Battaglia, G.; Briscoe, W.H. Stability of polymersomes prepared by size exclusion chromatography and extrusion. Colloids Surf. A Physicochem. Eng. Asp. 2016, 506, 739–746. [Google Scholar] [CrossRef]
- Bleul, R.; Thiermann, R.; Maskos, M. Techniques to Control Polymersome Size. Macromolecules 2015, 48, 7396–7409. [Google Scholar] [CrossRef]
- Arifin, D.R.; Palmer, A.F. Polymersome encapsulated hemoglobin: A novel type of oxygen carrier. Biomacromolecules 2005, 6, 2172–2181. [Google Scholar] [CrossRef] [PubMed]
- Winzen, S.; Bernhardt, M.; Schaeffel, D.; Koch, A.; Kappl, M.; Koynov, K.; Landfester, K.; Kroeger, A. Submicron hybrid vesicles consisting of polymer–lipid and polymer–cholesterol blends. Soft Matter 2013, 9, 5883–5890. [Google Scholar] [CrossRef]
- Robertson, J.D.; Yealland, G.; Avila-Olias, M.; Chierico, L.; Bandmann, O.; Renshaw, S.A.; Battaglia, G. pH-sensitive tubular polymersomes: Formation and applications in cellular delivery. ACS Nano 2014, 8, 4650–4661. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, S. DLS and zeta potential—What they are and what they are not? J. Control. Release 2016, 235, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Maulucci, G.; De Spirito, M.; Arcovito, G.; Boffi, F.; Castellano, A.C.; Briganti, G. Particle size distribution in DMPC vesicles solutions undergoing different sonication times. Biophys. J. 2005, 88, 3545–3550. [Google Scholar] [CrossRef] [PubMed]
- Smart, T.P.; Fernyhough, C.; Ryan, A.J.; Battaglia, G. Controlling Fusion and Aggregation in Polymersome Dispersions. Macromol. Rapid Commun. 2008, 29, 1855–1860. [Google Scholar] [CrossRef]
- Battaglia, G.; Ryan, A.J. Pathways of polymeric vesicle formation. J. Phys. Chem. B 2006, 110, 10272–10279. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, G.; Ryan, A.J. The evolution of vesicles from bulk lamellar gels. Nat. Mater. 2005, 4, 869–876. [Google Scholar] [CrossRef] [PubMed]
- Scarpa, E.; Bailey, J.L.; Janeczek, A.A.; Stumpf, P.S.; Johnston, A.H.; Oreffo, R.O.C.; Woo, Y.L.; Cheong, Y.C.; Evans, N.D.; Newman, T.A. Quantification of intracellular payload release from polymersome nanoparticles. Sci. Rep. 2016, 6, 29460. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chierico, L.; Little, D.; Patikarnmonthon, N.; Yang, Z.; Azzouz, M.; Madsen, J.; Armes, S.P.; Battaglia, G. Encapsulation of biomacromolecules within polymersomes by electroporation. Angew. Chem. Int. Ed. 2012, 51, 11122–11125. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Copolymer | PEG-PLA Mn a | Glass Transition Temperature of Hydrophobic Block b | fPEG | PDI c |
|---|---|---|---|---|
| PEG45PLA69 | PEG 2000: PLA 5000 | 23 °C | 0.28 | 1.20 |
| PEG114PLA153 | PEG 5000: PLA 11,000 | 39 °C | 0.30 | 1.15 |
| PEG114PLA180 | PEG 5000: PLA 13,000 | 40 °C | 0.27 | 1.16 |
| Systems | Hydrodynamic diameter by intensity (nm) | PDI |
|---|---|---|
| P1 2.0 | 836.7 (65.1%) and 213.5 (34.9%) | 0.962 |
| P1 2.20 | 300 (71.9%) and 1878 (28.1%) | 1 |
| P1 2.50 | 467.6 (94.8%) and 66.28 (5.2%) | 1 |
| P1 ON.0 | 217 (61%) and 664 (39%) | 0.749 |
| P1 ON.20 | 378.7 (69.7%) and 173.6 (30.3%) | 0.673 |
| P1 ON.50 | 443.9 (80.3%) and 146.6 (19.7%) | 0.807 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Apolinário, A.C.; Magoń, M.S.; Pessoa Jr., A.; Rangel-Yagui, C.D.O. Challenges for the Self-Assembly of Poly(Ethylene Glycol)–Poly(Lactic Acid) (PEG-PLA) into Polymersomes: Beyond the Theoretical Paradigms. Nanomaterials 2018, 8, 373. https://doi.org/10.3390/nano8060373
Apolinário AC, Magoń MS, Pessoa Jr. A, Rangel-Yagui CDO. Challenges for the Self-Assembly of Poly(Ethylene Glycol)–Poly(Lactic Acid) (PEG-PLA) into Polymersomes: Beyond the Theoretical Paradigms. Nanomaterials. 2018; 8(6):373. https://doi.org/10.3390/nano8060373
Chicago/Turabian StyleApolinário, Alexsandra Conceição, Monika S. Magoń, Adalberto Pessoa Jr., and Carlota De Oliveira Rangel-Yagui. 2018. "Challenges for the Self-Assembly of Poly(Ethylene Glycol)–Poly(Lactic Acid) (PEG-PLA) into Polymersomes: Beyond the Theoretical Paradigms" Nanomaterials 8, no. 6: 373. https://doi.org/10.3390/nano8060373
APA StyleApolinário, A. C., Magoń, M. S., Pessoa Jr., A., & Rangel-Yagui, C. D. O. (2018). Challenges for the Self-Assembly of Poly(Ethylene Glycol)–Poly(Lactic Acid) (PEG-PLA) into Polymersomes: Beyond the Theoretical Paradigms. Nanomaterials, 8(6), 373. https://doi.org/10.3390/nano8060373