1. Introduction
Photodynamic therapy (PDT) has emerged as an innovative therapeutic method for treating various oncological diseases, including gastrointestinal, esophagus, bladder and cervical cancer [
1,
2,
3]. This therapeutic modality is considered superior to traditional therapies such as surgery, radiotherapy and chemotherapy because it involves a minimally-invasive clinical procedure. In addition, traditional chemotherapy usually has serious side effects, and its clinical application is confined by multidrug resistance [
2]. However, PDT is expected to overcome multidrug resistance because the PDT cytotoxicity mechanism for cancer cells differs from that of traditional chemotherapy.
PDT is based on the accumulation of a photosensitizer in specific target cells, followed by selective optical irradiation of an appropriate wavelength to generate a highly reactive oxygen species (ROS), such as singlet oxygen. Increasing the ROS above a critical concentration threshold allows it to react directly with the biological substrate, oxidizing vital cellular components and inducing an acute cell-stress response, culminating in cellular death, mainly by apoptosis and/or necrosis [
4]. Therefore, the efficacy of PDT depends primarily on the intracellular accumulation of the photosensitizer in the target cells, in which the intracellular accumulation of the photosensitizer is affected by its chemical properties.
Unfortunately, such photosensitizers are limited in clinical use because of prolonged photosensitivity, nonspecific phototoxicity for normal cells due to insufficient selective accumulation around the specific target cells and hydrophobicity [
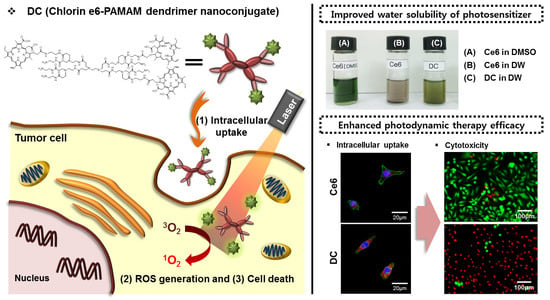
5]. In particular, hydrophobic photosensitizers can easily precipitate out or adhere to normal cell surfaces in the biological environment, losing their PDT efficacy and causing severe side effects. To address this situation, various photosensitizer-delivery systems have been investigated, such as polymeric micelles and conjugates, which have provided highly soluble photosensitizers in aqueous solutions and have improved tumor specificity [
6,
7]. One such system, photosensitizer-loaded cationic polymer complexes, combines with negatively-charged cell membranes, resulting in accelerated cellular internalization of photosensitizers [
5,
8].
Chlorins are a class of plant-derived tetrapyrroles and are prospective photosensitizers because of their high phototoxic potential and remarkably strong absorption in the red [
9]. These characteristics of chlorins allow for deeper tissue photodamage, which is particularly essential for treating cancer. However, in general, chlorins exhibit high hydrophobicity and low solubility in aqueous solutions, causing them to accumulate in the skin. In addition, they have prolonged light sensitivity, on the order of weeks, so that they continue to be active long after PDT treatment. Poly(amidoamine) (PAMAM) dendrimers are nanoscale monodisperse macromolecules with a hyperbranched three-dimensional architecture and a large number of terminal function groups [
5,
10]. PAMAM dendrimers have been used in various drug-delivery systems because they not only spatially distribute the drug, but also maintain superior dispersion stability even under physiological conditions [
10].
Cervical cancer is the third most common cancer among women worldwide, and the causal factor of cervical cancer is infection by the human papilloma virus [
11]. This persistent viral infection leads to intraepithelial transformations of an insidious and progressive nature that culminates in a cancer if left untreated. PDT may be an alternative to the traditional invasive treatment of cervical cancer [
12,
13,
14]. Therefore, the development of hydrophilic chlorin conjugates may lead to a successful treatment for cervical cancer. Motivated by this goal, the main purpose of the present study is to enhance PDT efficacy of chlorin e6 (Ce6) by enhancing its water solubility and cellular internalization. Toward this end, we prepared a hydrophilic Ce6-PAMAM dendrimer nanoconjugate (DC) (
Figure 1). The morphology, chemical structure and hydrophilicity of the prepared DC were systematically examined, and the cellular internalization and phototoxicity of DC were evaluated on the human cervical cancer cell line (HeLa) by using fluorescence microscopy and MTT assays.
2. Materials and Methods
2.1. Materials
Ce6 was purchased from Frontier Scientific (Logan, UT, USA). Poly(amidoamine) (PAMAM) dendrimer (ethylenediamine core, Generation 1.0), N,N’-dicyclohexylcarbodiimide (DCC), 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT), 3-amino-7-dimethylamino-2-methylphenazine hydrochloride (neutral red), bisbenzimide H 33258 (Hoechst 33258, H-33258), 9,10-dimethylnathracene (DMA), N,N-dimethylformamide (DMF) and dimethyl sulfoxide (DMSO) were obtained from Sigma-Aldrich (St. Louis, MO, USA) and used without further purification. The human cervical cancer cell line (HeLa) was obtained from the Korean Cell Line Bank (KCLB, Seoul, Korea). Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS), penicillin-streptomycin and Dulbecco’s phosphate-buffered saline (DPBS, pH 7.4) were obtained from Gibco BRL (Waltham, MA, USA). SlowFade Gold antifade mountant, LIVE/DEAD Viability/Cytotoxicity Assay Kit and the Image-iT LIVE Green Reactive Oxygen Species Detection Kit were purchased from Molecular Probes (Eugene, OR, USA). The Actin Cytoskeleton and Focal Adhesion Staining Kit was purchased from Merck Millipore (Burlington, MA, USA). Other reagents and solvents were commercially available and used as received.
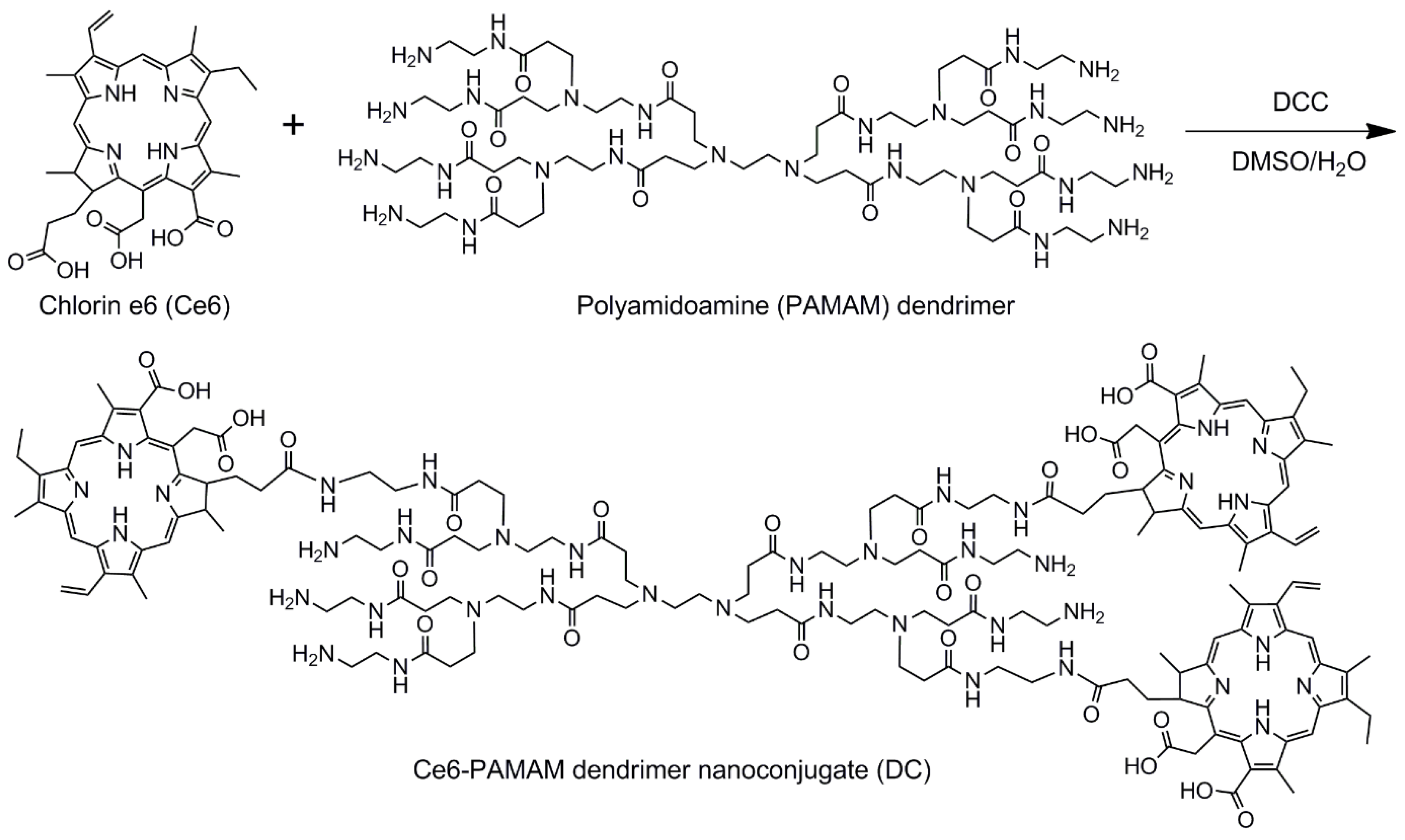
2.2. Synthesis of Ce6-PAMAM Dendrimer Nanoconjugate
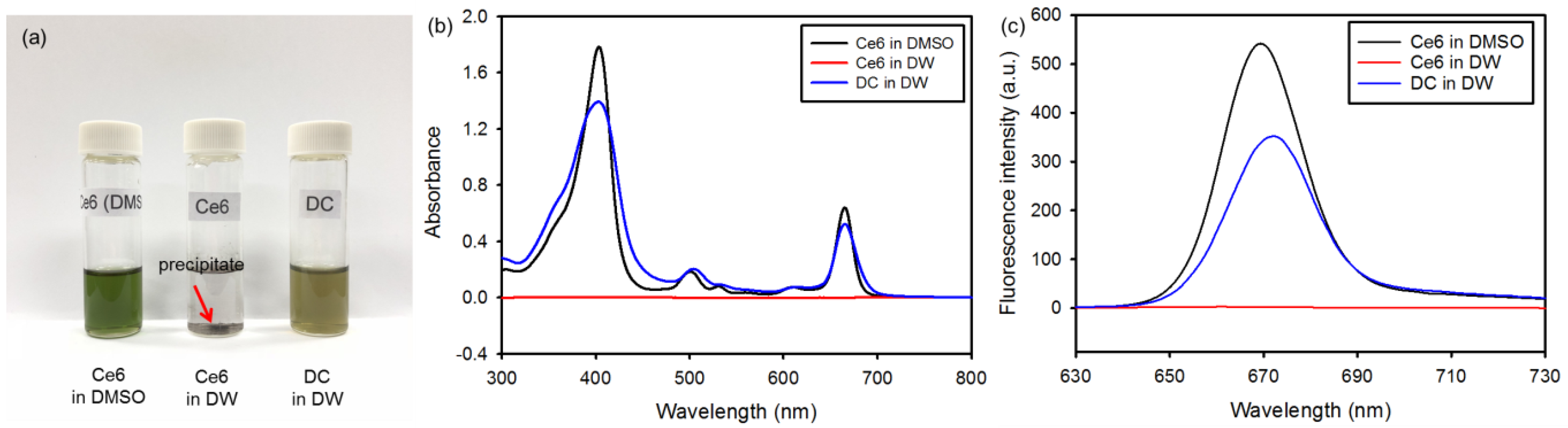
The hydrophilic Ce6-PAMAM dendrimer nanoconjugate (DC) was synthesized as follows. First, 98.5 mg Ce6 were dissolved in 20 mL DMSO, followed by the addition of 51.1 mg DCC to activate the carboxylic groups. After adding to the mixture 357.5 mg PAMAM dendrimer in 30 mL DMSO/distilled water (DW) at a ratio of 7:3, the reaction was allowed to progress at room temperature for 24 h to produce DC. The products were filtered and isolated by using a tubular membrane dialysis in a mixture of DMSO and DW for 48 h to remove unreacted agents, followed by lipophilization in vacuo. To determine the amount of Ce6 conjugated to DC, the absorbance was measured at 404 nm by using an ultraviolet-visible (UV-visible) spectrometer according to the previous studies [
7,
15]. Free Ce6 dissolved in DMSO was prepared at different concentrations (1, 2, 4, 8, 12 and 20 μg/mL) to generate a standard curve, and the amount of Ce6 conjugated to PAMAM dendrimer was measured as the absorbance at 404 nm by using a UV-visible spectrometer.
1H NMR (400 MHz, DMSO-d6), δ (ppm) = 9.47, 9.69, 9.11 (m, H-meso), 8.33 (d, CH=CH2), 6.39, 6.16 (d, CH=CH2), 5.59 (d, chlorin-CH2CO), 4.56, 4.47 (s, CCH), 3.83 (m, CH2CH3), 3.53, 3.33 (s, chlorin-CH3), 2.58 (m, CH2CH2CO), 2.32 (m, chlorin-CH2CH2CO), 1.60 (t, chlorin-CH2CH3) for Ce6, 3.03 (m, NHCH2CH2N) 2.84 (m, NCH2CH2CO), 2.63 (m, NHCH2CH2N), 2.18 (NCH2CH2CO) for the PAMAM dendrimer.
2.3. Characterization of Ce6-PAMAM Dendrimer Nanoconjugate
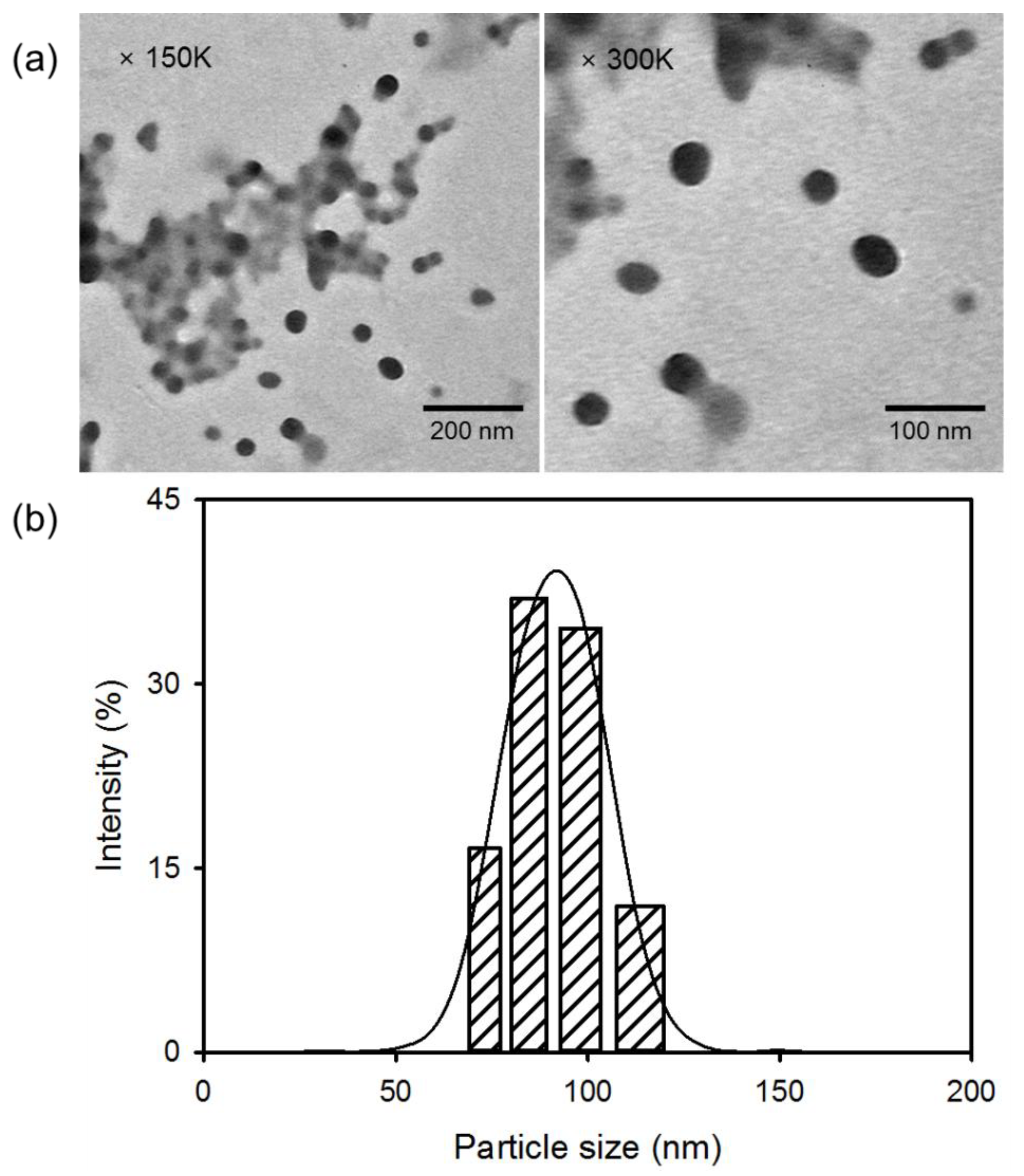
The structure of DC and its degree of Ce6 conjugation were determined by 1H nuclear magnetic resonance (NMR) spectroscopy (ADVANCE III 400, Bruker, Billerica, MA, USA), and its morphology was determined by transmission electron microscopy (TEM, H-7600, Hitachi, Tokyo, Japan). The average diameter of nanoconjugates was determined by analyzing the TEM images with Image-Pro Plus (Media Cybernetics Inc., Rockville, MD, USA). The particle size distribution was also measured by the dynamic light scattering (DLS) technique using a Zetasizer Nano ZS (Malvern Instruments, Malvern, UK). UV-visible spectra were recorded on a Hitachi U-2900 spectrometer (Tokyo, Japan), and the fluorescence emission spectra were measured with a PerkinElmer LS55 spectrofluorophotometer (Waltham, MA, USA) at 25 °C.
2.4. Singlet Oxygen Detection
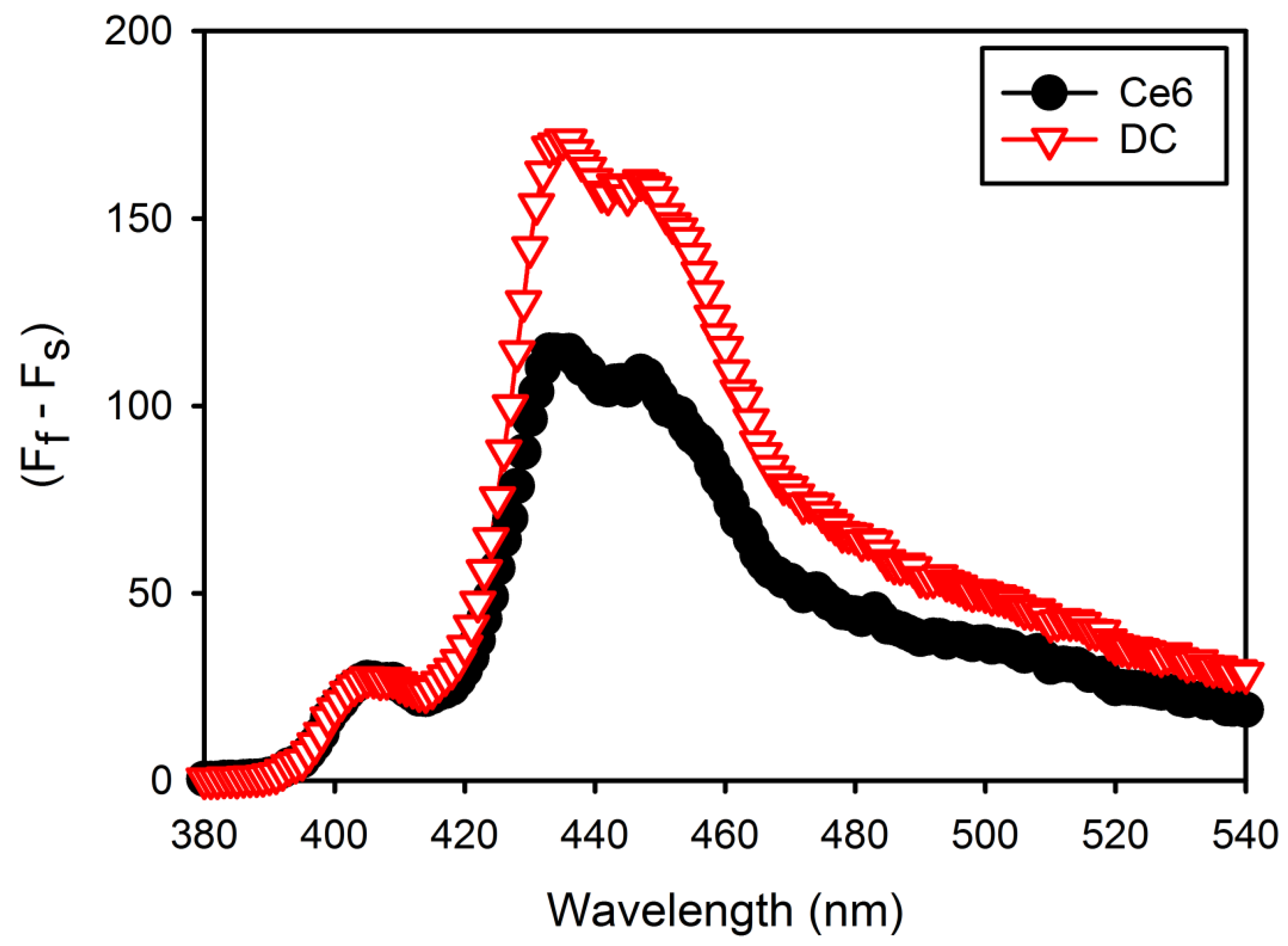
The singlet oxygen (
1O
2) generation was measured by an indirect method using a chemical probe [
16]. In this study,
1O
2 generation from free Ce6 or DC was detected using DMA as the
1O
2 probe. Free Ce6 or DC (1 mg/mL Ce6) was first dissolved in DMSO to prepare stock solution. This stock solution was dispersed in DPBS (pH 7.4) to obtain a concentration of 4 μg/mL Ce6, and then, DMA stock solution (20 mM in DMF) was added to give a final concentration of 20 μM DMA. Samples containing drug and DMA were irradiated at a light intensity with a 2.5-J/cm
2, 671-nm laser beam (LVI Technologies, Anyang, Korea). The fluorescence spectra of DMA (excitation, 360 nm; emission, 380–540 nm) as a result of the photosensitization reaction were monitored with a Perkin-Elmer LS55 spectrofluorophotometer (Waltham, MA, USA). The change in DMA fluorescence intensity (F
f − F
s) was plotted after subtracting each sample fluorescence intensity (F
s) from the full DMA fluorescence intensity (without free Ce6 or DC, indicating no singlet oxygen, F
f).
2.5. Cell Culture and Incubation Conditions
All experiments used the human cervical cancer cell line (HeLa). HeLa cells were cultured in DMEM containing 10% FBS and 0.5% penicillin-streptomycin and were incubated at 37 °C in a humidified 5% CO2 atmosphere. When the cells reached 80% confluence, they were harvested by using 0.25% trypsin-ethylenediaminetetraacetic acid (EDTA) and seeded in a new tissue culture plate to produce a subculture. DC and free Ce6 were dispersed in a serum-free medium. Untreated cells or cells that were maintained in the dark were used as reference cells.
2.6. Intracellular Uptake and Distribution Tests
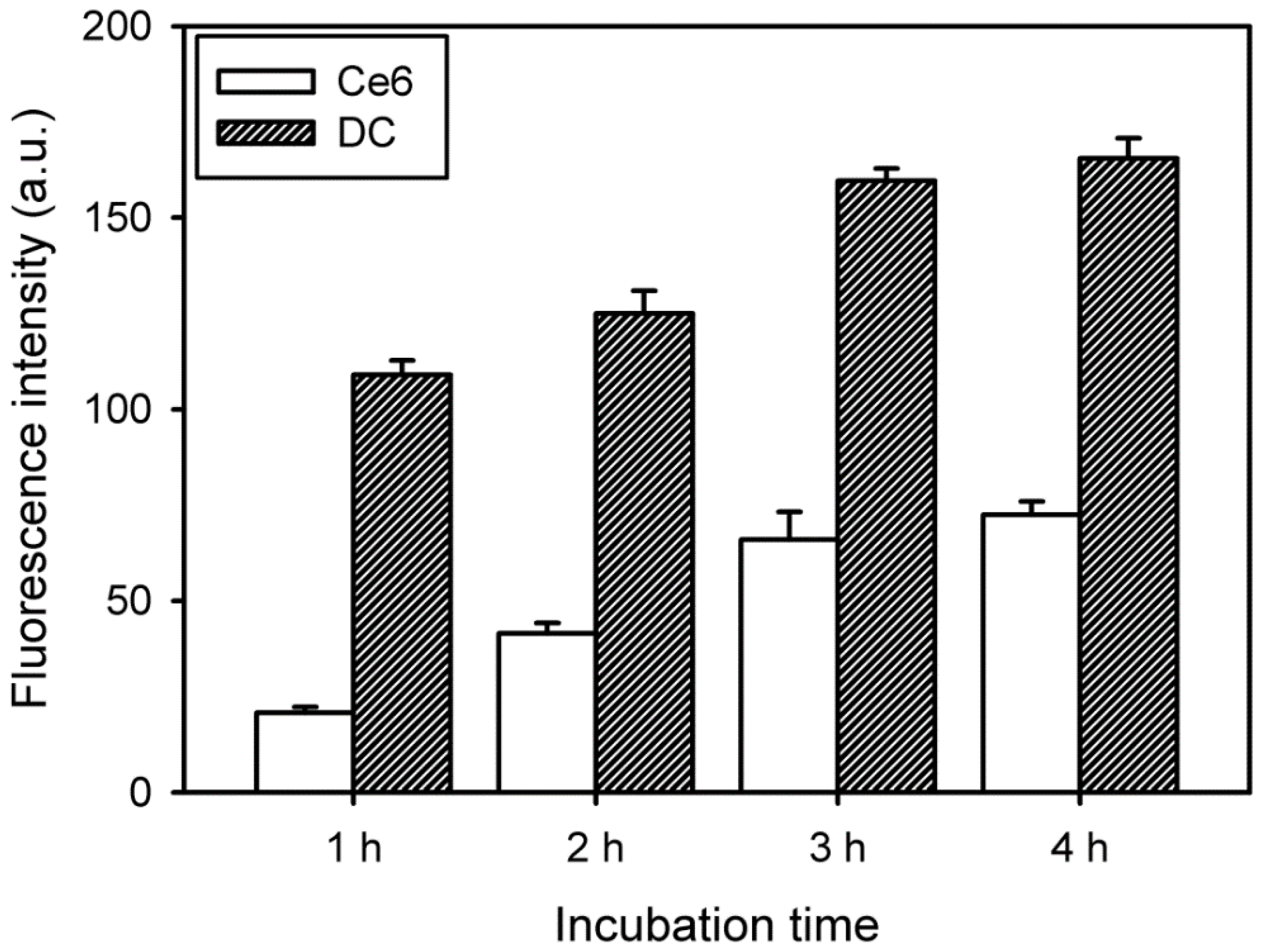
A multimode microplate reader (VICTOR3, PerkinElmer, Waltham, MA, USA) equipped with n excitation filter of 405 nm and an emission filter of 665 nm was used to quantify the intracellular uptake of free Ce6 and DC. HeLa cells (1 × 104 cells/well) were seeded into 96-well plates in 1 mL of culture medium containing 10% FBS and were incubated for 24 h. These cells were treated with free Ce6 or DC (4 μg/mL Ce6) for 1–4 h, following which the cells were washed twice with DPBS. After optically exciting the cell samples, the fluorescence emission of Ce6 or DC was measured by using the multimode microplate reader.
The intracellular uptake of drugs in cancer cells was determined by using confocal laser scanning microscopy. HeLa cells (1 × 104 cells/well) were seeded into an 8-well chamber slide for 24 h before being treated with the drug. The cells were incubated with free Ce6 or DC (4 μg/mL Ce6) for 2 h and rinsed twice with DPBS. Next, the cells were fixed in 4% paraformaldehyde solution for 10 min and permeabilized by applying 1% triton X-100 for 3 min. The cells were treated with 10 μg/mL H-33258 to stain the cell nuclei and, to label actin, with 3 μg/mL phalloidin-tetramethylrhodamine (TRITC) for 30 min at room temperature in the dark. The intracellular uptake and distribution of drugs were determined by using an inverted LSM 700 confocal laser scanning microscope (Carl Zeiss, Oberkochen, Germany).
2.7. Cell Phototoxicity Assay
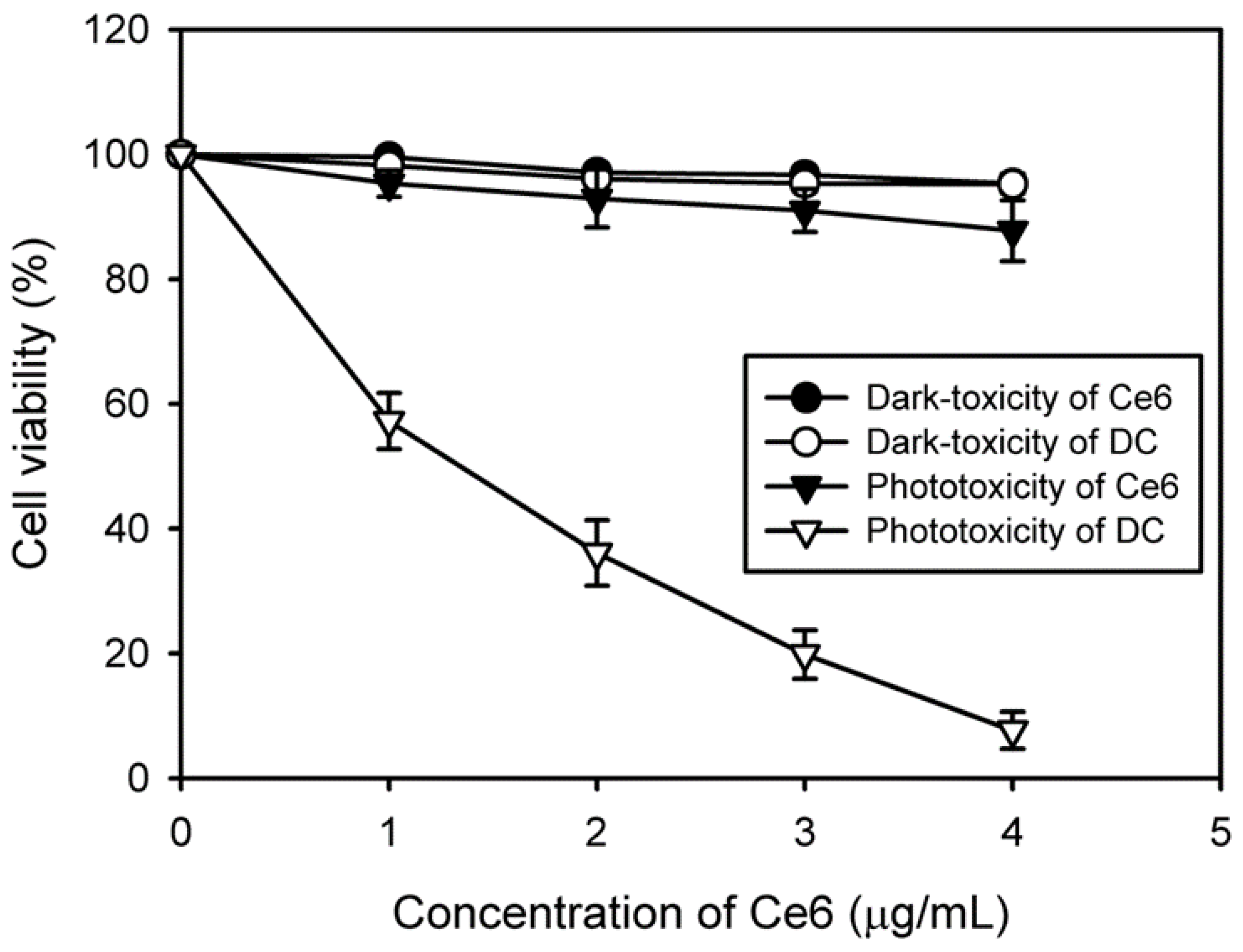
To determine cell viability in the dark, HeLa cells (1 × 104 cells/well) were seeded into 96-well plates and incubated for 24 h at 37 °C. After cell stabilization, the culture medium was replaced with 200 μL of culture medium containing free Ce6 or DC (0–4 μg/mL Ce6), followed by incubation for 2 h. The cells were then washed twice with a serum-free medium, and cell viability was evaluated via an MTT assay after 24 h.
To determine in vitro phototoxicity after laser irradiation, HeLa cells (1 × 104 cells/well) were seeded into 96-well plates and incubated for 24 h at 37 °C. These cells were then treated with free Ce6 or DC (0–4 μg/mL Ce6). After incubation for 2 h, the cells were washed twice with a serum-free medium and irradiated with a 2.5-J/cm2, 671-nm laser beam. After incubation for 24 h, the viability of irradiated cells was evaluated via an MTT assay.
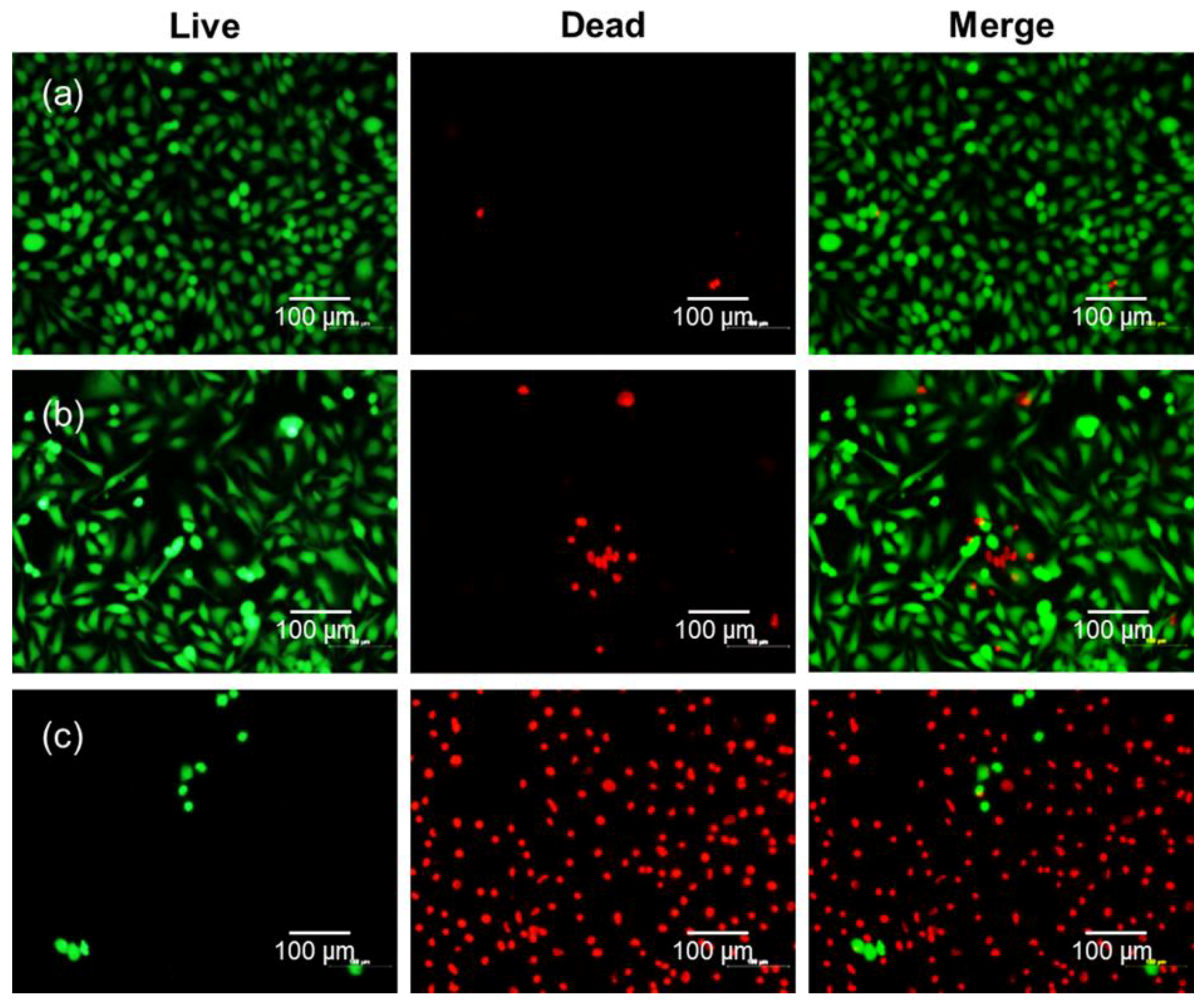
A qualitative cell viability assay was done by using the LIVE/DEAD Viability/Cytotoxicity Assay Kit. The kit contains calcein AM and ethidium homodimer-1 (EthD-1). Calcein AM stains live cells green, whereas EthD-1 stains dead cells red [
17]. HeLa cells (5 × 10
4 cells/well) were seeded into an8-well chamber slide and incubated for 24 h at 37 °C. Next, these cells were treated with free Ce6 or DC (4 μg/mL of Ce6). After incubation for 2 h, the cellular layers on the sample surface were rinsed twice with DPBS and irradiated with a 2.5-J/cm
2, 671-nm diode laser. Afterwards, the cells were treated for 20 min at 37 °C with 1 μM of calcein AM and 2 μM of EthD-1 to determine cell viability after 24 h of incubation. Finally, the cells were observed by using an inverted fluorescence microscope (Eclipse TS100, FITC-G2A filters, Nikon, Tokyo, Japan) equipped with a cooled charge-coupled device (CCD) camera (DS-U2, Nikon, Japan) and with NIS-Elements Imaging Software.
2.8. Generation of Reactive Oxygen Species
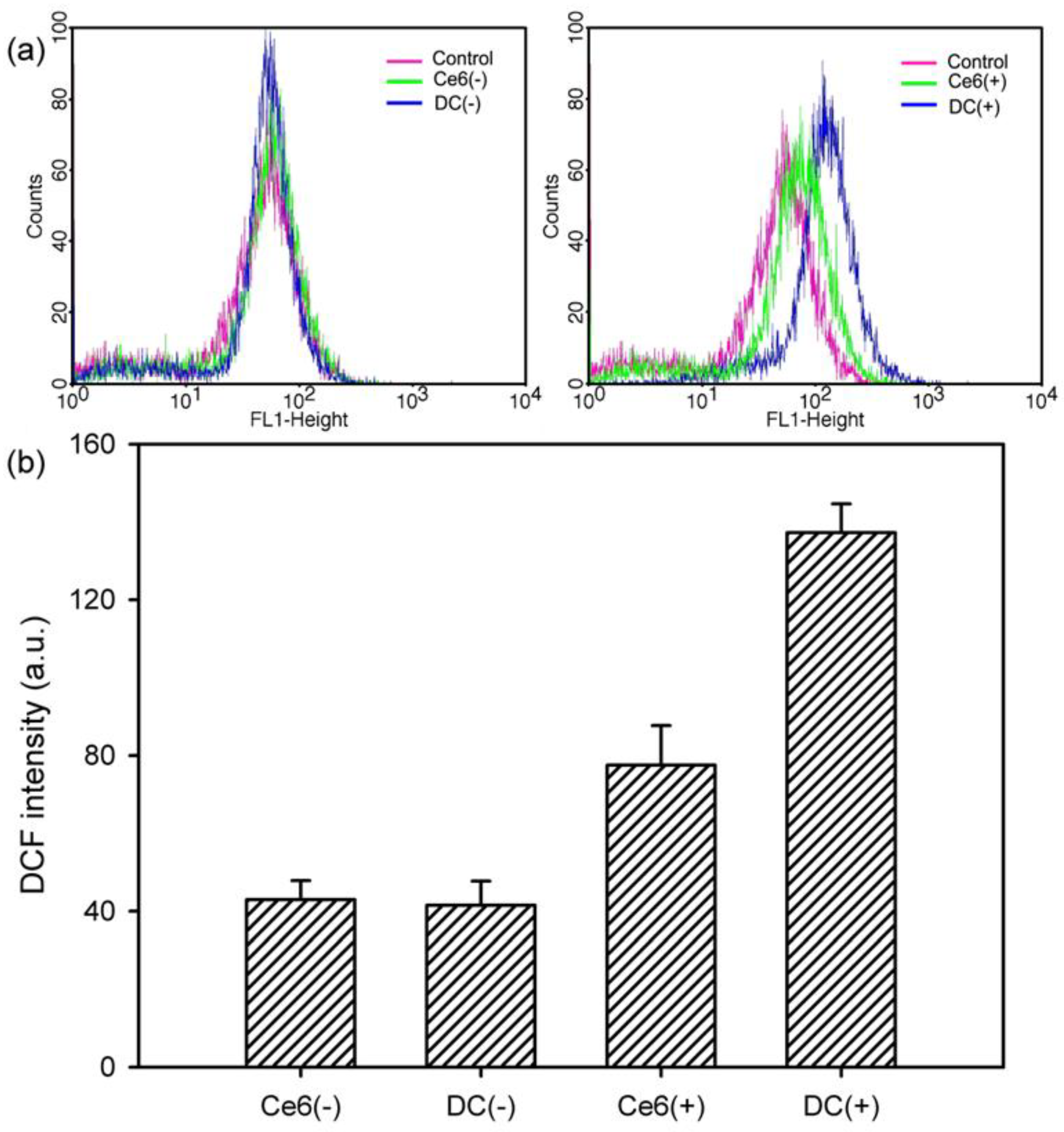
The generation of the ROS was monitored by using the Image-iT LIVE Green Reactive Oxygen Species Detection Kit, which contains 5-(and-6)-carboxy-2′,7′-dichlorodihydrofluorescein diacetate (carboxy-H2DCFDA). H2DCFDA is a nonfluorescent penetrant that permeates live cells and is hydrolyzed by intracellular esterases to form H2DCF. In the presence of the ROS, H2DCF oxidizes in the cytoplasm and is converted to DCF, which can emit green fluorescence [
2]. HeLa cells (5 × 10
4 cells/well) were seeded into 24-well plates and incubated for 24 h at 37 °C, following which, they were treated with Ce6 or DC (4 μg/mL Ce6). After incubation for 2 h, the cells were rinsed twice with Hank’s balanced salt solution and irradiated with a 2.5-J/cm
2, 671-nm diode laser. Next, the cells were treated with 5 μM carboxy-H2DCFDA for 30 min at 37 °C, and the concentration of intracellular ROS trapped by DCF was determined quantitatively by using a flow cytometer (FACSCalibur™, BD Biosciences, Franklin Lakes, NJ, USA). Data were analyzed by using Cellquest Software (BD Biosciences, Franklin Lakes, NJ, USA).
2.9. Apoptotic Analysis
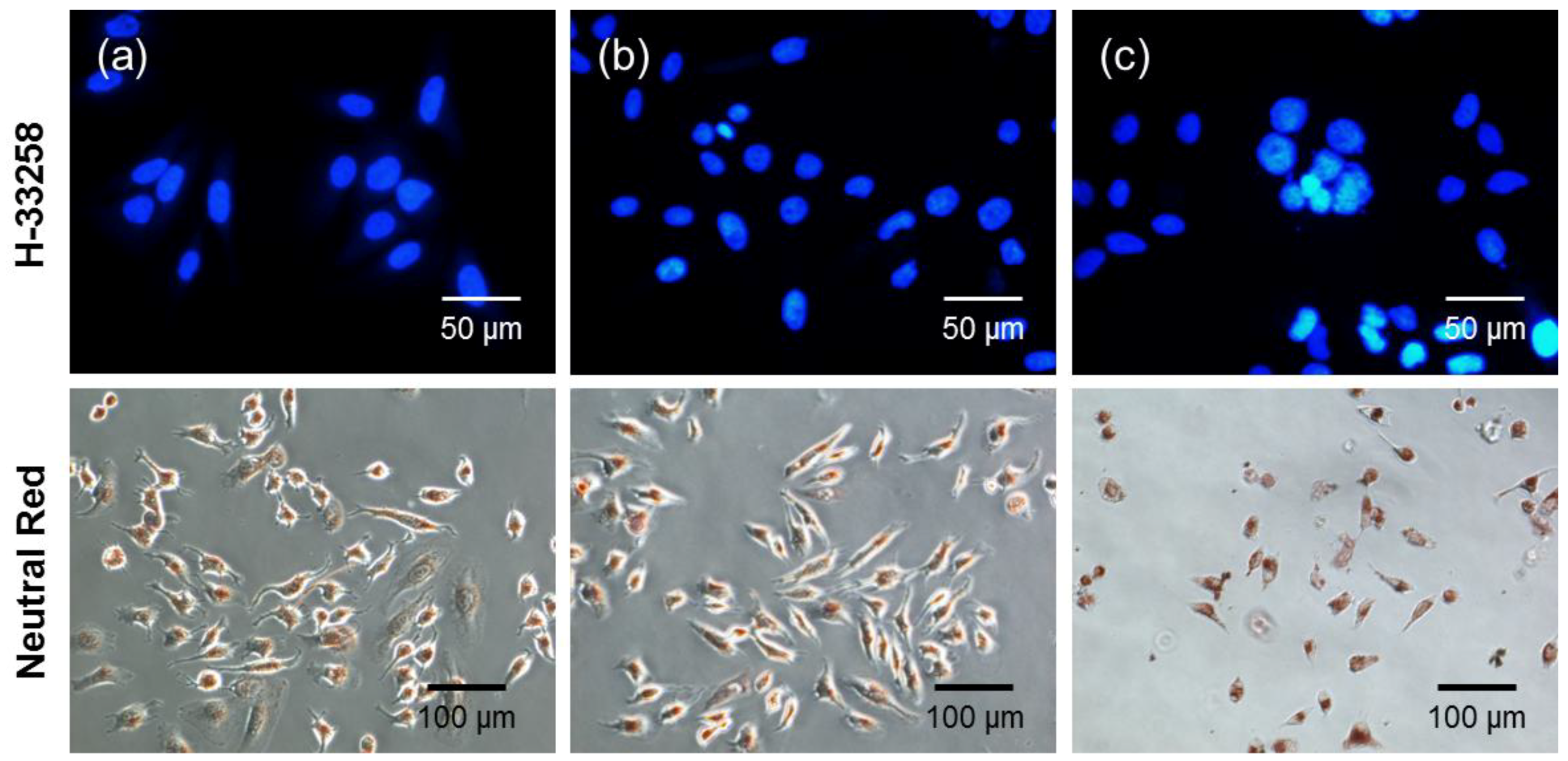
Morphological changes after photodynamic treatment were assessed by visualizing the control and drug-treated cells by using fluorescence microscopy. HeLa cells (2 × 104 cells/well) were seeded into an 8-well chamber slide for 24 h before being treated with the drug. The cells were incubated with free Ce6 or DC (4 μg/mL Ce6) for 2 h, rinsed twice with DPBS and illuminated with a 2.5-J/cm2, 671-nm diode laser. After 24 h, the cells were fixed in 4% paraformaldehyde solution for 10 min and stained with l mL of neutral red (30 μg/mL) or with 1 mL of H-33258 (10 μg/mL) for 30 min at room temperature in the dark. After washing and air drying, a coverslip was mounted on a microscope slide with a drop of antifade mounting solution to reduce fluorescence photobleaching. The cells were observed by using an inverted fluorescence microscope.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









