Thermal and Guest-Assisted Structural Transition in the NH2-MIL-53(Al) Metal Organic Framework: A Molecular Dynamics Simulation Investigation

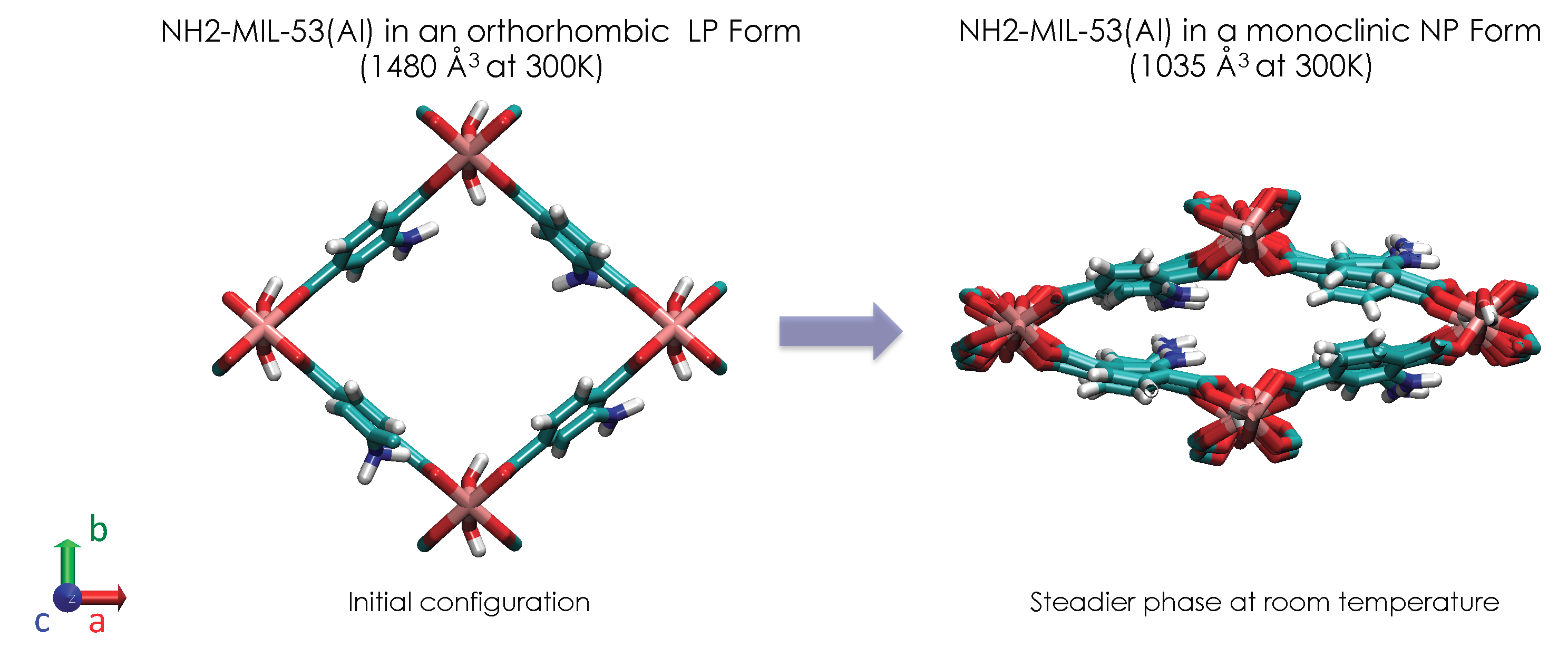{kind=link}
{kind=link}
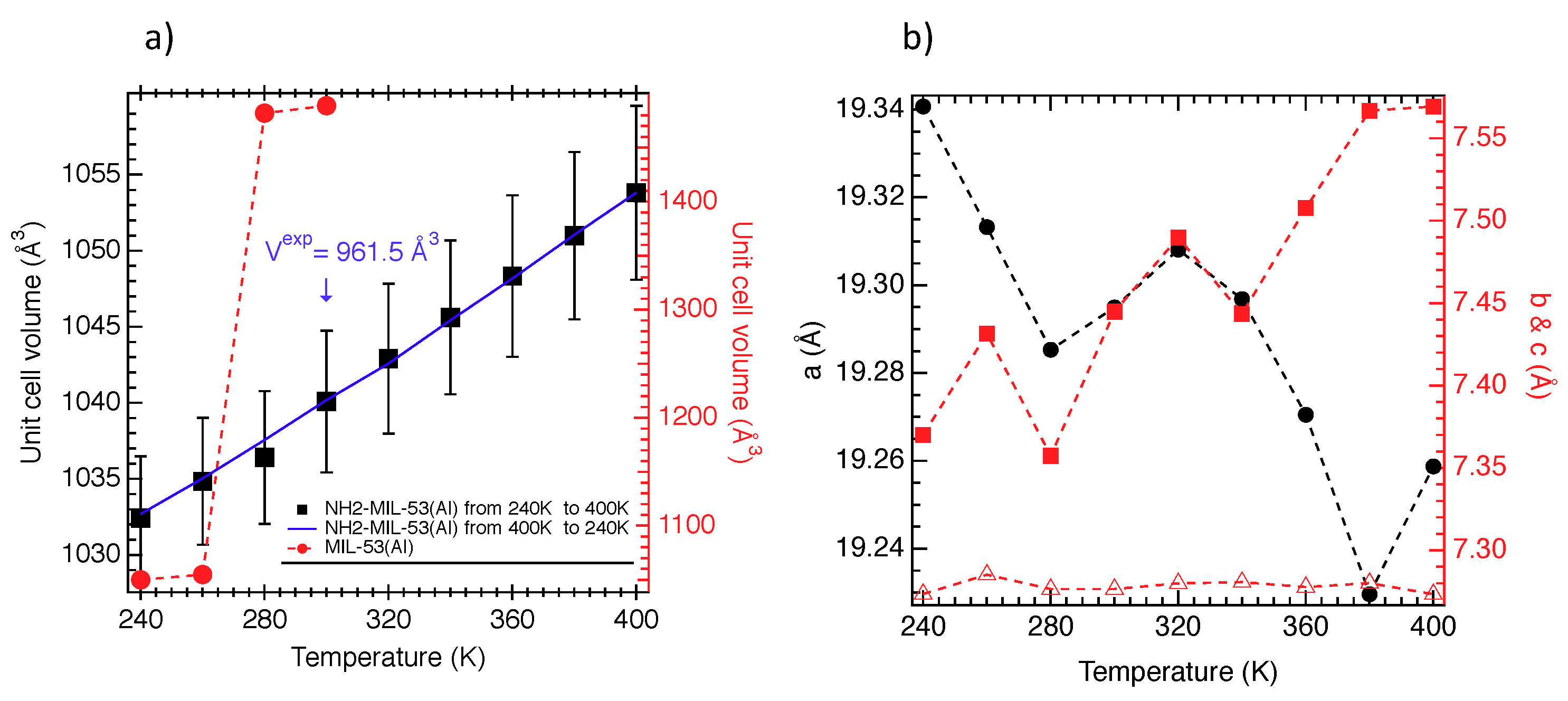{kind=link}
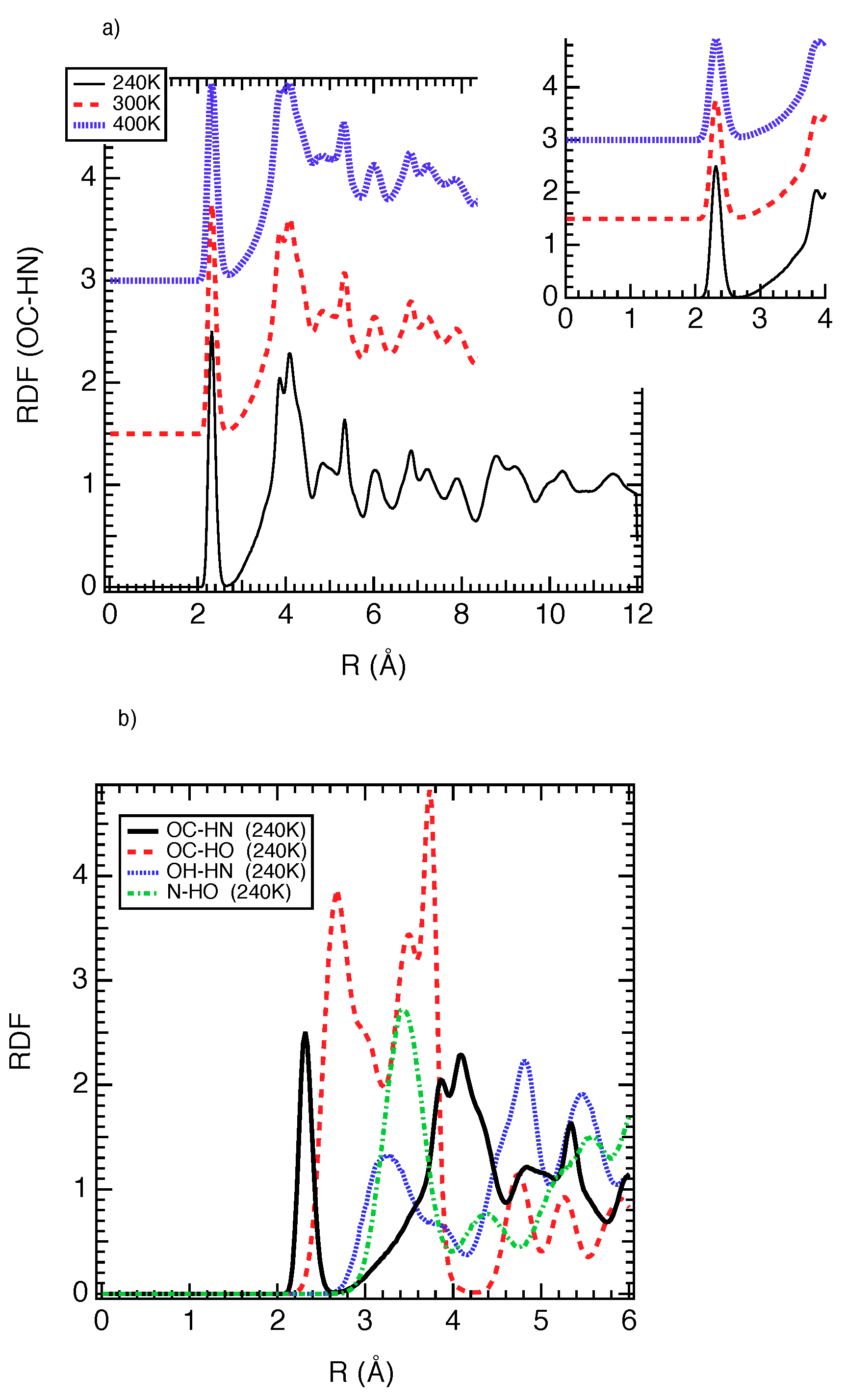{kind=link}
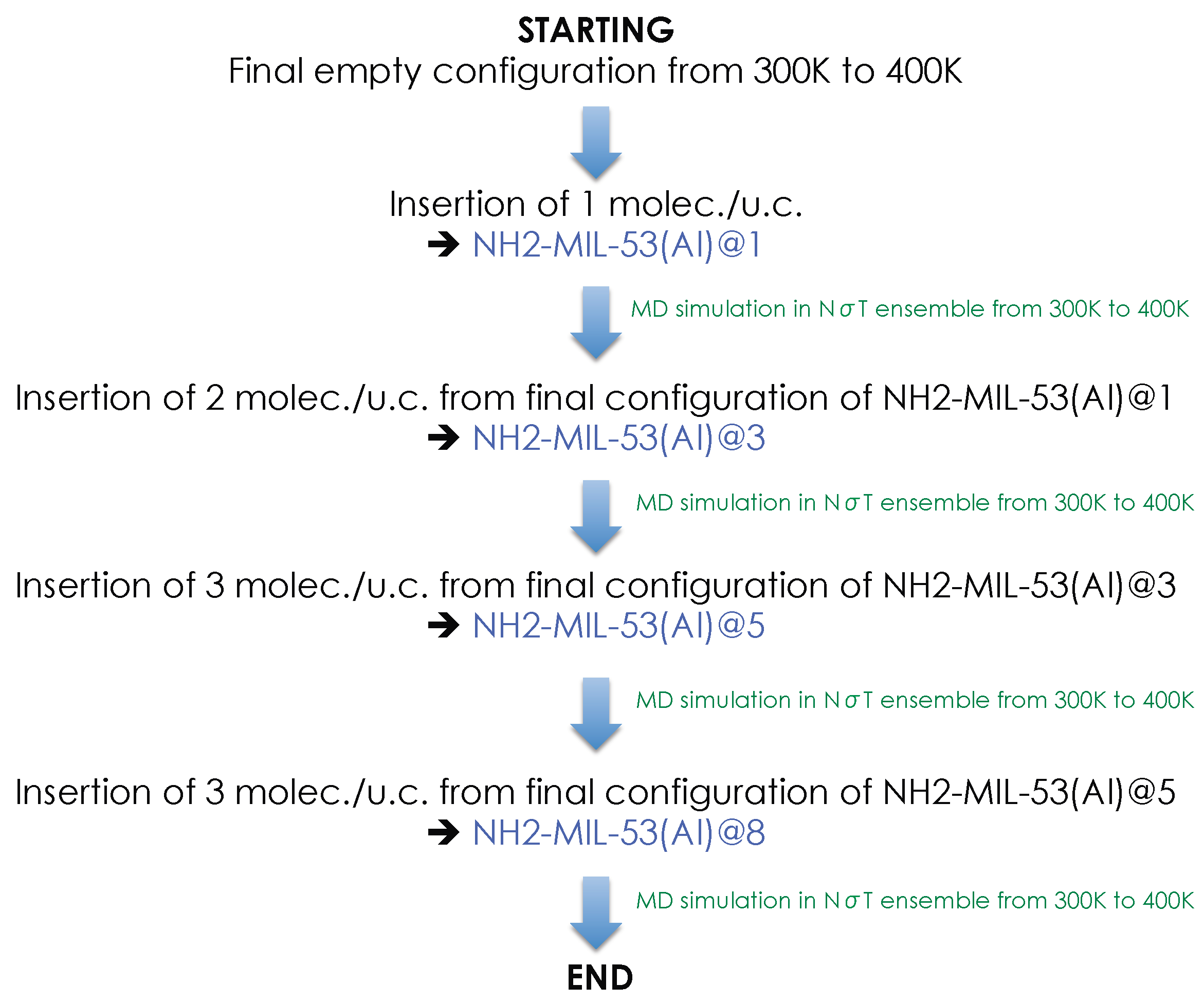{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ferey, G. Giant flexibility of crystallized organic–inorganic porous solids: Facts, reasons, effects and applications. New J. Chem. 2016, 40, 3950–3967. [Google Scholar] [CrossRef]
- Serre, C.; Férey, G. Large breathing effects in three-dimensional porous hybrid matter: Facts, analyses, rules and consequences. Chem. Soc. Rev. 2009, 38, 1380–1399. [Google Scholar]
- Horike, S.; Shimomora, S.; Kitagawa, S. Soft porous crystals. Nat. Chem. 2009, 1, 695. [Google Scholar] [CrossRef] [PubMed]
- Férey, G.; Serre, C.; Devic, T.; Maurin, G.; Jobic, H.; Llewellyn, P.; Weireld, G.D.; Vimont, A.; Daturi, M.; Chang, J. Why hybrid porous solids capture greenhouse gases? Chem. Soc. Rev. 2011, 40, 550–562. [Google Scholar] [CrossRef] [PubMed]
- Kitawaga, S.; Kitaura, R.; Noro, S. Functional Porous Coordination Polymers. Angew. Chem. Int. Ed. 2004, 43, 2334–2375. [Google Scholar] [CrossRef] [PubMed]
- Schneemann, A.; Bon, V.; Schwedler, I.; Senkovska, I.; Kaskel, S.; Fischer, R. Flexible metal-organicframeworks. Chem. Soc. Rev. 2014, 16, 6062–6096. [Google Scholar] [CrossRef] [PubMed]
- Modrow, A.; Zargarani, D.; Herges, R.; Stock, N. The first porous MOF with photoswitchable linker molecules. Dalton Trans. 2011, 40, 4217–4222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanai, N.; Uemura, T.; Inoue, M.; Matsuda, R.; Fukushima, T.; Tsujimoto, T.; Isoda, S.; Kitagawa, S. Guest-to-host transmission of structural changes for stimuli-response adsorption property. J. Am. Chem. Soc. 2012, 14, 4501–4504. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.W.; Henderson, B.L.; Kiesz, M.D.; Whalley, A.C.; Morris, W.; Grunder, S.; Deng, H.; Furukawa, H.; Zink, J.; Stoddart, J.; et al. Photophysical pore control in an azobenzene containing metal-organic framework. Chem. Sci. 2013, 4, 2858–2864. [Google Scholar] [CrossRef]
- Ghoufi, A.; Benhamed, K.; Bouki-Hacene, L.; Maurin, G. Electrically induced Breathing of the MIL-53(Cr) Metal-Organic Framework. ACS Cent. Sci. 2017, 3, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Knebel, A.; Geppert, B.; Volgmann, K.; Kolokolov, D.; Stepanov, A.; Twiefel, J.; Heitjans, P.; Volkmer, D.; Caro, J. Defibrillation of soft porous metal-organic frameworks with electric fields. Science 2017, 358, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Yot, P.; Boudene, Z.; Macia, J.; Granier, D.; Vanduyfhuys, L.; Verstraelen, T.; Speybroeck, V.V.; Devic, T.; Serre, C.; Férey, G.; et al. Metal-organic framework as potential shock absorbers, the case of the highly flexible MIL-53(Al). Chem. Commun. 2014, 50, 9462–9464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, J.; Beurroies, I.; Loiseau, T.; Denoyel, R.; Llewellyn, P. The direct heat measurement of mechanical energy storage metal-organic frameworks. Angew. Chem. Int. Ed. 2015, 54, 4626–4630. [Google Scholar] [CrossRef] [PubMed]
- Yot, P.; Vanduyfhus, L.; Alvarez, E.; Rodriguez, J.; Itié, J.; Fabry, P.; Guillou, N.; Devic, T.; Beurroies, I.; Llewellyn, P.; et al. Mechanical energy storage performance of an aluminum fumarate metal–organic framework. Chem. Sci. 2016, 7, 446–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salles, F.; Ghoufi, A.; Maurin, G.; Bell, R.; Mellot-Draznieks, C.; Llewellyn, P.; Serre, C.; Ferey, G. Molecular Dynamics Simulations of Breathing MOFs, Structural transformations of MIL-53(Cr) upon Thermal Activation and CO2 Adsorption. Angew. Chem. Int. Ed. 2008, 47, 8487–8491. [Google Scholar] [CrossRef] [PubMed]
- Ghoufi, A.; Maurin, G.; Férey, G. Physics Behind the Guest-Assisted Structural Transitions of a Porous Metal-Organic Framework Material. J. Phys. Chem. Lett. 2010, 1, 2810–2815. [Google Scholar] [CrossRef]
- Boutin, A.; Coudert, F.; Springuel-Huet, M.; Neimark, A.; Férey, G.; Fuchs, A. The behavior of Flexible MIL-53(Al) upon CH4 and CO2 Adsorption. J. Phys. Chem. C 2010, 114, 22237–22244. [Google Scholar] [CrossRef]
- Gaab, M.; Trukhan, N.; Maurer, S.; Gummaraju, R.; Mu¨ller, U. The progression of Al-based metal-organic frameworks—From academic research to industrial production and applications. Microporous Mesoporous Mater. 2012, 157, 131–136. [Google Scholar] [CrossRef]
- Ghoufi, A.; Subercaze, A.; Ma, Q.; Yot, P.; Ke, Y.; Puente-Orench, I.; Devic, T.; Guillerm, V.; Zhong, C.; Serre, C.; et al. Comparative Guest, Thermal, and Mechanical Breathing of the Porous Metal Organic Framework MIL-53(Cr): A Computational Exploration Supported by Experiments. J. Phys. Chem. C 2012, 116, 13289–13295. [Google Scholar] [CrossRef] [Green Version]
- Ma, Q.; Yang, Q.; Ghoufi, A.; Férey, G.; Zhong, C.; Maurin, G. Guest dependent pressure behavior of the flexible MIL-53(Cr), A computational approach. Dalton Trans. 2012, 41, 3915–3919. [Google Scholar] [CrossRef] [PubMed]
- Mason, J.; Oktawiec, J.; Taylor, M.; Hudson, R.; Rodriguez, J.; Bachman, J.; Gonzalez, M.; Cervellino, A.; Guagliardi, A.; Brown, C.; et al. Methane storage in flexible metal-organic frameworks with intrinsic thermal management. Nature 2016, 527, 357. [Google Scholar] [CrossRef] [PubMed]
- Bon, V.; Kavoosi, N.; Senkovska, I.; Kaskel, S. Tolerance of Flexible MOFs toward Repeated Adsorption Sress. ACS Appl. Mater. Interfaces 2015, 7, 22292–22300. [Google Scholar] [CrossRef] [PubMed]
- Krause, S.; Bon, V.; Senkovska, I.; Stoeck, U.; Wallacher, D.; Tobbens, D.; Zander, S.; Pillai, R.; Maurin, G.; Coudert, F.; et al. A pressure-amplifying framework material with negative gas adsorption transitions. Nature 2016, 532, 348. [Google Scholar] [CrossRef] [PubMed]
- Serre, C.; Millange, F.; Thouvenot, C.; Noguès, M.; Marsolier, G.; Louer, D.; Férey, G. Very High Breathing effect in the First Nanoporous Chromium(III)-based Solids: MIL-53 or Cr(OH).O2C-C6H4- CO2.HO2C-C6H4-CO2Hx.H2Oy. J. Am. Chem. Soc. 2002, 124, 13519–13526. [Google Scholar] [CrossRef] [PubMed]
- Serre, C.; Bourrelly, S.; Vimont, A.; Ramsahye, N.; Maurin, G.; Llewellyn, P.; Daturi, M.; Filinchuk, Y.; Leynaud, O.; Garnes, P.; et al. An Explanation for the Very Large Breating Effect of a Metal-Organic Framework during CO2 Adsorption. Adv. Mater. 2007, 19, 2246–2251. [Google Scholar] [CrossRef]
- Devic, T.; Horcajada, P.; Serre, C.; Salles, F.; Maurin, G.; Moulin, B.; Heurtaux, D.; Clet, G.; Vimont, A.; Grenèche, J.; et al. Functionalization in Flexible Porous Solids: Effects on the Pore Opening and the Host-Guest Interactions. J. Am. Chem. Soc. 2010, 132, 1127–1136. [Google Scholar] [CrossRef] [PubMed]
- Munn, A.; Pillai, R.; Biswas, S.; Stock, N.; Maurin, G.; Walton, R. The flexibility of modified-linker MIL-53 materials. Dalton Trans. 2016, 45, 4162–4168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lescouet, T.; Kockrick, E.; Bergeret, G.; Pera-Titus, M.; Farrusseng, D. Engineering MIL-53(Al) flexibility by controlling amino tags. Dalton Trans. 2011, 40, 11359–11361. [Google Scholar] [CrossRef] [PubMed]
- Ahnfelt, T.; Gunzelmann, D.; Loiseau, T.; Hirsemann, D.; Senker, J.; Ferey, G.; Stock, N. Synthesis and Modification of a Functionalized 3D Open-Framework Structure with MIL-53 Topology. Inorg. Chem. 2009, 48, 3057–3064. [Google Scholar] [CrossRef] [PubMed]
- Stavitski, E.; Pidko, E.A.; Couck, S.; Remy, T.; Hensen, E.J.; Weckhuysen, B.; Denayer, J.; Gascon, J.; Kapteijn, F. Complexity behind CO2 Capture on NH2-MIL-53(Al). Langmuir 2011, 27, 3970–3976. [Google Scholar] [CrossRef] [PubMed]
- Todorov, I.; Smith, W.; Trachenko, K.; Dove, M. DLPOLY3: New dimensions in molecular dynamis simulations via massive parallelism. J. Mater. Chem. 2006, 16, 1911–1918. [Google Scholar] [CrossRef]
- Mayo, S.L.; Olafson, B.D.; Goddard, W. DREIDING: A generic force field for molecular simulations. J. Phys. Chem. 1990, 94, 8897–8909. [Google Scholar] [CrossRef]
- Cornell, W.; Cleplak, P.; Bayly, C.; Gould, I.; Merz, K.; Fergusson, D.; Spellmeyer, D.; Fox, T.; Caldwell, J.; Kollman, P. A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Pérez, E.; Serra-Crespo, P.; Hamad, S.; Kapteijn, F.; Gascon, J. Molecular simulation of gas adsorption and diffusion in a breathing MOF using a rigid force field. Phys. Chem. Chem. Phys. 2014, 16, 16060–16066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorgensen, W.; Maxwell, D.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef] [Green Version]








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boulé, R.; Roland, C.; Le Pollés, L.; Audebrand, N.; Ghoufi, A. Thermal and Guest-Assisted Structural Transition in the NH2-MIL-53(Al) Metal Organic Framework: A Molecular Dynamics Simulation Investigation. Nanomaterials 2018, 8, 531. https://doi.org/10.3390/nano8070531
Boulé R, Roland C, Le Pollés L, Audebrand N, Ghoufi A. Thermal and Guest-Assisted Structural Transition in the NH2-MIL-53(Al) Metal Organic Framework: A Molecular Dynamics Simulation Investigation. Nanomaterials. 2018; 8(7):531. https://doi.org/10.3390/nano8070531
Chicago/Turabian StyleBoulé, Roald, Claire Roland, Laurent Le Pollés, Nathalie Audebrand, and Aziz Ghoufi. 2018. "Thermal and Guest-Assisted Structural Transition in the NH2-MIL-53(Al) Metal Organic Framework: A Molecular Dynamics Simulation Investigation" Nanomaterials 8, no. 7: 531. https://doi.org/10.3390/nano8070531
APA StyleBoulé, R., Roland, C., Le Pollés, L., Audebrand, N., & Ghoufi, A. (2018). Thermal and Guest-Assisted Structural Transition in the NH2-MIL-53(Al) Metal Organic Framework: A Molecular Dynamics Simulation Investigation. Nanomaterials, 8(7), 531. https://doi.org/10.3390/nano8070531