1. Introduction
Malaysia, Indonesia, Thailand, African countries like Nigeria, Cameroon and several Southern provinces of China are among the top producers of palm oil in the world [
1]. As the leading producer and supplier of the oil palm, Malaysia keeps on increasing the production of palm oil by developing oil palm plantations beginning with only 54,000 hectares in the early 1960s, expected to increase to 5.1 million hectares by 2020 [
2]. The oil palm industries in Malaysia are producing about 90 × 10
6 tons of lignocellulosic biomass each year, of which empty fruit bunch (EFB), oil palm trunk (OPT) and oil palm fronds (OPF) are about 40 × 10
6 tons [
3]. It is an opportunity for oil palm industries to use these wastes to turn them into valuable products instead of discarding them by open burning [
4] which resulted in bad consequences for the environment. The increasing number of waste material from industrial development contributed to the deterioration of the earth through ozone depletion. Oil palm solid waste (OPSW) like palm kernel shell is a good candidate for the production of activated carbon as it contains a highly developed porosity and surface area because of its high carbon content and relatively low price [
5].
Palm kernel shell is a sustainable source of materials included in the economic sector which is responsible for the breakdown of global greenhouse gas emissions and climate change. Many industries, academics, and governmental agencies are now focusing on the green chemistry and engineering technologies to minimize the negative impacts towards the environment [
6]. The manufacturing processes are now restructured by the application of green solvents and reagents, energy conservations, waste minimization, and utilization of natural resources.
Consisting of about 87% to 97% of carbon and other elements such as hydrogen, oxygen, sulfur and nitrogen, activated carbon has a very high adsorption capability by having the highest volume of adsorbing porosity [
7]. Activated carbon with highly developed porosity, has a large surface area, a high micropore volume (V
mic), a favourable pore size distribution, and thermal stability [
8]. The surface area of activated carbon is usually more than 1000 m
2/g. Activated carbon has a random imperfect structure which consists of a broad range of pore sizes—micro, meso, and macro—that makes activated carbon different from graphite [
7] or other carbon materials. The surface of the activated carbon can be used to accumulate contaminants as it contains the protonated (C–OH
2+), neutral (COH), or ionized (CO
−) groups [
9].
Activated carbon can be prepared using three main methods: chemical, physical and physicochemical activations. Researchers used the chemical method at the beginning of the activated carbon production era, but recently the physical and physicochemical methods are more preferred for the production of activated carbon, especially from oil palm solid waste. Chemical activation is the most widely used for the production as it provides a superior quality, high surface area, high porosity, and higher carbon yield [
10]. Activating agents such as KOH, ZnCl
2, K
2CO
3, and H
3PO
4 are usually used as they help to develop the pore structure of the activated carbon [
11]. The most popular chemical activating agents that were used for the activation of activated carbon are ZnCl
2 and H
3PO
4. It inhibits the tar formation and widens the porous structure of the activated carbon by increasing the carbon yield. The chemical activation by KOH and K
2CO
3 show different mechanism as the reaction with carbon causes carbon gasification and the formation of hydrogen, which will not contribute to the increase in carbon yield [
12].
Controlling the activation process by chemical treatment will enable one to enhance and provide higher specific surface properties to the resulting activated carbon [
13]. In chemical activation, an acidic or basic solution is used as the activating agent to produce a higher surface area and porosity [
8]. There are several activating agents which are usually used in treating the OPSW such as H
3PO
4, K
2CO
3, KOH, ZnCl
2, and H
2SO
4. The synthesis method will determine the resulting physicochemical characteristics of the activated carbon as it depends on the activating agent, the amount of the precursor, the condition of the activation process, and the raw materials used [
7]. Proper chemical management is needed to discharge the activating agent after sample treatment in order to prevent pollutions. There are several methods that can be adapted to dispose of this highly polluting effluent, such as the treatment system based on membrane technology [
14], in situ solvent and reagent recycle by Nanofiltration [
15], or by solvent recycle with imperfect membranes by the separation method [
16].
Thermal energy storage is an energy storage device that functions to develop a new energy source which is very important to help in conserving energy which, in turn, reduces the negative impact towards the polluted environment [
17]. Phase change material (PCM) is the medium for latent heat thermal energy storage in which the energy stored will be used based on energy supply and demand when there is a change of phase caused by the freezing or melting processes [
18]. Solid-liquid PCM such as n-octadecane is very useful as it can store a large quantity of energy with small changes of volume. In addition, it also possesses desirable properties such as a high latent heat, chemical inertness, no phase segregation, and it is commercially available [
19]. In the preparation of the SSPCM nano-composite, the PCM was encapsulated into the activated carbon pores or inorganic frameworks to prevent any leakage during the solid-liquid phase transition.
In this work, the preparation of activated carbon from palm kernel shell will be studied and the resulting activated carbon will be subsequently used as inorganic frameworks for the preparation of shape-stabilized phase change material (SSPCM) for the thermal energy storage (TES) application. Parametric optimization for the preparation of activated carbon was done in order to obtain the highest graphitic content with the highest surface area and porosity. Three different parameters, namely, activation temperature, activation holding time, and concentration of H3PO4 were optimized.
2. Materials and Methods
2.1. List of Materials
Palm kernel shell (PKS), deionized water, orthophosphoric acid (85%) (SystermChemAR, Shah Alam, Malaysia), ethyl alcohol (99.7%) (R&M Chemicals, Semenyih, Malaysia), and octadecane (99%) (Aldrich Chemistry, St. Louis, MO, USA) were used in this work.
2.2. Pre-Treatment of Palm Kernel Shell
The palm kernel shell (PKS) samples were collected from the Seri Ulu Langat Palm Oil Mill, Dengkil, Selangor, to be used as the precursor for the activated carbon production. The PKS was cleaned using water, followed by de-ionized water. The sample was crushed into powder using a stable arm grinder before it was treated with H3PO4. About 20 g of the precursor was weighted and treated with 100 mL of freshly prepared H3PO4 at various concentrations of H3PO4: 0%, 5%, 10%, 20%, 30%, and 40% (v/v) using 85% H3PO4. Treatment was done in a conical flask with the ratio of 1:5 of 20 g of PKS and 100 mL of freshly prepared H3PO4. After 24 h of treatment, the liquid was filtered off using a filter paper and the sample was dried in an oven at 70 °C for 24 h before it was used as the precursor for AC preparation.
2.3. Sample Activation
An electrical tubular furnace was used in this one step activation with a constant flow rate of nitrogen gas at 150 cm3 min−1. The sample was activated in three different conditions based on the parameters to be optimized and this will be described in the following section.
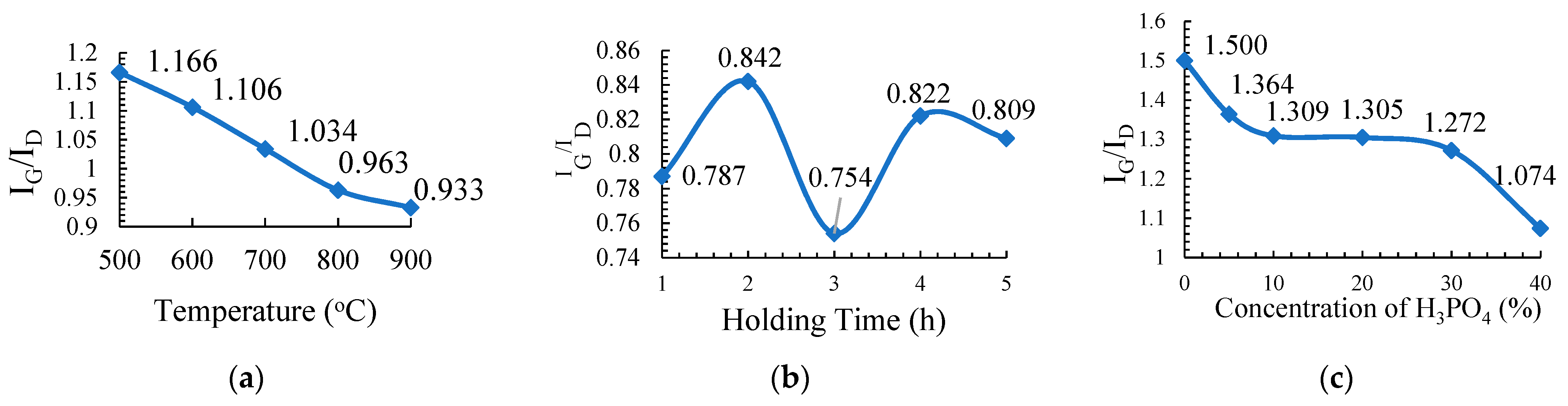
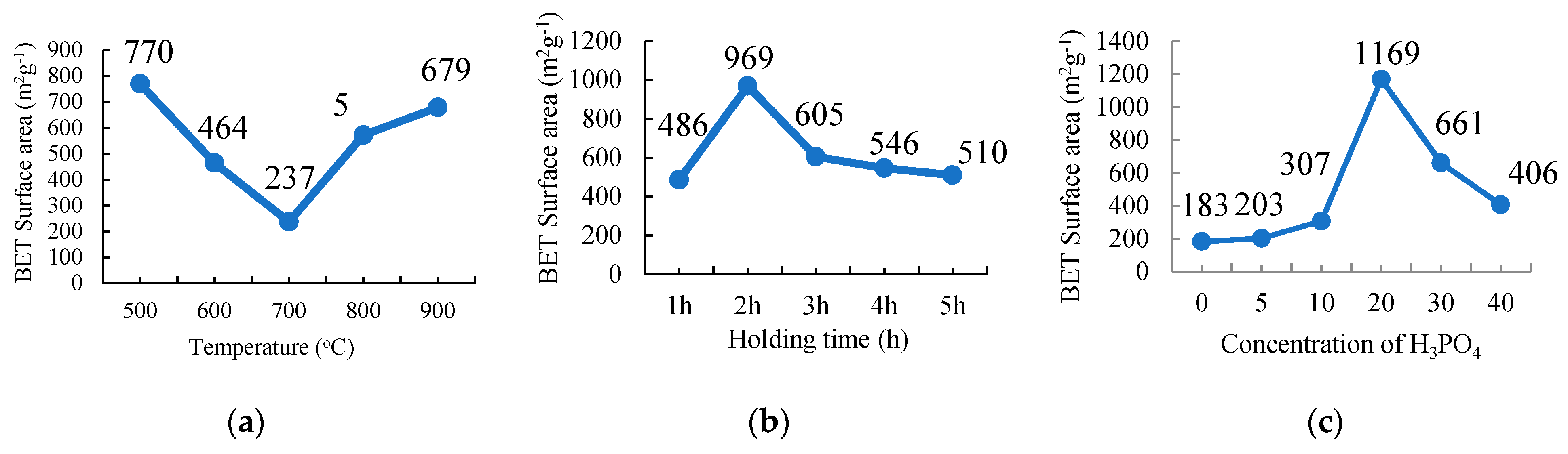
2.3.1. Carbonization Temperature
The PKS samples of about 5 g were carbonized using a tubular furnace under a nitrogen gas environment. The sample treated with 20% H3PO4 was chosen to study the effect of the carbonization temperature on the physicochemical properties of the activated carbons. The sample was carbonized at different temperatures: 500 °C, 600 °C, 700 °C, 800 °C, and 900 °C for 2 h with a holding time at a 10 °C min−1 heating rate. The resulting PKSACs prepared were crushed using a mortar and pestle before they were cleaned. The cleaned samples were oven-dried at 110 °C for 24 h to remove any remaining internal moisture. The samples were weighted and recorded again before they were kept in vials and labelled as T500, T600, T700, T800, and T900 for the sample prepared at carbonization temperatures of 500 °C, 600 °C, 700 °C, 800 °C, and 900 °C, respectively, for further used and various analyses.
2.3.2. Carbonization Holding Time
For this experiment, the sample that was treated with 20% of H3PO4 was subsequently chosen and carbonized at 500 °C at different holding times: 1 h, 2 h, 3 h, 4 h, and 5 h at a 10 °C min−1 heating rate. The as-produced activated carbons were crushed using a mortar and pestle before they were further cleaned. The cleaned samples were oven dried at 110 °C for 24 h to remove any internal moisture left during the preparation. The samples were then weighted and recorded again before they were kept into vials and labelled as H1, H2, H3, H4, and H5 for holding times of 1 h, 2 h, 3 h, 4 h, and 5 h, respectively, for further used and various analyses.
2.3.3. Concentration of the Activating Agent, H3PO4
Each treated sample that was prepared at different concentrations of H3PO4 was carbonized at 500 °C for 2 h of a holding time and at a 10 °C min−1 heating rate. The resulting PKSACs were crushed using a mortar and pestle before it was cleaned. The cleaned samples were then oven-dried at 110 °C for 24 h to remove any remaining internal moisture. The samples were then weighted and recorded again before they were kept into vials and labelled as C0, C5, C10, C20, C30 and C40 for treatment with H3PO4 at 0%, 5%, 10%, 20%, 30%, and 40%, respectively for further used and various analyses.
2.4. Preparation of Shape-Stabilized Phase Change Materials
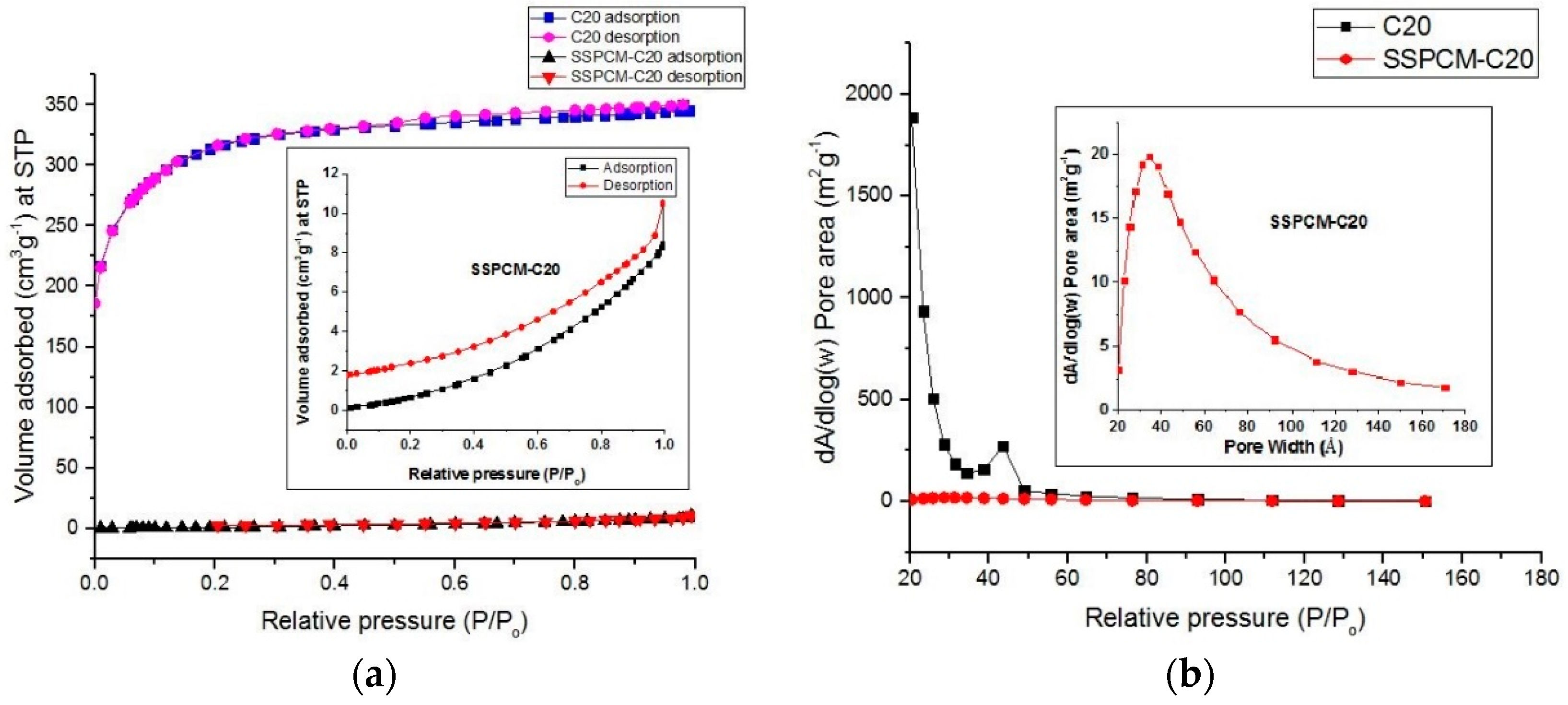
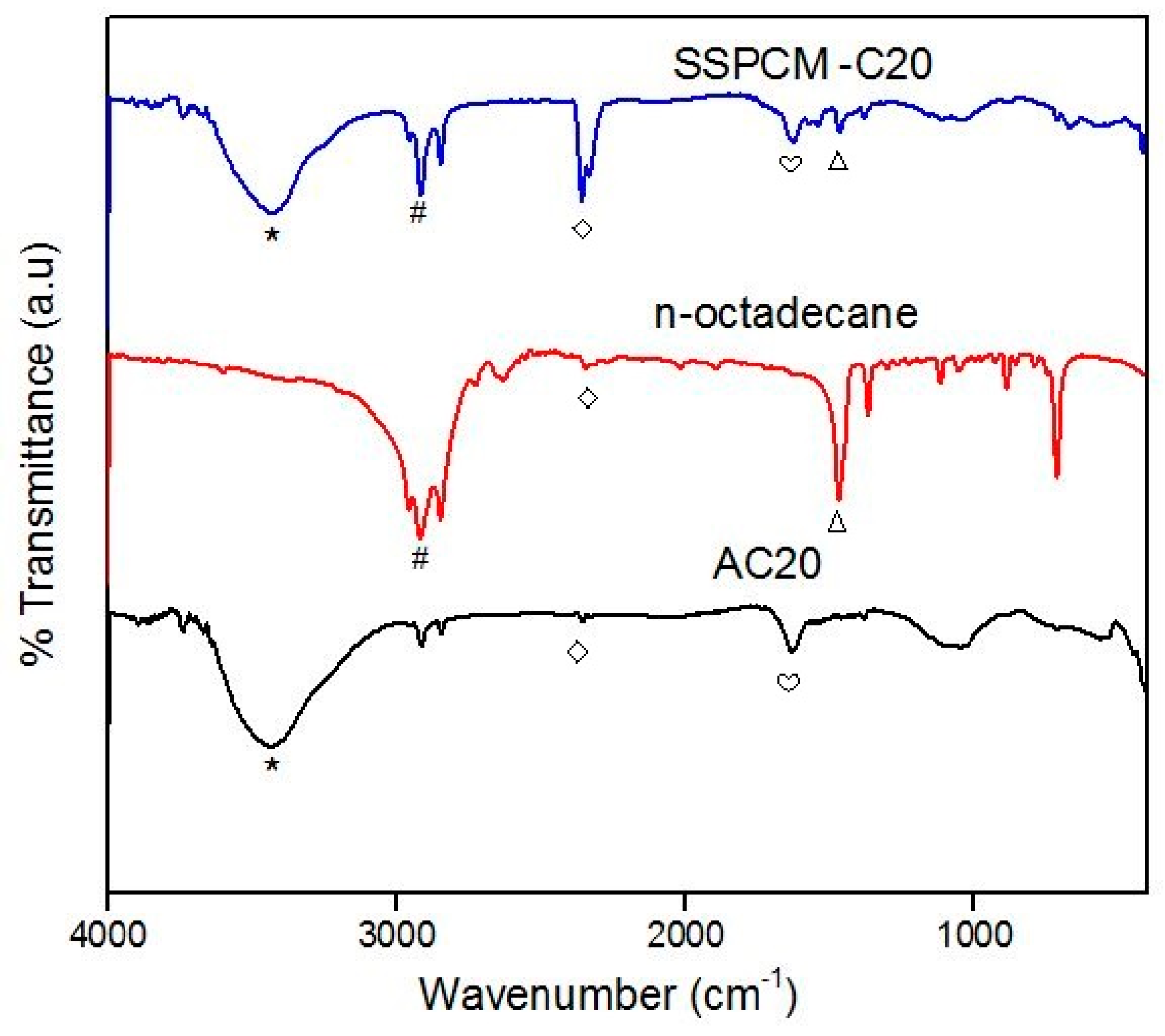
A shape-stabilized phase change material (SSPCM) nano-composite was prepared by a simple impregnation method. About 30 mL of absolute ethanol was used to dissolve melted n-octadecane by heating above the melting temperature of n-octadecane above 28–30 °C. The prepared PKSAC, C20 was added into the n-octadecane solution and the solution was then stirred at 600 rpm for 4 h. The mixture was oven-dried at 80 °C for 48 h or until all the excess ethanol was evaporated. The SSPCM-C20 prepared was stored in a sample bottle for further used and characterization.
2.5. Characterization Method
The chemical characterization of PKSAC was carried out using the Raman spectroscopic method with a WiTec Raman spectrometer (WiTec, Ulm, Germany) using a 514 nm laser. The intensity ratio between the D-line (~1350 cm−1) and the G-line (~1597 cm−1) of the Raman spectra was used to evaluate the graphitic character of the PKSAC. In this study, characterizations were done for all the PKSACs prepared from the three different parameters: carbonization temperature, carbonization holding time, and concentration of the activating agent.
The surface area and porosity of PKSAC and SSPCM nano-composites were determined using the BET nitrogen gas adsorption–desorption method at 77 K using a Micromeratics Tristar II plus (Micromeratics, Norcross, GA, USA). The method was also conducted to identify whether the PCM, n-octadecane, can be impregnated into the newly prepared PKSAC pores or only adsorbed on the surface. The samples were degassed at 290 °C for 9 h under vacuum before the measurements and the specific surface area and pore size distribution of the samples was determined using the Brunauer-Emmet-Teller (BET) and BJH (Barret-Joyner-Halenda) Equations, respectively.
The chemical properties of the PKSAC and SSPCM nano-composites were carried out using the fourier-transform infrared spectroscopy (FTIR) on a Perkin Elmer BX FTIR spectrophotometer (Waltham, MA, USA) with the KBr method at room temperature. The absorbance was recorded from 400–4000 cm−1.
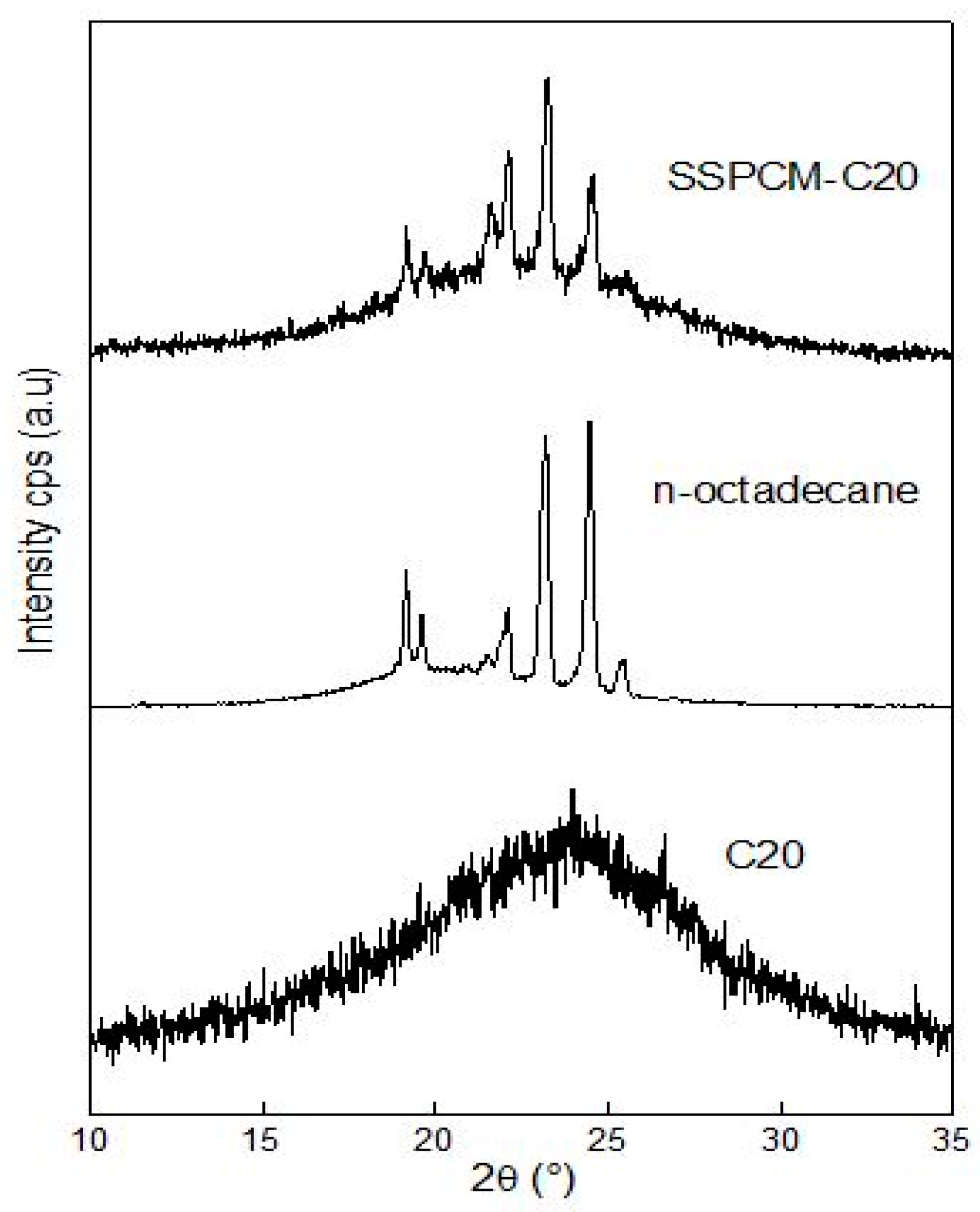
The powder X-ray diffraction (PXRD) patterns of PKSAC, n-octadecane, and SSPCM were obtained using a Shimadzu XRD-6000 PXRD (Kyoto, Japan) at room temperature with a scanning range of 10–35° (2θ) and a scanning rate of 4° min−1 for all samples.
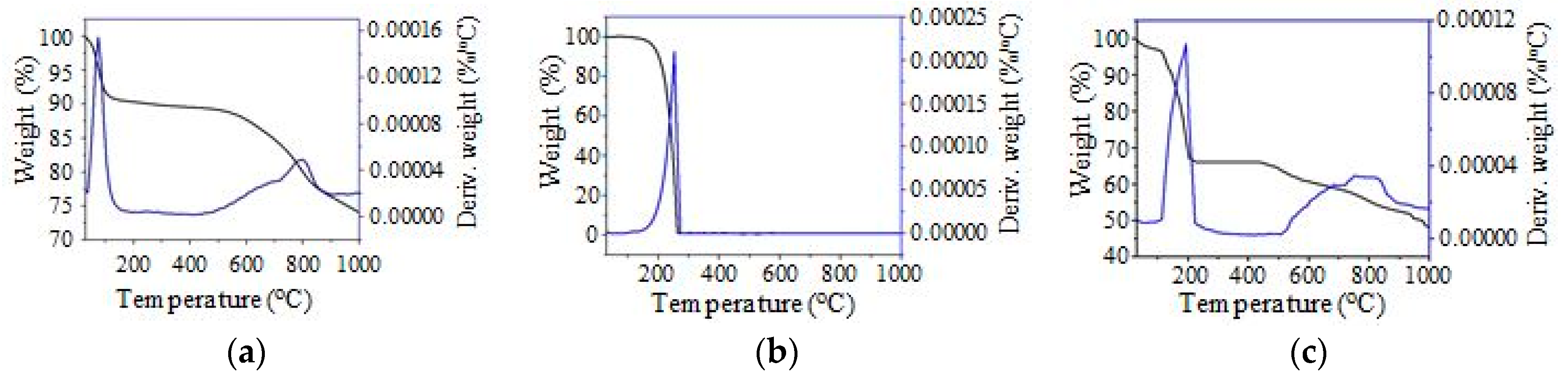
TGA/DTG thermal analyses were obtained using a Q500 V20.13 Build 39 (TA Instruments, Lukens Drive, New Castle, DE, USA) to investigate the thermal stability of the PKSAC, n-octadecane and SSPCM. The experiment was conducted using 10 mg of samples and heated under the nitrogen atmosphere at a 5 °C min−1 heating rate from room temperature to 1000 °C.
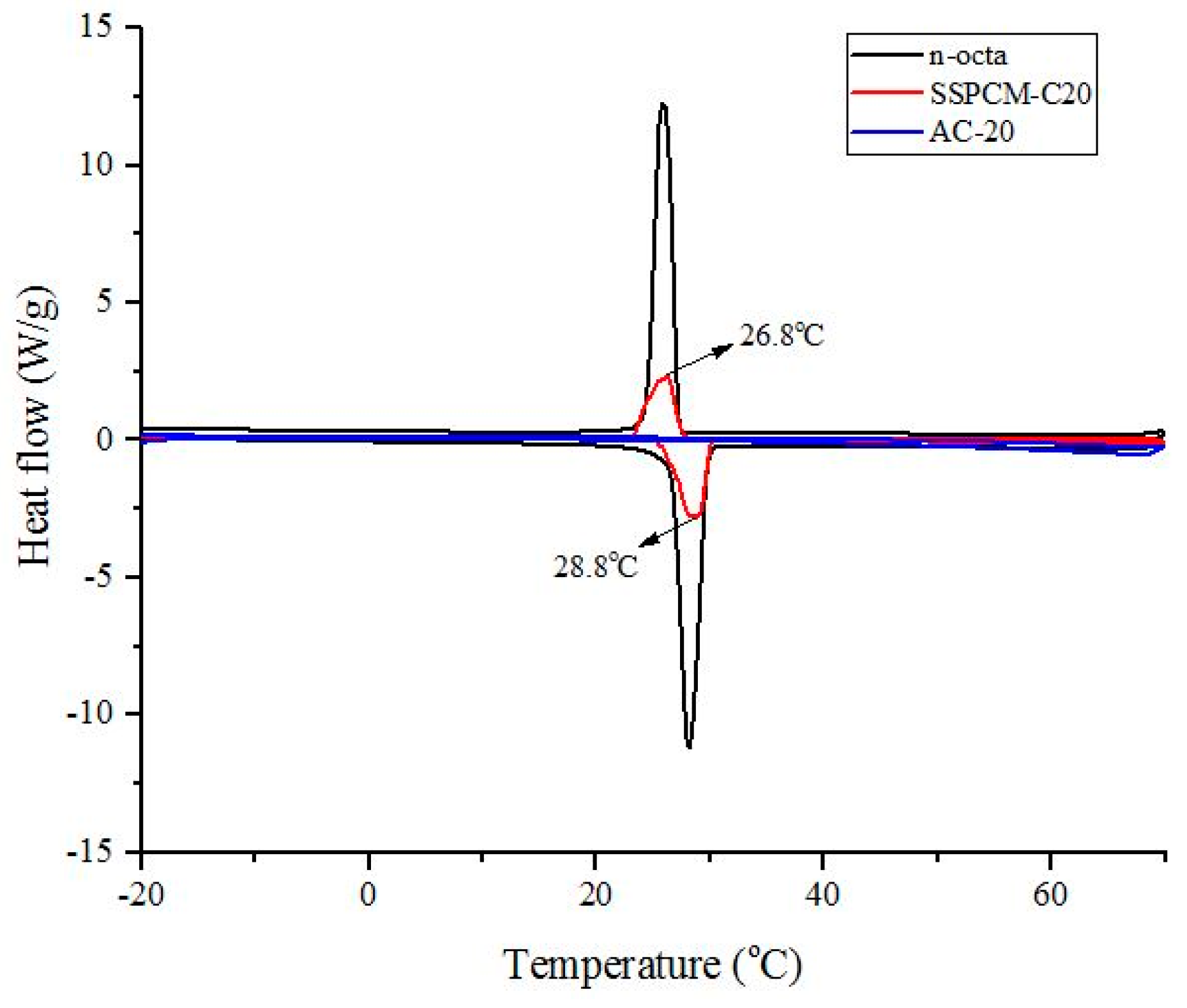
The thermal storage properties, such as the melting temperature, freezing temperature and enthalpy (latent heat) of the pure n-octadecane and SSPCM nano-composite were measured by a differential scanning calorimeter (DSC), 822e, Mettler Toledo equipped with a refrigerated cooling system. About 6 ± 1 mg of sample was weighed into an aluminium pan. Under a constant flow of nitrogen atmosphere with the flow rate of 60 mL/min, the measurements were performed at −20 to 70 °C for the heating cycle stage and 70 to −20 °C for the cooling cycle stage. The encapsulation efficiency of n-octadecane in the nano-composite s was calculated based on the enthalpy of pure n-octadecane using the following equation:
where ΔH
m is the enthalpy of melting of the SSPCM nano-composite (Jg
−1) and ∆H
PCM is the enthalpy of melting for the pure n-octadecane (Jg
−1).
The external surface morphology and microstructure of the PKS, PKSAC and SSPCM nano-composites were obtained using a Nova Nanosem 230 field emission scanning electron microscope (FESEM). The dried samples were dispersed on a conductive carbon adhesive tape surface that was attached to a FESEM stub and then gold-coated prior to the analysis.
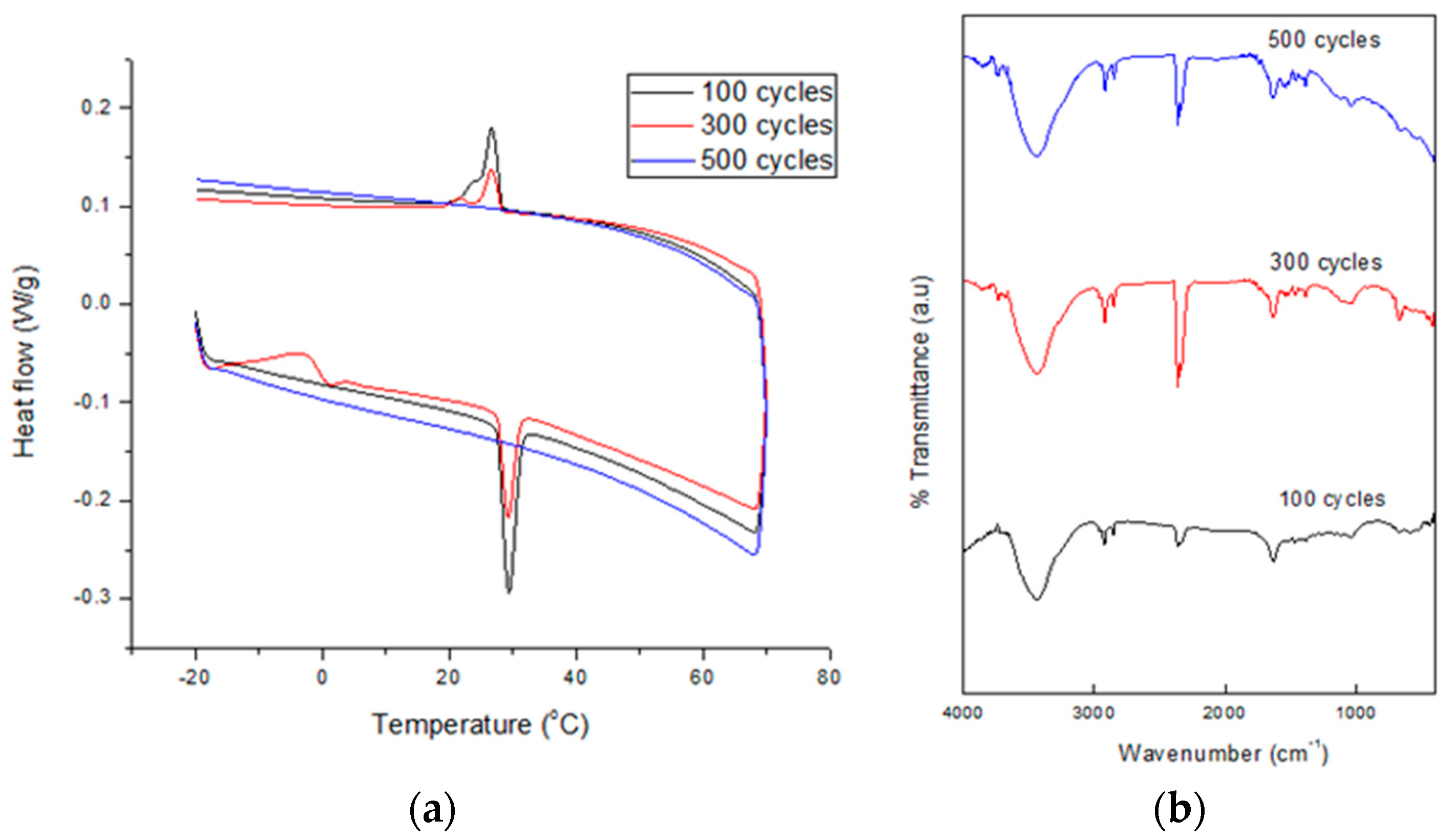
To measure the chemical and thermal stability of the SSPCM nano-composite s to be used as a TES material, a thermal cycling test was conducted. This test is very important in determining a material’s service lifetime. The test was performed at a relative humidity of 70% which was maintained constant using a climatic chamber (Weiss Umwelt Technik, UK) equipped with a K-type thermocouple in the middle [
20]. About 5 g of the dried SSPCM nano-composite was placed in contact with the thermocouple in a chamber. The accuracy of the temperature measurement was 0.1 °C. The nano-composite was subjected to 1000 thermal cycles at temperatures below and above the melting point of n-octadecane. Sampling was performed at 100, 300, 500, 700, and 1000 cycles. Changes in the samples’ thermal properties and chemical stability after the thermal cycling test were determined using the DSC and FTIR technique, respectively.
The leakage study was performed by keeping the SSPCM nano-composite inside an oven at 80 ± 5 °C for 3 days (72 h). Before that, about 1 g of the nano-composite was weighed on a filter paper and exposed to 30 °C for 8 h to study the ability of the C20 to hold n-octadecane during the melting phase. Then the samples were directly placed into an oven at 80 ± 5 °C for 3 days. The weight and the latent heat of the sample after the exposure period were recorded.













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}