1. Introduction
Noble metal nanoparticles have been extensively explored for application in catalysis [
1,
2,
3,
4,
5]. Being small, they offer unique physicochemical properties compared with their bulk counterparts [
6,
7,
8]. The small size means a large surface energy. Unfortunately, these nanoparticles tend to agglomerate to reduce their surface-to-volume ratio and their catalytic activity, and thus are undesirable for industrial applications [
9,
10].
Nanoparticles can be used directly as a colloid form for catalytic reactions [
11,
12]. In this case, surface-capping ligands are indispensable for the colloidal stability. However, these ligands often limit surface accessibility of the catalyst by blocking surface active sites, and decrease selectivity and activity by interacting with intermediates [
13,
14,
15,
16,
17]. The presence of undesirable co-existing ions and natural organic matters may cause their agglomeration as well. Furthermore, the recycling of these nanoscale catalysts from the reaction medium is of importance [
18]. Current established post-separation methods such as centrifugation and filtration are unsuitable for these nanoparticles and often lead to serious agglomeration [
19].
To address the recycling issue, nanocatalysts can be anchored onto low-cost support materials. In this respect, carbon material [
13,
20,
21,
22], metallic oxide [
23,
24], and polymer, among others [
25,
26,
27], have been employed as effective support materials to accommodate these metallic nanocatalysts. It is noteworthy that the resulting catalytic properties of these nanocomposite materials depend on both the characteristics of nanoparticles and the properties of support materials. For example, the nanoparticles must securely bonded with the support, and the support materials and/or protecting ligands should not block the active sites on the nanoparticles. Dispersing the supported composite catalysts in a reaction medium ensures their accessibility to substrates and allows for post-separation using a conventional filtration, centrifugation or magnetic process. However, the loss and agglomeration of the catalysts can still occur during this tedious process. An ideal way to fully utilize these nanocomposite materials is to load the nanocatalysts onto an immobilized support (e.g., column reactor and fix bed reactor) via continuous flow catalysis [
28]. Such a design is attractive for industrial applications due to its high operability and easy controllability. Unfortunately, mass transport-limited kinetics within either column reactors or fix bed reactors packed with immobilized catalysts is still not desirable, which greatly restricts the wide application of these processes. This limitation may be due to the pores that are blocked by the catalyst reactants and/or products. Alternatively, a more promising design is to construct a catalytic membrane reactor by integrating the catalytic nanoparticles with a desirable membrane material [
29]. These nanoscale catalysts can be effectively immobilized onto or into a support matrix without inhibiting access to catalytic sites [
30]. Such a catalytic membrane reactor ensures that separation and catalytic reactions occur in a single unit and can overcome the sluggish kinetics and improve the mass transport performance [
31]. Meanwhile, transforming the unique characteristics of individual nanoscale components into macroscopic materials such as membranes or sheets remains a challenge, as the engineering of these structures often compromises their intrinsic properties [
32].
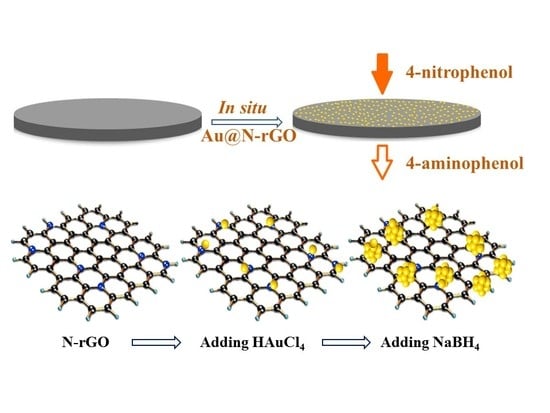
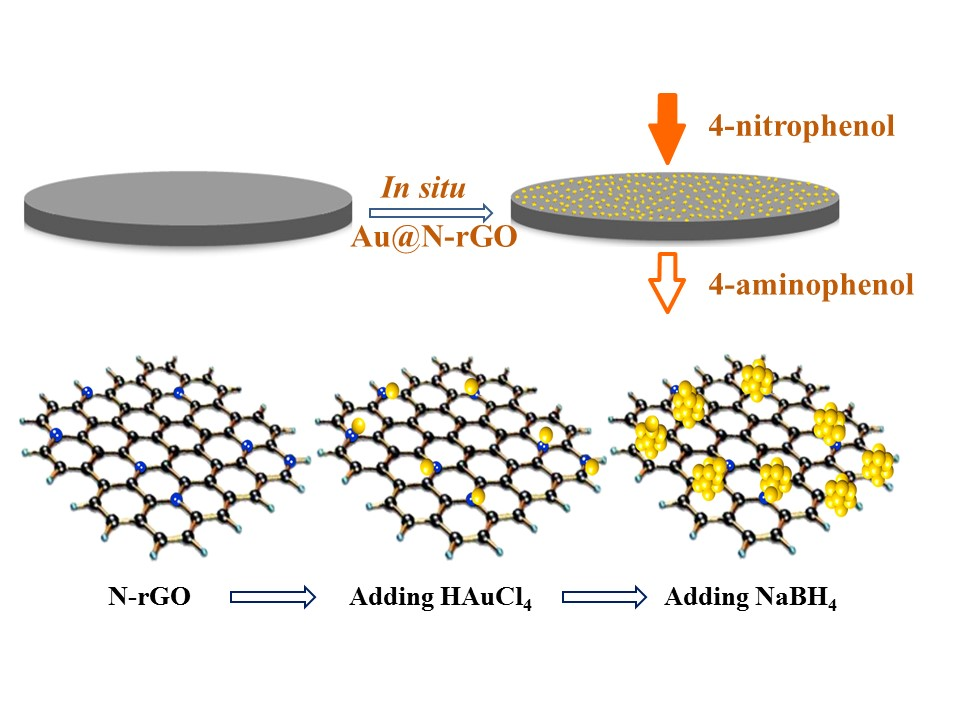
For this contribution to research, ligand-free nanoscale gold (nano-Au) catalysts with a tunable size were grown in situ onto a nitrogen-doped graphene (N-rGO) support membrane for continuous flow catalysis applications. To the best of the authors’ knowledge, only a very limited number of reports are available on the integration of two-dimensional materials with ligand-free nano-Au for continuous flow catalytic applications [
33]. Both ligand-free nano-Au and N-rGO supports played an indispensable role in this design. In particular, the nano-Au served as a high-performance catalyst and provided sufficient active sites for catalytic reactions. The graphene or rGO served as a promising candidate for next-generation membrane material. After doping with nitrogen, the as-produced N-rGO could effectively anchor the Au precursors carrying an opposite charge via electrostatic attraction. The catalytic performance was evaluated using the reduction of 4-nitrophenol (4-NP) to 4-aminophenol (4-AP) as a model reaction due to its affirmatory characterization protocol and simplicity. The catalytic activities of the as-fabricated filters were comparatively studied with a conventional batch reactor and the impact of several operational parameters were identified. Finally, a few 4-NP spiked real water samples were challenged with the catalytic filter.
2. Experimental Section
2.1. Chemicals and Materials
All the reagents were of analytical grade and used without further purification. Sodium borohydride (NaBH4, ≥98%) and gold (III) chloride trihydrate (HAuCl4·3H2O, ≥49% Au basis) were purchased from Sigma-Aldrich (Shanghai, China). 4-Nitrophenol (4-NP, ≥99%) was purchased from Alfar Aesar (Haverhill, MA, USA). The ammonia solution (NH3·H2O, 25–28%), N-methyl-2-pyrrolidone (NMP, ≥99.5%), and ethyl alcohol were purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). Graphene oxide powder was provided by Graphene Nanotech Co., Ltd. (Suzhou, China). Deionized water (DI-H2O) with a resistivity of 18.2 MΩ·cm was produced from a Milli-Q Direct 8 purification system (Merck, Darmstadt, Germany).
2.2. Catalytic Membrane Reactor Fabrication
The N-rGO filters were fabricated according to a reported protocol [
34]. Briefly, N-rGO hydrogel was firstly prepared by hydrothermal treatment of a 2.0 mg/mL GO and 2.0 mL NH
3·H
2O solution at 180 °C for 20 h. The N-rGO filter was then fabricated by dispersing 0.25 mg/mL of the as-prepared N-rGO hydrogel in NMP by ultrasonication, followed by vacuum filtration of the dispersion solution onto a 5 μm Millipore JMWP PTFE filter (Merck, Darmstadt, Germany). Then, the as-fabricated N-rGO filter was washed sequentially with 100 mL ethanol and 100 mL DI-H
2O. As a control, an rGO filter was fabricated in a similar procedure without the addition of NH
3·H
2O during the hydrothermal process.
The Au/N-rGO catalytic filters were fabricated by the in situ reduction of gold salt. In a typical synthetic process, a 50 mL of 0.04 mmol/L HAuCl4 solution and a 50 mL of 0.50 mmol/L NaBH4 solution were sequentially passed through the as-fabricated N-rGO filter using a 4-channel Ismatec REGLO digital peristaltic pump (Wertheim, Germany) at a flow rate of 1.0 mL/min.
2.3. Characterizations
The morphologies of the as-fabricated samples were examined on a JSM-6700F field emission scanning electron microscopy (FESEM, JEOL, Peabody, MA, USA) and a 2100 transmission electron microscopy (TEM, JEOL, Peabody, MA, USA). X-ray photoelectron spectroscopy (XPS) spectra were performed on a Escalab 250Xi XPS system (Thermo Fisher Scientific, Waltham, MA, USA), where the analysis chamber was 10−9 Torr and the bonding energies were calibrated and referenced to C 1s line at 284.8 eV. The Raman characterization of the filters was recorded on a LabRAM HR Evolution Raman spectrometer (Horiba, Kyoto Japan) excited by a 633 nm radiation from an He-Ne laser. The crystallinity of the filters was characterized by a SmartLab X-ray diffraction (XRD, Rigaku, Tokyo, Japan).
2.4. Catalytic Activity Evaluation
The catalytic activity of the catalytic filters was evaluated using the conversion of 4-nitrophenol (4-NP) to 4-aminophenol as a model reaction. The catalytic filters were first loaded into a polycarbonate Whatman filter casing (D = 47 mm, Piscataway, NJ). The effective active area of the catalytic membrane was 7.1 cm
2. A 4-channel Ismatec REGLO digital peristaltic pump (Cole-Parmer GmbH, Germany) was used to pump freshly-prepared 4-NP and NaBH
4 solution through the filter. The liquid residency time within the catalytic reactor was only 0.3 s. A photograph of the operational device is shown in
Figure S1. Effluents were collected and analyzed by a UV-2600 UV-vis spectrometer (Shimadzu, Kyoto, Japan). As a comparison, this reaction was performed in a conventional batch reactor system using the same catalytic filter in 60 mL of 0.2 mmol/L 4-NP and 37.5 mmol/L freshly-prepared NaBH
4 solution. The 4-NP conversion was calculated using pseudo-first-order kinetics due to the large excess of NaBH
4 in the reaction medium.
3. Results and Discussion
Using the typical synthesis process of a catalytic filter, a certain amount of Au precursors was passed through a pre-fabricated N-rGO filter. An in situ reduction of the Au precursors was then carried out by flowing an excess amount of NaBH
4 solution to yield nano-Au catalysts.
Figure 1 displays the N-rGO filter and Au/N-rGO filters prepared with different Au precursor concentrations. As has been previously reported, the N-rGO filter shows a crumpled sheet-like surface, typical of graphene [
35]. The space between the sheets provides microchannels for the passage of fluids. As can be seen, the particle size and the loading amount of nano-Au can be tuned by varying the concentration of Au precursors. At a limited Au/C weight ratio of 0.1% and 0.2%, no visible particles were observed from the FESEM characterizations. However, a TEM characterization of these same samples showed that well-defined nano-Au with an average diameter of 2.0 ± 0.5 nm and 6.2 ± 1.1 nm, respectively, were formed and both uniformly distributed onto the N-rGO surface (see
Figure 1c,d). Aggregates of Au particles appeared when the Au/C ratio reached ≥0.5% (see
Figure S2, Supplementary Materials). For example, the average diameter of Au particles was identified as 75 ± 16 nm and 350 ± 35 nm, respectively, at an Au/C ratio of 2.0% and 5.0%. This result showed that supported ligand-free Au nanoparticles with tunable size could be prepared in situ onto an N-rGO support. The obtained ligand-free AuNPs had more active sites exposed and were expected to be exceptionally active in selected catalytic reactions. Moreover, the wrinkled surface morphologies of the N-rGO remained after loading the Au catalyst. This suggested that the Au loading process through in situ reduction did not greatly affect the N-rGO’s microstructure. Energy-dispersive spectroscopy (EDS) analysis also supports the successful loading of nano-Au onto N-rGO filters, with a typical Au atomic ratio of 0.5–3.3% at an Au/C ratio of 1–5% (see
Table S1, Supplementary Materials).
Another interesting finding is that more nano-Au catalysts were produced on an N-rGO surface compared with that of an rGO counterpart. This may be due to the nucleation of nanocatalysts on the support, which is related to some extent to electrostatic interactions. To confirm this hypothesis, a zeta potential measurement of the N-rGO aqueous solution was performed. Indeed, a positive zeta potential of ζ = +0.4 mV was obtained, whereas a negative zeta potential of rGO (ζ = −1.2 mV) was obtained under similar conditions. This can be ascribed to the successful doping of nitrogen into the rGO. It is well known that graphene oxide is negatively charged due to its many oxygen-containing functional groups such as carbonyl, hydroxyl, and carboxyl [
36]. A positive zeta potential of the N-rGO was obtained after hydrothermal treatment of the GO dispersion solution, employing overheated supercritical water and NH
3.H
2O acting as a reducing agent. However, the as-produced rGO was still negatively charged due to the presence of the remaining oxy-functional groups. This also suggests that the GO reduction using ammonia and supercritical water as reducing agents (N-rGO case) is more thorough than using supercritical water alone (rGO case). Therefore, the electrostatic attraction between the positively charged nitrogenous groups in N-rGO and the negatively charged [AuCl
4] ions provided an effective driving force for the formation and stabilization of these Au precursors. After doping with N, more nucleation sites were available on the graphene surface. This favors the anchoring of Au precursors via electrostatic attraction during their passing through the N-rGO filter, and the further formation of nano-Au by NaBH
4 reduction. Previous studies have reported that N-rGO can act as the electron donors or as reductants to produce metal nanoparticles from precursors [
14]. However, the reductive capability of N-rGO alone is rather limited and cannot completely convert Au precursors into nanoparticles within a short reaction time (
τ = 1.0 s) at ambient temperature. The presence of excess NaBH
4 could compensate this reduction power, leading to the generation of nano-Au catalysts immediately after coming into contact with the previously anchored Au salts.
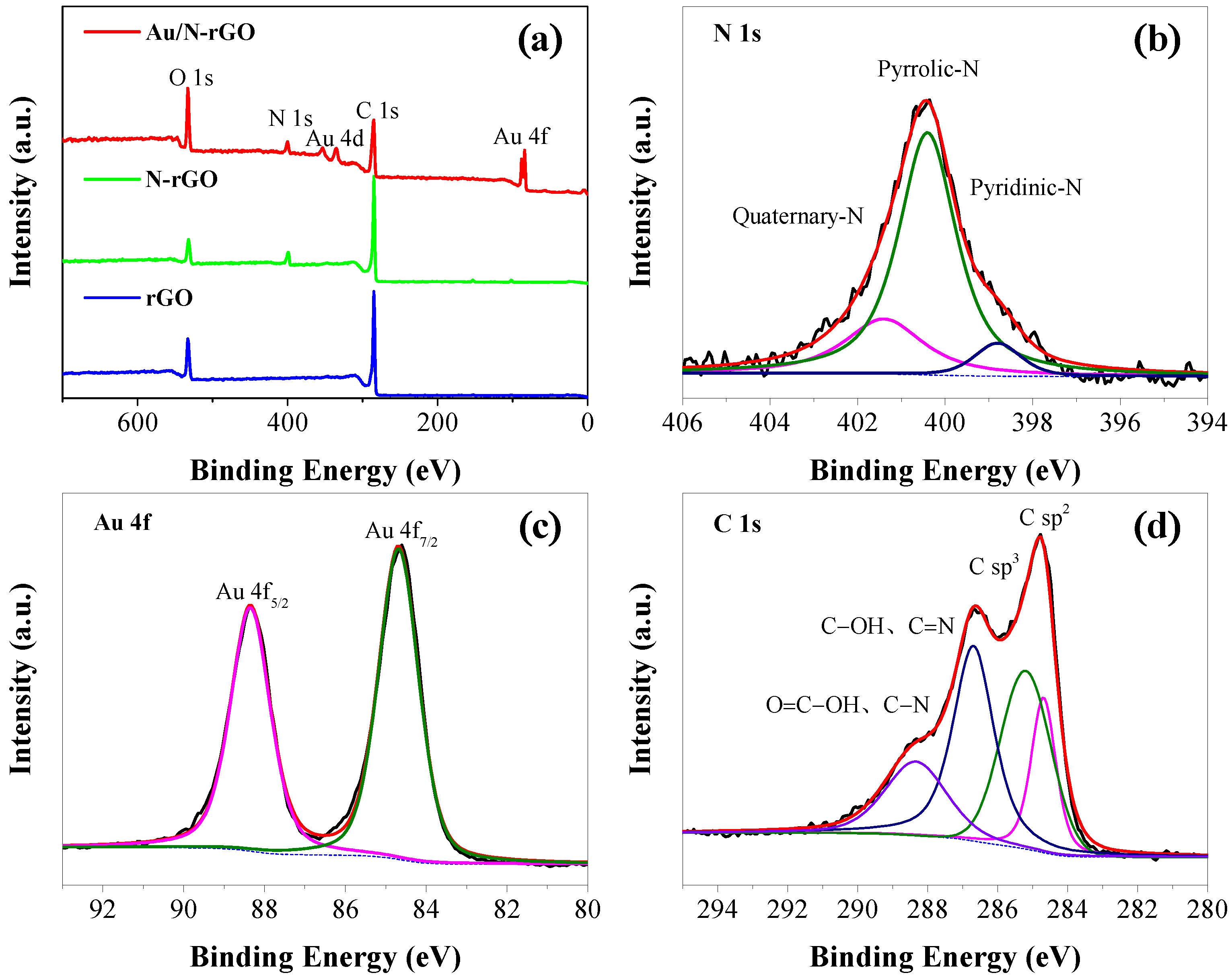
XPS was further used to confirm the change in elemental compositions and the oxidation state after N-doping and Au loading. As displayed in
Figure 2a, characteristic peaks of C 1s, O 1s, and N 1s were identified from the survey pattern of an N-rGO filter sample, whereas only C 1s and O 1s were observed for the rGO filter. These data provided supportive evidence for the successful doping of N. A decrease in the oxygen content and an increase in the carbon content were observed after Au loading onto the N-rGO. This can be due to the reduction of the oxylated functional groups by NaBH
4. As determined from the XPS data, the atomic ratio of N in the N-rGO filter was 7.8%, within the typical doping range of 1–15% [
37,
38,
39,
40]. From the high-resolution N 1s scan (see
Figure 2b), the signal could be further deconvoluted into pyridine nitrogen, pyrrolic nitrogen, and quaternary nitrogen. Based on the result of the peak area integration, the pyrrolic N centered at 399.6 eV (70.6%) was found to be the dominant N in the Au/N-rGO filter, which was higher than that of quaternary N at 401.5 eV (22.5%) and pyridinic N at 398.4 eV (6.9%). The ratio of pyrrolic N:pyridinic N:quaternary N is determined to be 1:0.32:0.10 based on the XPS data. This indicated that the dominant N state was pyrrolic N in the Au/N-rGO filter. Introducing N atoms into rGO could produce more defects and active sites onto which the Au precursors could be easily attached. The atomic ratio of Au was determined to be 0.13%, in agreement with the feeding amount of Au precursors (Au/C = 0.2%). Specifically, the high-resolution scan of Au 4f indicates Au 4f
7/2 and Au 4f
5/2, respectively, centered at a binding energy of 84.7 eV and 88.4 eV (peak-to-peak distance was 3.7 eV). This suggested the conversion of Au(III) to Au (0) by NaBH
4 reduction. The asymmetric C1s spectrum of N-rGO (see
Figure 2d) could be further deconvoluted into four characteristic peaks. Among them, the peaks centered at 284.7, 285.3, 286.5, and 288.3 eV corresponded to the C sp
2, C sp
3, C–OH, and C=N bonds, as well as O=C–OH and C–N bonds, respectively.
Catalytic Performance Evaluation
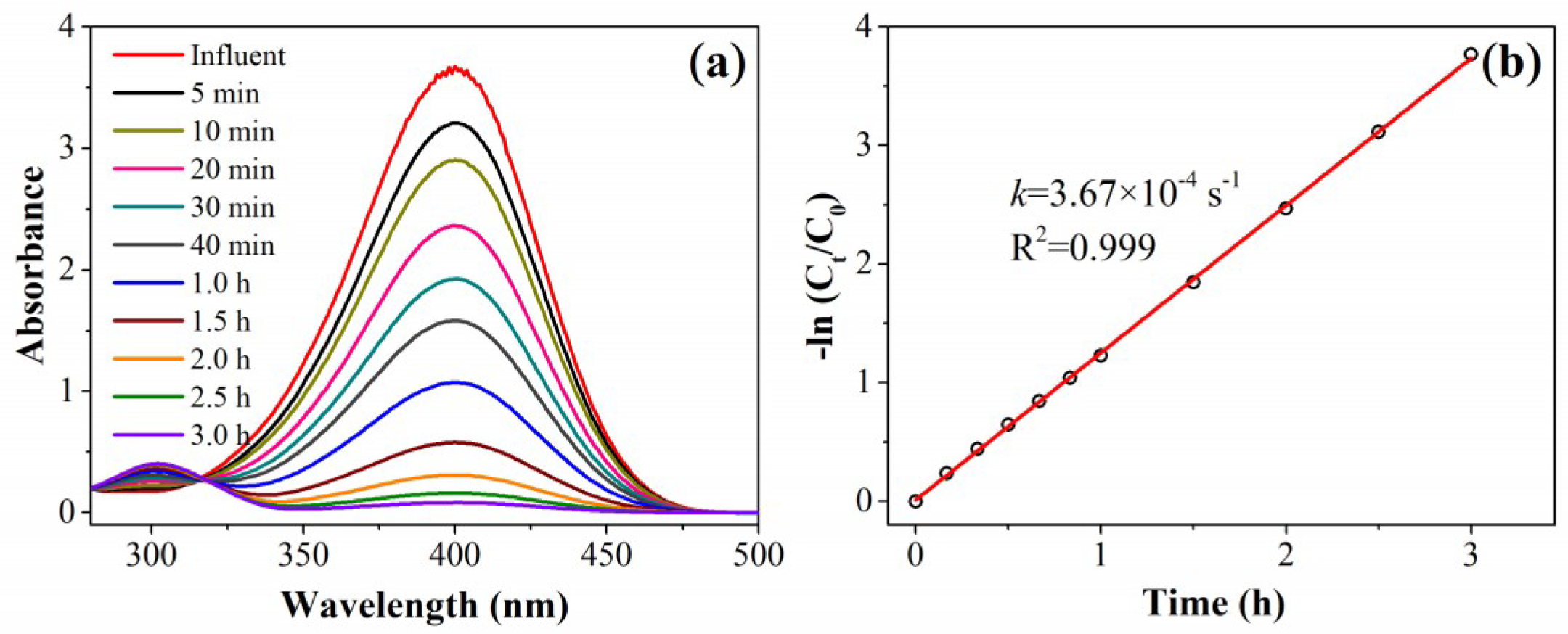
To evaluate the performance of the as-designed catalytic filter, the authors used the conversion of 4-nitrophenol (4-NP) into 4-aminophenol (4-AP; using NaBH
4 as the reductant) as a model catalytic reaction. As a control experiment, the time-dependent UV-vis absorption spectra of 4-NP in the presence of a typical Au/N-rGO filter (prepared by using 50 mL 0.04 mmol/L HAuCl
4 solution and 50 mL 0.5 mmol/L NaBH
4 solution passed through the 5 mg of N-rGO filter sequentially) and excess NaBH
4 (250 excess times) in a conventional batch reactor are shown in
Figure 3. The characteristic 4-NP absorption peak, centered at 400 nm, gradually decreased with time due to the continuous catalytic conversion to 4-AP in the presence of an Au/N-rGO catalyst. Consequently, the characteristic 4-AP absorption peak, centered at 300 nm, emerged and increased with time. The yellow 4-NP solution was completely converted to colorless 4-AP after 3 h continuous reaction. A linear relationship between ln(
C/
C0) and reaction time was obtained. These data led to pseudo-first-order kinetics with a rate constant,
kapp, calculated to be 3.67 × 10
−4 s
−1. In this study, the nano-Au catalysts were embedded onto/into the N-rGO filter surface. The reactants needed more time to diffuse and get in contact with these active sites, whereas the interaction of AuNPs and 4-NP molecules was much easier by Brownian motion in a homogeneous system. This could explain why the
kapp obtained in this study was only 24% of that obtained employing the AuNPs catalyst in a homogeneous batch system [
20].
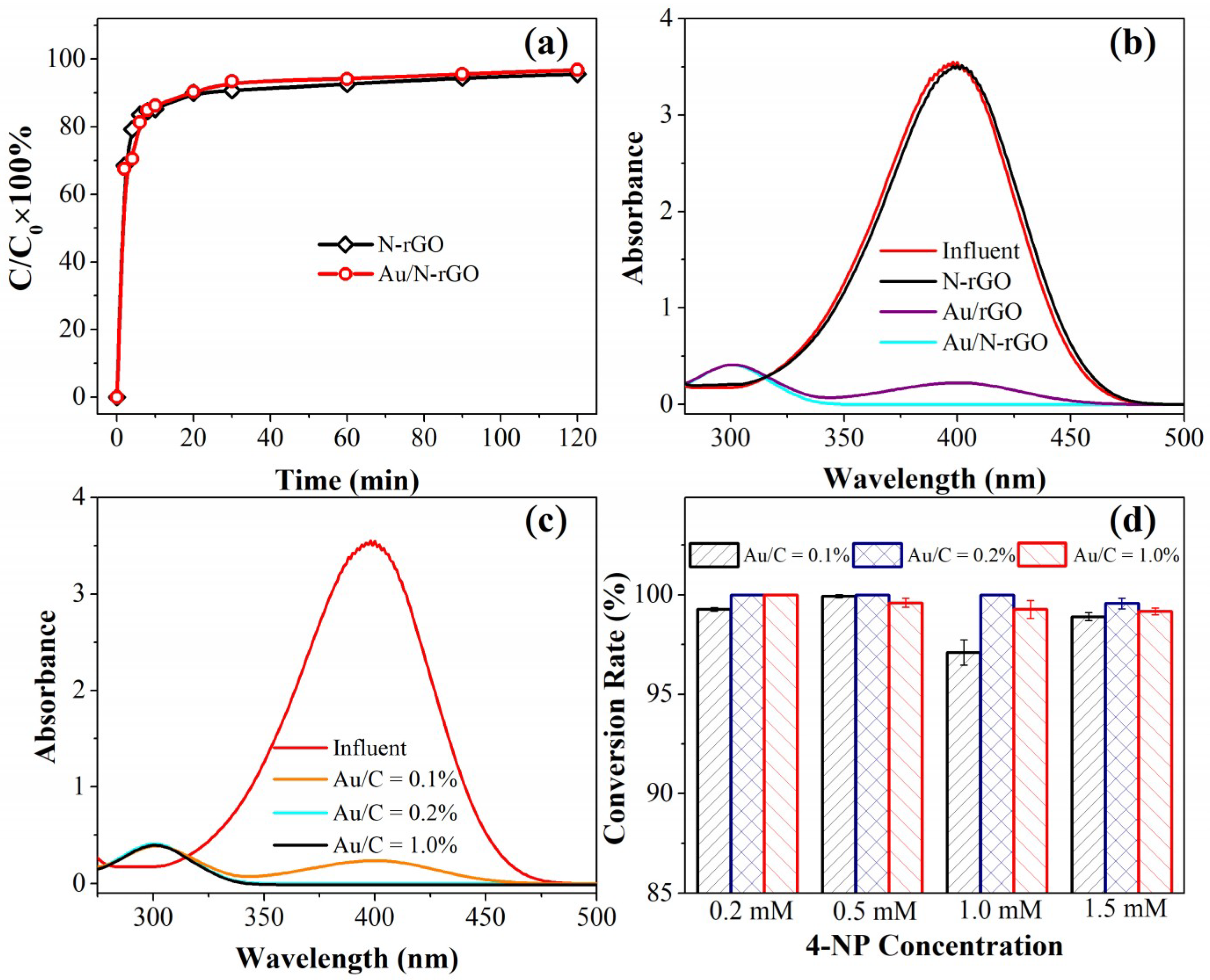
For comparison, the authors performed the conversion of 4-NP in a continuous flow catalytic filter system. A continuous flow catalytic filter design is more advantageous and attractive compared with a conventional batch reactor. To eliminate adsorption on the removal of 4-NP, the adsorption saturation of 4-NP was first performed by passing 0.2 mmol/L 4-NP alone through the filter at a flow rate of 1.0 mL/min. As shown in
Figure 4a, similar breakthrough curves were obtained for N-rGO and Au/N-rGO filters. This indicated that nano-Au loading posed a negligible effect on the adsorption behavior of the N-rGO filters as well as on the mass transfer-controlled sorption process. Because of the limited thickness (4.0 ± 0.3 µm, as shown in
Figure S3) of the N-rGO filter and the convection-enhanced mass transport, the effluent 4-NP concentration increased rapidly in the first 10 min and a complete breakthrough occurred over 40 min continuous filtration for both filters. Another reason for this rapid 4-NP sorption may be due to the strong π–π interactions between the two-dimensional planar hexagonal carbon structure of rGO and the aromatic molecular structure of 4-NP.
After the 4-NP sorption saturation, a certain amount of 4-NP passed through the catalytic filter together with NaBH
4. Some control experiments were performed to clarify the contribution of nano-Au and/or doped N on the 4-NP conversion. As shown in
Figure 4b, one control experiment using the N-rGO filter alone exhibited negligible 4-NP conversion under similar conditions. N-rGO has been reported to possess good catalytic activity toward selected oxidation and reduction reactions [
41,
42,
43]. However, the N-rGO only demonstrated limited activity toward the 4-NP conversion reaction, and this confirmed the essential role of the nano-Au catalyst. Another control experiment using the Au/rGO filter with an Au/C ratio of 0.2% highlighted the important role of doped N. Under similar experimental conditions, an Au/rGO filter could only partially convert 0.2 mmol/L of 4-NP, whereas an Au/N-rGO filter could completely convert 4-NP to 4-AP by a single pass (
τ = 1.0 s) through the catalytic filter. After N doping, more Au precursors carrying opposite charges could be adsorbed, and a uniform Au distribution could be expected on the N-rGO surface due to the uniform doping of nitrogen. This could provide more active sites on the catalytic filter. Compared to a batch reactor system, the continuous flow catalysis significantly promoted the catalytic performance even when employing the same amount of Au catalyst. The evidently enhanced catalytic kinetics in the catalytic filter could be ascribed to the convectively-enhanced mass transport during filtration. The high-performance nanoscale catalysts were strongly anchored onto the hosting support, ensuring a long-term stability and catalytic activity. In a conventional batch system, the activity of the nano-Au catalysts were predominantly controlled by diffusion, which greatly limits the mass transport performance and leads to sluggish catalytic kinetics. Alternatively, the catalytic reaction and separation of the catalyst from the solution (including the product) take place simultaneously by employing a catalytic filter design. Thus, the post-separation of catalysts can be avoided and the mass transport in a continuous-flow design can be enhanced convectively using an external peristatic pump.
Some key operational parameters were further optimized. First, the nano-Au loading amount was an important parameter in the current catalytic filter system. As shown in
Figure 4c, under the experimental conditions of 1.0 mL/min flow rate, 0.2 mmol/L 4-NP, and 50 mmol/L NaBH
4, the complete 4-NP conversion was obtained once the Au/C ratio was higher than 0.2%. Only a partial conversion of 4-NP was obtained at a lower Au/C ratio of 0.1%, indicating that sufficient active sites were available for the catalytic conversion of 4-NP. These results were expected, as the Au nanoparticles in the filter provided active sites for the reaction. In general, a higher Au loading amount could produce more catalysts for the system. Furthermore, increasing the 4-NP concentration could also lead to an incomplete 4-NP conversion. For example, at an Au/C ratio of 2.0%, only an incomplete conversion was observed at a 4-NP concentration of 1.5 mmol/L. However, at an Au/C ratio of 5.0%, a complete 4-NP conversion was only be obtained at 0.2 mmol/L of 4-NP. The effect of the 4-NP concentration could be explained by the mass transport limitation in the current catalytic filter system. A higher substrate concentration may lead to insufficient contact between 4-NP and the active sites due to a limited contact time between active sites (i.e., nano-Au catalyst) and substrate (i.e., 4-NP). Furthermore, a proper Au loading was necessary since the catalytic performance at an Au/C ratio of 0.2% was found to be higher than that of 0.5%. This phenomenon could be explained by the size effect of the nano-Au catalyst. As suggested by the FESEM and TEM characterizations of the Au/N-rGO filters with different Au loadings, the nano-Au size increased with the Au precursor concentration, and agglomeration occurred at the higher Au loading (e.g., Au/C ratio of 0.5%). Generally, the catalytic activity of nano-Au was opposite to their particle size. Although the particle size at 0.1% Au/C ratio is the smallest, the limited amount of nano-Au produces insufficient catalytic activity of the catalytic filter. To further verify the stability of the as-prepared catalytic filters, 5 h of continuous filtration of 1.0 mM 4-NP demonstrated a negligible decline in conversion efficiency, indicating an excellent catalytic activity and stability of the catalytic filters (as shown in
Figure S5).
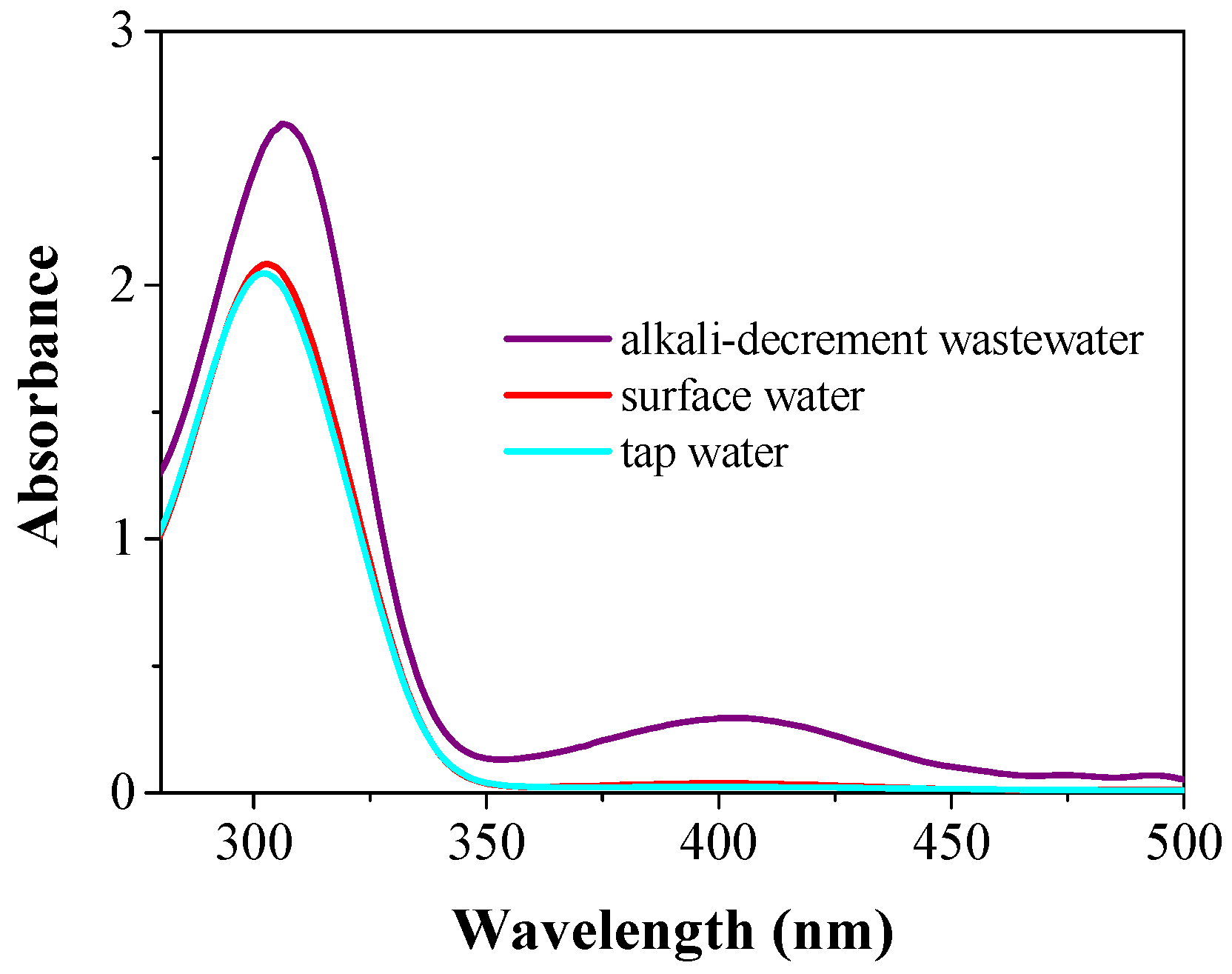
To further evaluate the performance of the catalytic membrane system with respect to real water samples, 1.0 mmol/L 4-NP and 250-fold NaBH
4 (compared with 4-NP) were spiked into real water samples collected from a local tap, a local river, and industrial dyeing wastewater (alkali-decrement wastewater, diluted 1000 times), respectively, before filtration. The influent spectra of the three 4-NP spiked real wastewater samples are available in the
Supplementary Materials (see
Figure S6). As shown in
Figure 5, the catalytic kinetics in all three real water samples displayed a similar behavior to that in DI-H
2O, and a complete 4-NP conversion was achieved in tap water and reservoir water. A >98.3% 4-NP conversion was also achieved under the dyeing wastewater conditions (COD
Cr: 37,668.4 mg/L). It is noteworthy that the three real water samples had totally different compositions and characteristics. These results suggested an excellent catalytic activity in the as-developed catalytic membrane system. In addition, hydrogen may be produced from the reaction of NaBH
4 and H
2O and a large quantity of hydrogen gas may block the filter pores and significantly increase the internal pressure within the filtration device. To slow down the kinetics for hydrogen gas production, the 4-NP solution was stored in ice water before the addition of NaBH
4. The catalytic activity of the filter may also be affected once it is allowed to dry out. The shrinkage of the support materials may deteriorate the catalytic performance, and these engineering issues should be resolved before implementing practical engineering applications.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




