Tunable Electronic Properties of Nitrogen and Sulfur Doped Graphene: Density Functional Theory Approach


 and
and 
Abstract
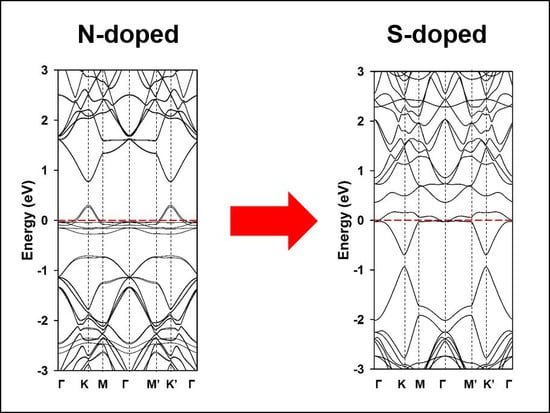
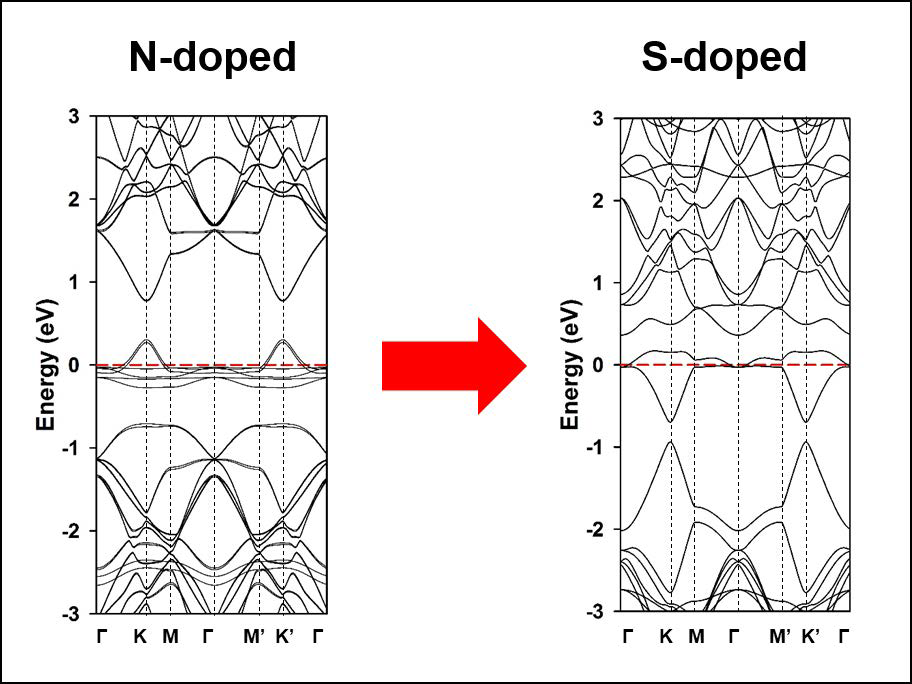
:
1. Introduction
2. Computational Details
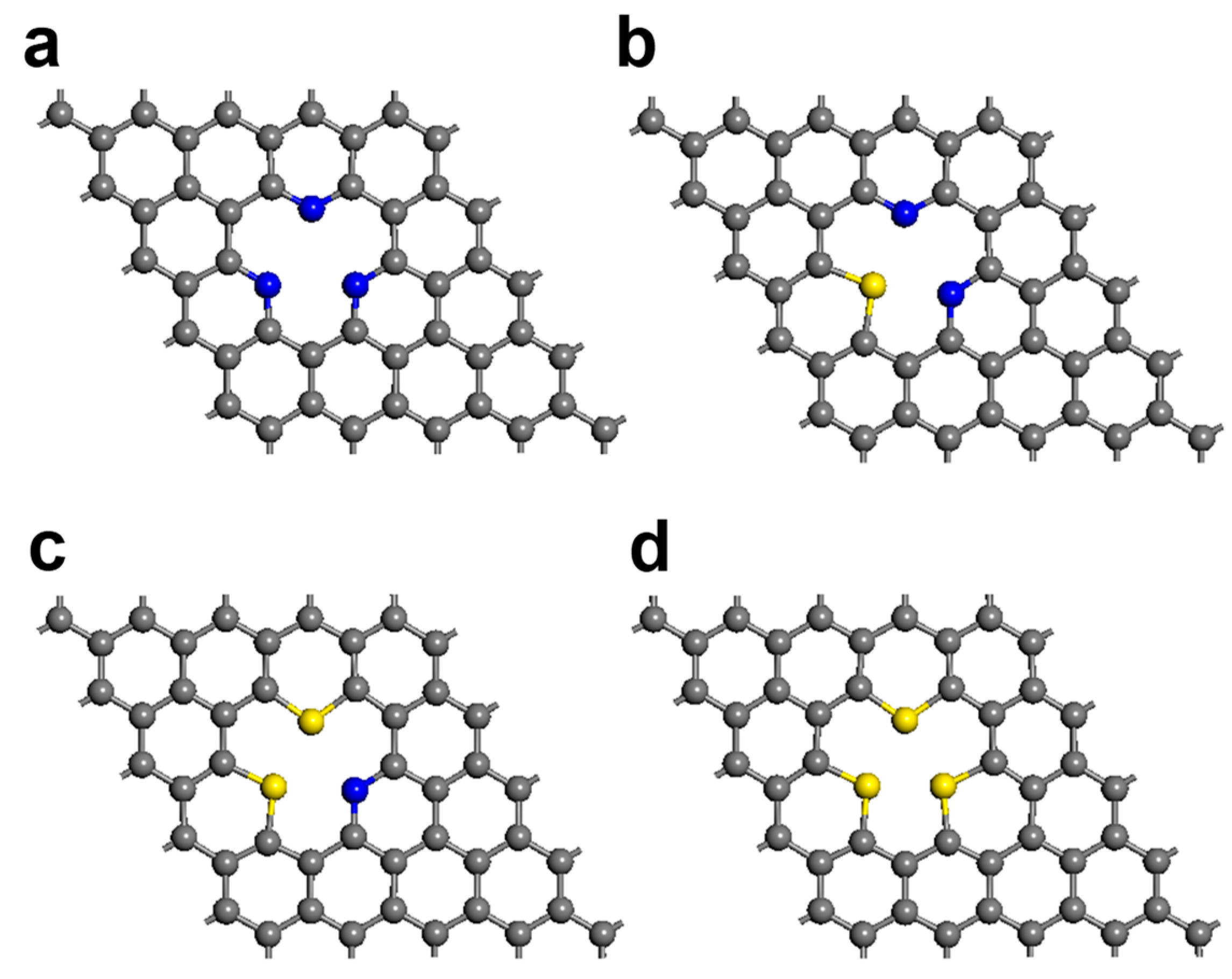
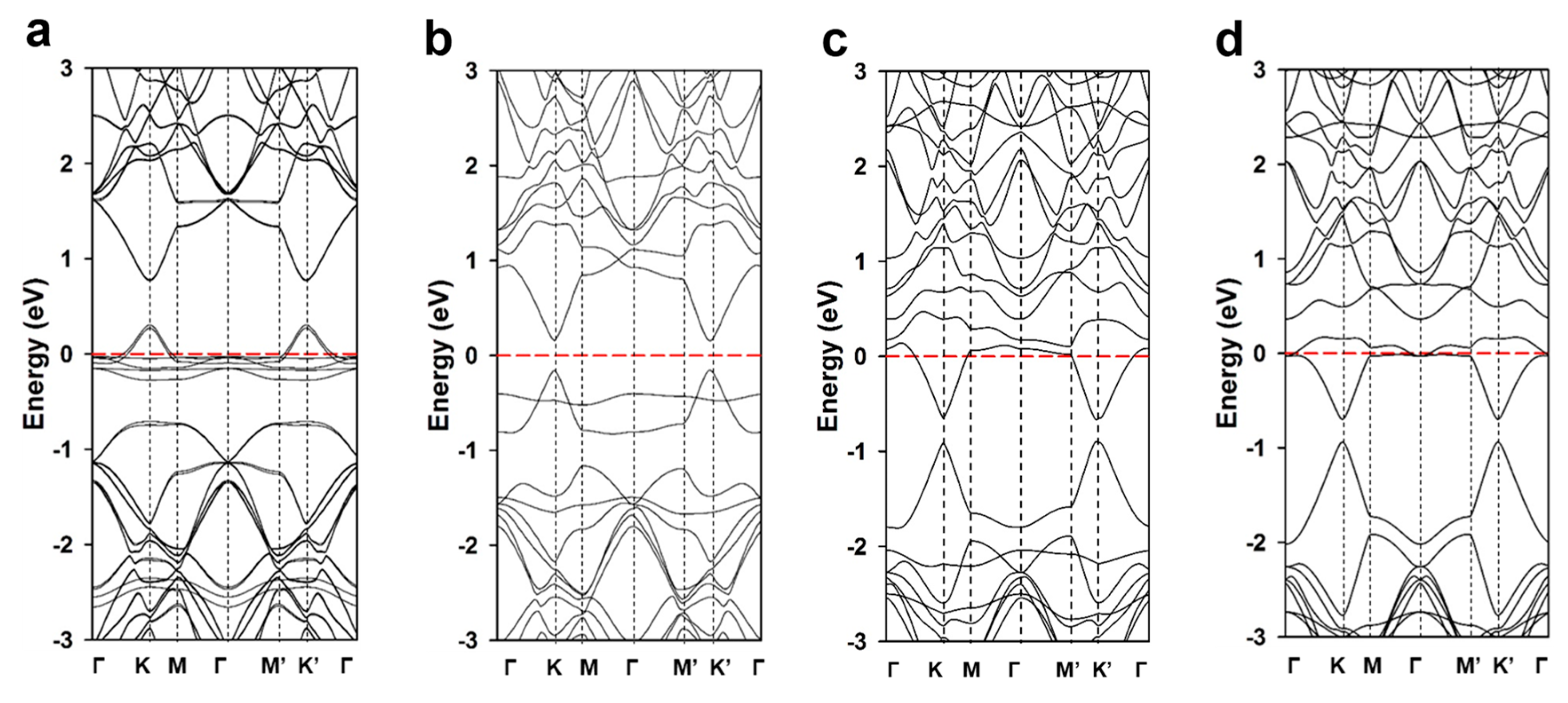
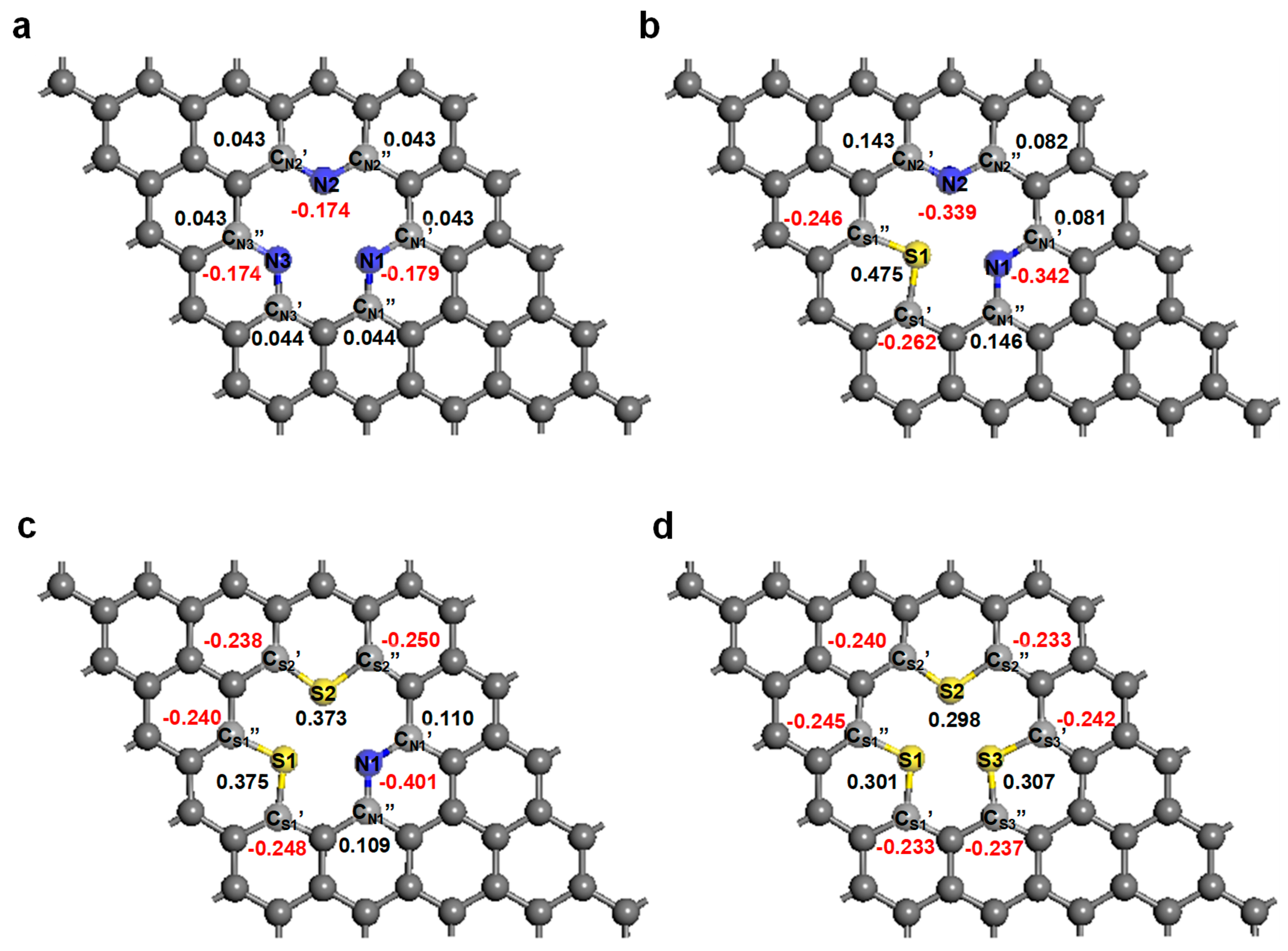
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Geim, A.K.; Novoselov, K.S. The rise of graphene. Nat. Mater. 2007, 6, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Castro Neto, A.H.; Guinea, F.; Peres, N.M.R.; Novoselov, K.S.; Geim, A.K. The electronic properties of graphene. Rev. Modern Phys. 2009, 81, 109–162. [Google Scholar] [CrossRef]
- Geim, A.K. Graphene: Status and Prospects. Science 2009, 324, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Stankovich, S.; Dikin, D.A.; Dommett, G.H.B.; Kohlhaas, K.M.; Zimney, E.J.; Stach, E.A.; Piner, R.D.; Nguyen, S.T.; Ruoff, R.S. Graphene-based composite materials. Nature 2006, 442, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Peigney, A.; Laurent, C.; Flahaut, E.; Bacsa, R.R.; Rousset, A. Specific surface area of carbon nanotubes and bundles of carbon nanotubes. Carbon 2001, 39, 507–514. [Google Scholar] [CrossRef]
- Zhu, Y.W.; Murali, S.; Stoller, M.D.; Ganesh, K.J.; Cai, W.W.; Ferreira, P.J.; Pirkle, A.; Wallace, R.M.; Cychosz, K.A.; Thommes, M.; et al. Carbon-Based Supercapacitors Produced by Activation of Graphene. Science 2011, 332, 1537–1541. [Google Scholar] [CrossRef] [PubMed]
- Bolotin, K.I.; Sikes, K.J.; Jiang, Z.; Klima, M.; Fudenberg, G.; Hone, J.; Kim, P.; Stormer, H.L. Ultrahigh electron mobility in suspended graphene. Solid State Commun. 2008, 146, 351–355. [Google Scholar] [CrossRef]
- Balandin, A.A.; Ghosh, S.; Bao, W.Z.; Calizo, I.; Teweldebrhan, D.; Miao, F.; Lau, C.N. Superior thermal conductivity of single-layer graphene. Nano Lett. 2008, 8, 902–907. [Google Scholar] [CrossRef]
- Lee, C.; Wei, X.D.; Kysar, J.W.; Hone, J. Measurement of the elastic properties and intrinsic strength of monolayer graphene. Science 2008, 321, 385–388. [Google Scholar] [CrossRef]
- Freitag, M. Graphene—Nanoelectronics goes flat out. Nat. Nanotechnol. 2008, 3, 455–457. [Google Scholar] [CrossRef]
- Bonaccorso, F.; Sun, Z.; Hasan, T.; Ferrari, A.C. Graphene photonics and optoelectronics. Nat. Photon. 2010, 4, 611–622. [Google Scholar] [CrossRef]
- Hou, J.B.; Shao, Y.Y.; Ellis, M.W.; Moore, R.B.; Yi, B.L. Graphene-based electrochemical energy conversion and storage: Fuel cells, supercapacitors and lithium ion batteries. Phys. Chem. Chem. Phys. 2011, 13, 15384–15402. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Jung, S.M.; Seo, J.M.; Chang, D.W.; Dai, L.M.; Baek, J.B. Graphene for energy conversion and storage in fuel cells and supercapacitors. Nano Energy 2012, 1, 534–551. [Google Scholar] [CrossRef]
- Bonaccorso, F.; Colombo, L.; Yu, G.H.; Stoller, M.; Tozzini, V.; Ferrari, A.C.; Ruoff, R.S.; Pellegrini, V. Graphene, related two-dimensional crystals, and hybrid systems for energy conversion and storage. Science 2015, 347, 1246501. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.Y.; Wang, J.; Wu, H.; Liu, J.; Aksay, I.A.; Lin, Y.H. Graphene Based Electrochemical Sensors and Biosensors: A Review. Electroanalysis 2010, 22, 1027–1036. [Google Scholar] [CrossRef]
- Huang, C.C.; Li, C.; Shi, G.Q. Graphene based catalysts. Energy Environ. Sci. 2012, 5, 8848–8868. [Google Scholar] [CrossRef]
- Liu, H.T.; Liu, Y.Q.; Zhu, D.B. Chemical doping of graphene. J. Mater. Chem. 2011, 21, 3335–3345. [Google Scholar] [CrossRef]
- Nigar, S.; Zhou, Z.F.; Wang, H.; Imtiaz, M. Modulating the electronic and magnetic properties of graphene. RSC Adv. 2017, 7, 51546–51580. [Google Scholar] [CrossRef]
- Wang, X.W.; Sun, G.Z.; Routh, P.; Kim, D.H.; Huang, W.; Chen, P. Heteroatom-doped graphene materials: Syntheses, properties and applications. Chem. Soc. Rev. 2014, 43, 7067–7098. [Google Scholar] [CrossRef]
- Liu, L.L.; Qing, M.Q.; Wang, Y.B.; Chen, S.M. Defects in Graphene: Generation, Healing, and Their Effects on the Properties of Graphene: A Review. J. Mater. Sci. Technol. 2015, 31, 599–606. [Google Scholar] [CrossRef]
- Banhart, F.; Kotakoski, J.; Krasheninnikov, A.V. Structural Defects in Graphene. ACS Nano 2011, 5, 26–41. [Google Scholar] [CrossRef] [PubMed]
- Schiros, T.; Nordlund, D.; Palova, L.; Prezzi, D.; Zhao, L.Y.; Kim, K.S.; Wurstbauer, U.; Gutierrez, C.; Delongchamp, D.; Jaye, C.; et al. Connecting Dopant Bond Type with Electronic Structure in N-Doped Graphene. Nano Lett. 2012, 12, 4025–4031. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, Y.; Saito, S. Formation, stabilities, and electronic properties of nitrogen defects in graphene. Phys. Rev. B 2011, 84, 245446. [Google Scholar] [CrossRef]
- Hou, Z.F.; Wang, X.L.; Ikeda, T.; Terakura, K.; Oshima, M.; Kakimoto, M. Electronic structure of N-doped graphene with native point defects. Phys. Rev. B 2013, 87, 165401. [Google Scholar] [CrossRef]
- Wang, T.; Wang, L.X.; Wu, D.L.; Xia, W.; Jia, D.Z. Interaction between Nitrogen and Sulfur in Co-Doped Graphene and Synergetic Effect in Supercapacitor. Sci. Rep. 2015, 5, 9591. [Google Scholar] [CrossRef]
- Xu, J.X.; Dong, G.F.; Jin, C.H.; Huang, M.H.; Guan, L.H. Sulfur and Nitrogen Co-Doped, Few-Layered Graphene Oxide as a Highly Efficient Electrocatalyst for the Oxygen-Reduction Reaction. ChemSusChem 2013, 6, 493–499. [Google Scholar] [CrossRef]
- Wu, Z.S.; Winter, A.; Chen, L.; Sun, Y.; Turchanin, A.; Feng, X.L.; Mullen, K. Three-Dimensional Nitrogen and Boron Co-doped Graphene for High-Performance All-Solid-State Supercapacitors. Adv. Mater. 2012, 24, 5130–5135. [Google Scholar] [CrossRef]
- Panchokarla, L.S.; Subrahmanyam, K.S.; Saha, S.K.; Govindaraj, A.; Krishnamurthy, H.R.; Waghmare, U.V.; Rao, C.N.R. Synthesis, Structure, and Properties of Boron- and Nitrogen-Doped Graphene. Adv. Mater. 2009, 21, 4726–4730. [Google Scholar] [CrossRef]
- Reddy, A.L.M.; Srivastava, A.; Gowda, S.R.; Gullapalli, H.; Dubey, M.; Ajayan, P.M. Synthesis Of Nitrogen-Doped Graphene Films For Lithium Battery Application. ACS Nano 2010, 4, 6337–6342. [Google Scholar] [CrossRef]
- Zhang, C.Z.; Mahmood, N.; Yin, H.; Liu, F.; Hou, Y.L. Synthesis of Phosphorus-Doped Graphene and its Multifunctional Applications for Oxygen Reduction Reaction and Lithium Ion Batteries. Adv. Mater. 2013, 25, 4932–4937. [Google Scholar] [CrossRef]
- Choi, C.H.; Chung, M.W.; Kwon, H.C.; Park, S.H.; Woo, S.I. B, N- and P, N-doped graphene as highly active catalysts for oxygen reduction reactions in acidic media. J. Mater. Chem. A 2013, 1, 3694–3699. [Google Scholar] [CrossRef]
- Ma, X.L.; Ning, G.Q.; Qi, C.L.; Xu, C.G.; Gao, J.S. Phosphorus and Nitrogen Dual-Doped Few-Layered Porous Graphene: A High-Performance Anode Material for Lithium-Ion Batteries. ACS Appl. Mater. Interf. 2014, 6, 14415–14422. [Google Scholar] [CrossRef] [PubMed]
- Paraknowitsch, J.P.; Thomas, A. Doping carbons beyond nitrogen: An overview of advanced heteroatom doped carbons with boron, sulphur and phosphorus for energy applications. Energy Environ. Sci. 2013, 6, 2839–2855. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmuller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comp. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Lee, J.H.; Kang, S.G.; Moon, H.S.; Park, H.; Kim, I.T.; Lee, S.G. Adsorption mechanisms of lithium oxides (LixO2) on a graphene-based electrode: A density functional theory approach. App. Surf. Sci. 2015, 351, 193–202. [Google Scholar] [CrossRef]
- Pham, N.N.T.; Park, J.S.; Kim, H.-T.; Kim, H.-J.; Son, Y.-A.; Kang, S.G.; Lee, S.G. Catalytic performance of graphene quantum dot supported manganese phthalocyanine for highly efficient oxygen reduction: A DFT+U approach. New J. Chem. 2019, 43, 348–355. [Google Scholar] [CrossRef]
- Lee, H.W.; Moon, H.S.; Hur, J.; Kim, I.T.; Park, M.S.; Yun, J.M.; Kim, K.H.; Lee, S.G. Mechanism of sodium adsorption on N-doped graphene nanoribbons for sodium ion battery applications: A density functional theory approach. Carbon 2017, 119, 492–501. [Google Scholar] [CrossRef]
- Hwang, D.G.; Jeong, E.; Lee, S.G. Density functional theory study of CH4 and CO2 adsorption by fluorinated graphene. Carbon Lett. 2016, 20, 81–85. [Google Scholar] [CrossRef]
- Moon, H.S.; Yun, J.M.; Kim, K.H.; Jang, S.S.; Lee, S.G. Investigations of the band structures of edge-defect zigzag graphene nanoribbons using density functional theory. RSC Adv. 2016, 6, 39587–39594. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, Z.X.; Qiu, J.Y.C.; Lee, H.W. Design and synthesis of nitrogen and sulfur co-doped porous carbon via two-dimensional interlayer confinement for a high-performance anode material for lithium-ion batteries. J. Mater. Chem. A 2016, 4, 5802–5809. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Materials Studio; BIOVIA: San Diego, CA, USA, 2018.
- Mulliken, R.S. Citation Classic—Electronic Population Analysis on Lcao-Mo Molecular Wave-Functions. Curr. Contents/Eng. Technol. Appl. Sci. 1985, 18. [Google Scholar]
- Wang, H.B.; Maiyalagan, T.; Wang, X. Review on Recent Progress in Nitrogen-Doped Graphene: Synthesis, Characterization, and Its Potential Applications. ACS Catal. 2012, 2, 781–794. [Google Scholar] [CrossRef]
- Wu, J.J.; Ma, L.L.; Yadav, R.M.; Yang, Y.C.; Zhang, X.; Vajtai, R.; Lou, J.; Ajayan, P.M. Nitrogen-Doped Graphene with Pyridinic Dominance as a Highly Active and Stable Electrocatalyst for Oxygen Reduction. ACS Appl. Mater. Interf. 2015, 7, 14763–14769. [Google Scholar] [CrossRef]
- Li, L.J.; Dai, P.C.; Gu, X.; Wang, Y.; Yan, L.T.; Zhao, X.B. High oxygen reduction activity on a metal-organic framework derived carbon combined with high degree of graphitization and pyridinic-N dopants. J. Mater. Chem. A 2017, 5, 789–795. [Google Scholar] [CrossRef]
- Sun, D.F.; Yang, J.; Yan, X.B. Hierarchically porous and nitrogen, sulfur-codoped graphene-like microspheres as a high capacity anode for lithium ion batteries. Chem. Commun. 2015, 51, 2134–2137. [Google Scholar] [CrossRef]
- Wohlgemuth, S.A.; Vilela, F.; Titirici, M.M.; Antonietti, M. A one-pot hydrothermal synthesis of tunable dual heteroatom-doped carbon microspheres. Green Chem. 2012, 14, 741–749. [Google Scholar] [CrossRef]
- Qiu, Z.Z.; Lin, Y.M.; Xin, H.L.; Han, P.; Li, D.Z.; Yang, B.; Li, P.C.; Ullah, S.; Fan, H.S.; Zhu, C.Z.; et al. Ultrahigh level nitrogen/sulfur co-doped carbon as high performance anode materials for lithium-ion batteries. Carbon 2018, 126, 85–92. [Google Scholar] [CrossRef]
- Xiong, J.W.; Pan, Q.C.; Zheng, F.H.; Xiong, X.H.; Yang, C.H.; Hu, D.L.; Huang, C.L. N/S Co-doped Carbon Derived From Cotton as High Performance Anode Materials for Lithium Ion Batteries. Front. Chem. 2018, 6, 78. [Google Scholar] [CrossRef] [PubMed]
- Ruan, J.F.; Yuan, T.; Pang, Y.P.; Luo, S.N.; Peng, C.X.; Yang, J.H.; Zheng, S.Y. Nitrogen and sulfur dual-doped carbon films as flexible free-standing anodes for Li-ion and Na-ion batteries. Carbon 2018, 126, 9–16. [Google Scholar] [CrossRef]
- Wei, T.Y.; Wei, X.L.; Yang, L.W.; Xiao, H.P.; Gao, Y.; Li, H.M. A one-step moderate-explosion assisted carbonization strategy to sulfur and nitrogen dual-doped porous carbon nanosheets derived from camellia petals for energy storage. J. Power Sources 2016, 331, 373–381. [Google Scholar] [CrossRef]
- Cai, D.D.; Wang, C.S.; Shi, C.Y.; Tan, N. Facile synthesis of N and S co-doped graphene sheets as anode materials for high-performance lithium-ion batteries. J. Alloys Compd. 2018, 731, 235–242. [Google Scholar] [CrossRef]
- Ai, W.; Luo, Z.M.; Jiang, J.; Zhu, J.H.; Du, Z.Z.; Fan, Z.X.; Xie, L.H.; Zhang, H.; Huang, W.; Yu, T. Nitrogen and Sulfur Codoped Graphene: Multifunctional Electrode Materials for High-Performance Li-Ion Batteries and Oxygen Reduction Reaction. Adv. Mater. 2014, 26, 6186–6192. [Google Scholar] [CrossRef]
- Xing, L.B.; Xi, K.; Li, Q.Y.; Su, Z.; Lai, C.; Zhao, X.S.; Kumar, R.V. Nitrogen, sulfur-codoped graphene sponge as electroactive carbon interlayer for high-energy and -power lithium-sulfur batteries. J. Power Sources 2016, 303, 22–28. [Google Scholar] [CrossRef]
- Usachov, D.; Vilkov, O.; Gruneis, A.; Haberer, D.; Fedorov, A.; Adamchuk, V.K.; Preobrajenski, A.B.; Dudin, P.; Barinov, A.; Oehzelt, M.; et al. Nitrogen-Doped Graphene: Efficient Growth, Structure, and Electronic Properties. Nano Lett. 2011, 11, 5401–5407. [Google Scholar] [CrossRef]
- Yang, Y.F.; Jin, S.; Zhang, Z.; Du, Z.Z.; Liu, H.R.; Yang, J.; Xu, H.X.; Ji, H.X. Nitrogen-Doped Hollow Carbon Nanospheres for High-Performance Li-Ion Batteries. ACS Appl. Mater. Interf. 2017, 9, 14180–14186. [Google Scholar] [CrossRef]
- Guo, P.P.; Xiao, F.; Liu, Q.; Liu, H.F.; Guo, Y.L.; Gong, J.R.; Wang, S.; Liu, Y.Q. One-Pot Microbial Method to Synthesize Dual-Doped Graphene and Its Use as High-Performance Electrocatalyst. Sci. Rep. 2013, 3, 3499. [Google Scholar] [CrossRef]
- Yang, Z.; Nie, H.G.; Zhou, X.M.; Yao, Z.; Huang, S.M.; Chen, X.H. Investigation of Homologous Series as Precursory Hydrocarbons for Aligned Carbon Nanotube Formation by the Spray Pyrolysis Method. Nano 2011, 6, 205–213. [Google Scholar] [CrossRef]
- Ji, J.Y.; Zhang, G.H.; Chen, H.Y.; Wang, S.L.; Zhang, G.L.; Zhang, F.B.; Fan, X.B. Sulfonated graphene as water-tolerant solid acid catalyst. Chem. Sci. 2011, 2, 484–487. [Google Scholar] [CrossRef]
- Park, J.E.; Jang, Y.J.; Kim, Y.J.; Song, M.S.; Yoon, S.; Kim, D.H.; Kim, S.J. Sulfur-doped graphene as a potential alternative metal-free electrocatalyst and Pt-catalyst supporting material for oxygen reduction reaction. Phys. Chem. Chem. Phys. 2014, 16, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Pauling, L. Citation Classic—The Nature of the Chemical-Bond and the Structure of Molecules and Crystals—An Introduction to Modern Structural Chemistry. Curr. Contents/Phys. Chem. Earth Sci. 1985, 16. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 3N-gra | 2N1S-gra | 1N2S-gra | 3S-gra | |
|---|---|---|---|---|
| Bandgap (eV) | 0.473 | 0.350 | 0.275 | 0.255 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.H.; Kwon, S.H.; Kwon, S.; Cho, M.; Kim, K.H.; Han, T.H.; Lee, S.G. Tunable Electronic Properties of Nitrogen and Sulfur Doped Graphene: Density Functional Theory Approach. Nanomaterials 2019, 9, 268. https://doi.org/10.3390/nano9020268
Lee JH, Kwon SH, Kwon S, Cho M, Kim KH, Han TH, Lee SG. Tunable Electronic Properties of Nitrogen and Sulfur Doped Graphene: Density Functional Theory Approach. Nanomaterials. 2019; 9(2):268. https://doi.org/10.3390/nano9020268
Chicago/Turabian StyleLee, Ji Hye, Sung Hyun Kwon, Soonchul Kwon, Min Cho, Kwang Ho Kim, Tae Hee Han, and Seung Geol Lee. 2019. "Tunable Electronic Properties of Nitrogen and Sulfur Doped Graphene: Density Functional Theory Approach" Nanomaterials 9, no. 2: 268. https://doi.org/10.3390/nano9020268
APA StyleLee, J. H., Kwon, S. H., Kwon, S., Cho, M., Kim, K. H., Han, T. H., & Lee, S. G. (2019). Tunable Electronic Properties of Nitrogen and Sulfur Doped Graphene: Density Functional Theory Approach. Nanomaterials, 9(2), 268. https://doi.org/10.3390/nano9020268