Graphene Nanoplatelet-Reinforced Poly(vinylidene fluoride)/High Density Polyethylene Blend-Based Nanocomposites with Enhanced Thermal and Electrical Properties



Abstract
:
1. Introduction
2. Materials and Methods
3. Results
3.1. Phase Morphology and Selective Localization of GNP
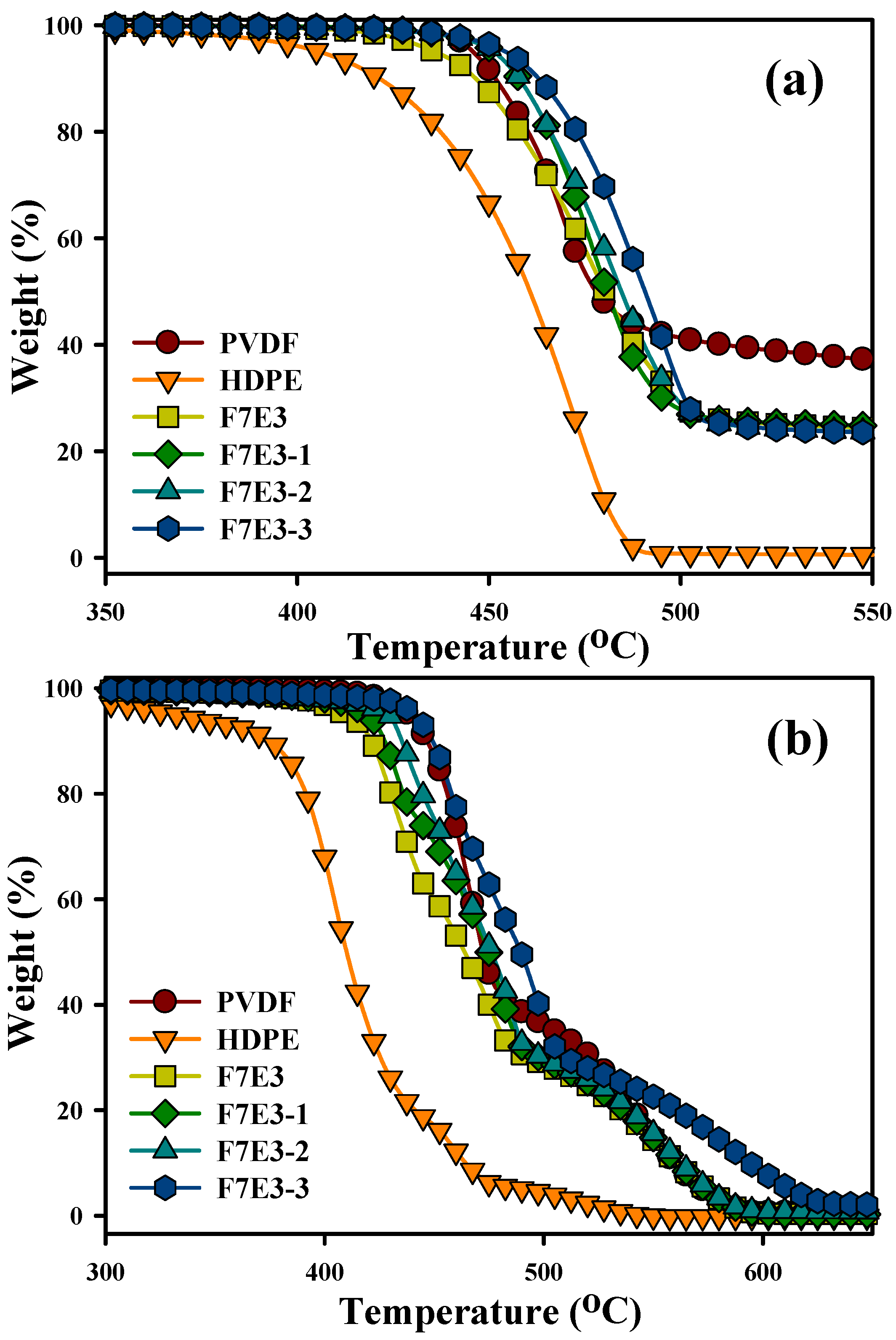
3.2. Thermal Stability
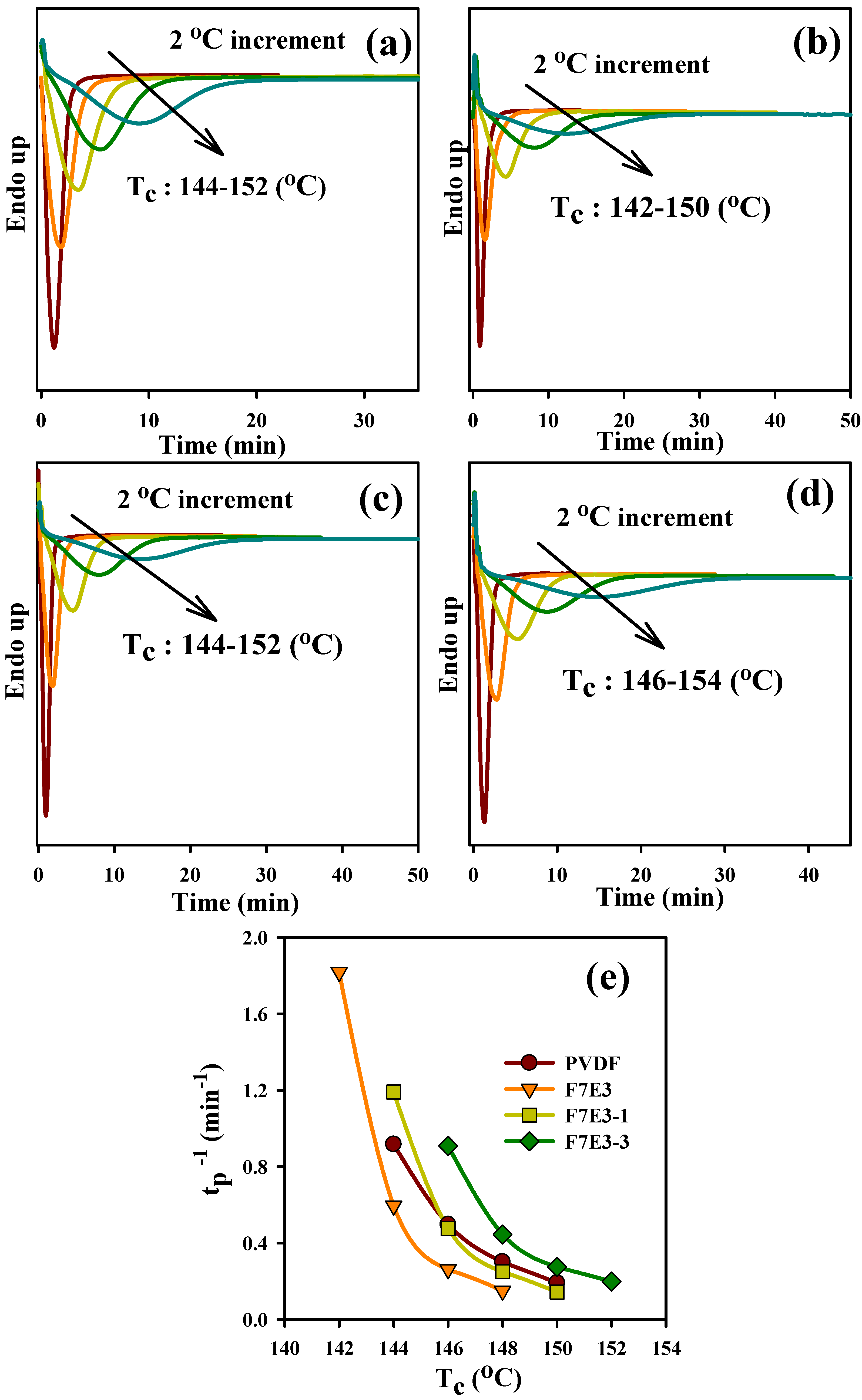
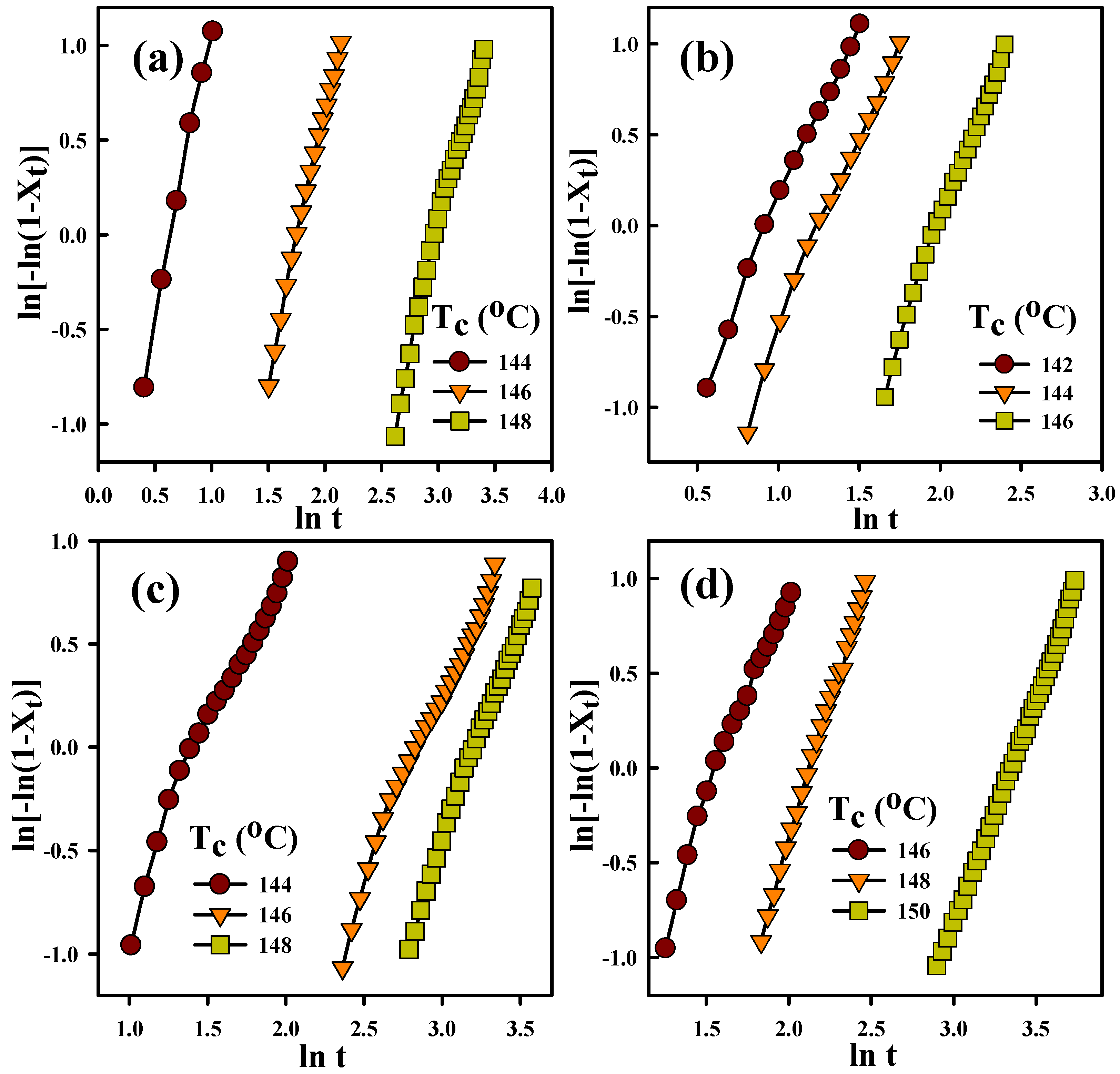
3.3. Crystallization Kinetics
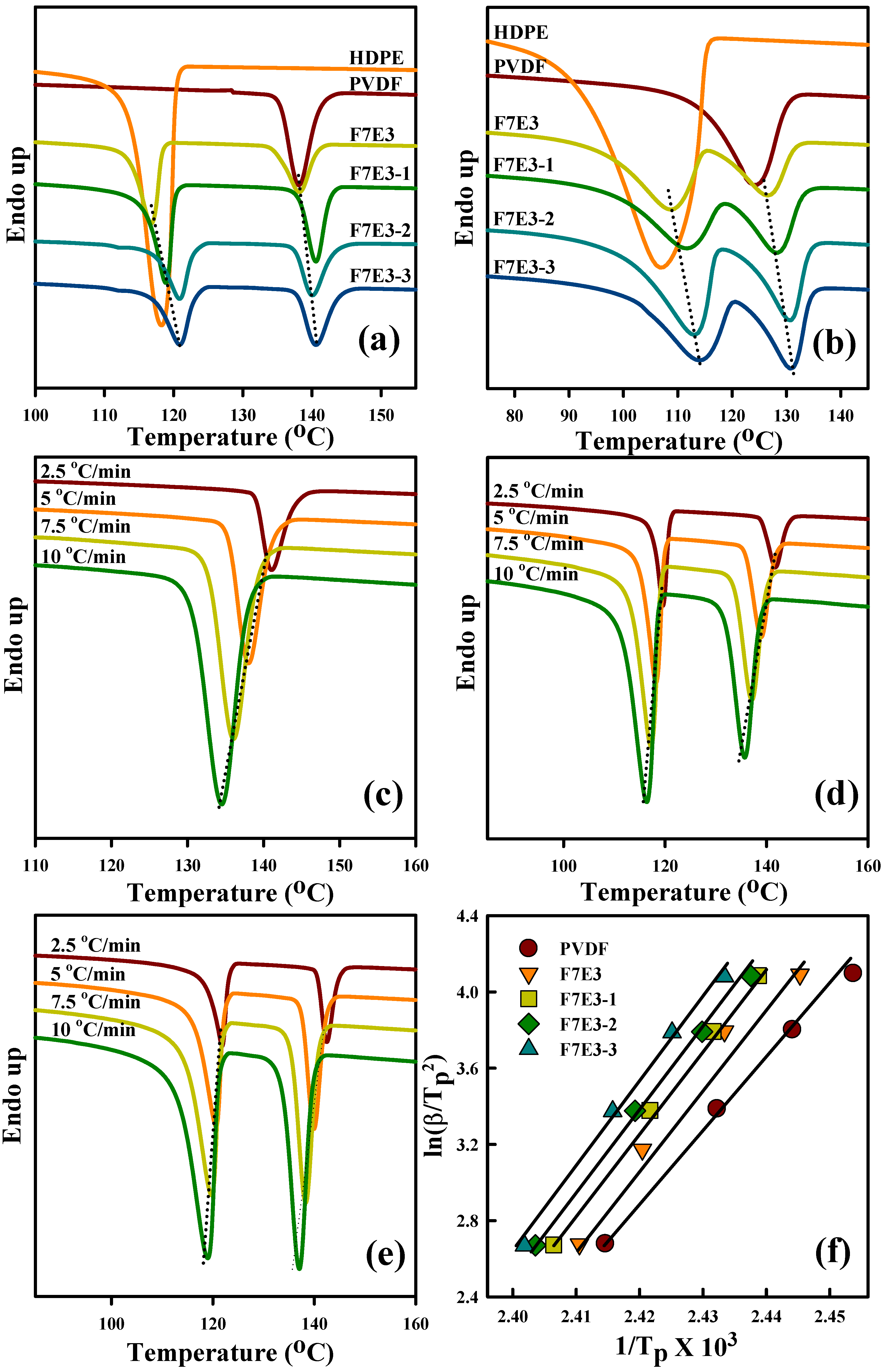
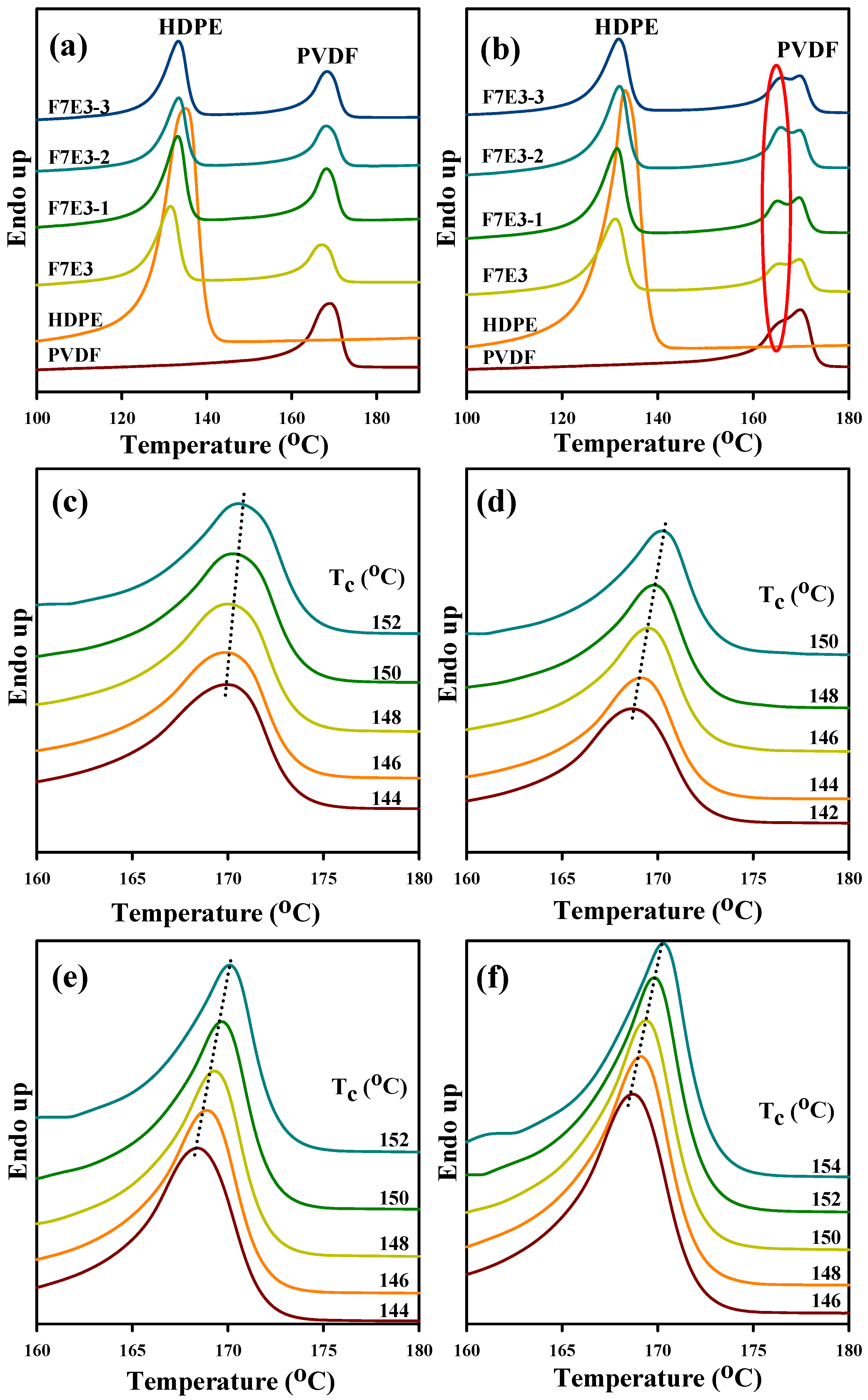
3.4. Melting Behavior
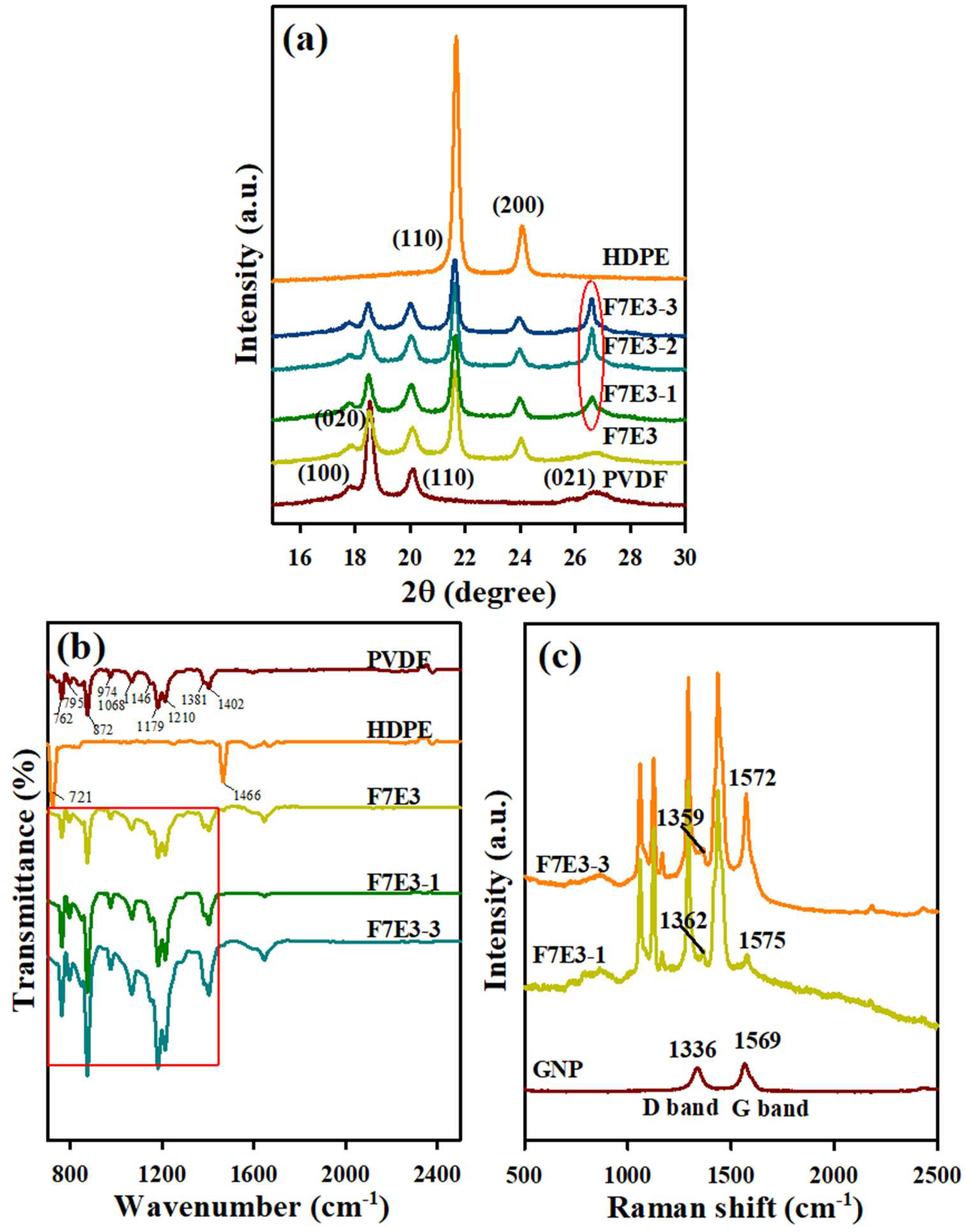
3.5. Crystal Structure, FTIR, and Raman Spectra
3.6. Melt Rheology
3.7. Mechanical Properties
3.8. Electrical Resistivity
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schaefer, D.W.; Justice, R.S. How nano are nanocomposites? Macromolecules 2007, 40, 8501–8517. [Google Scholar] [CrossRef]
- Kotal, M.; Bhowmick, A.K. Polymer nanocomposites from modified clays: Recent advances and challenges. Prog. Polym. Sci. 2015, 51, 127–187. [Google Scholar] [CrossRef]
- Sivanjineyulu, V.; Behera, K.; Chang, Y.H.; Chiu, F.C. Selective localization of carbon nanotube and organoclay in biodegradable poly(butylene succinate)/polylactide blend-based nanocomposites with enhanced rigidity, toughness and electrical conductivity. Compos. Part A Appl. Sci. Manuf. 2018, 114, 30–39. [Google Scholar] [CrossRef]
- Kim, H.S.; Lee, S.K.; Wang, M.; Kang, J.; Sun, Y.; Jung, J.W.; Kim, K.; Kim, S.M.; Nam, J.D.; Suhr, J. Experimental investigation on 3D graphene-CNT hybrid foams with different interactions. Nanomaterials 2018, 8, 694–703. [Google Scholar] [CrossRef] [PubMed]
- Punetha, V.D.; Rana, S.; Yoo, H.J.; Chaurasia, A.; McLeskey, J.T.; Ramasamy, M.S.; Sahoo, N.G.; Cho, J.W. Functionalization of carbon nanomaterials for advanced polymer nanocomposites: A comparison study between CNT and graphene. Prog. Polym. Sci. 2017, 67, 1–47. [Google Scholar] [CrossRef]
- Zhou, S.; Hrymak, A.N.; Kamal, M.R. Effect of hybrid carbon fillers on the electrical and morphological properties of polystyrene nanocomposites in microinjection molding. Nanomaterials 2018, 8, 779–794. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Abdala, A.A.; Macosko, C.W. Graphene/polymer nanocomposites. Macromolecules 2010, 43, 6515–6530. [Google Scholar] [CrossRef]
- Li, B.; Zhong, W.H. Review on polymer/graphite nanoplatelet nanocomposites. J. Mater. Sci. 2011, 46, 5595–5614. [Google Scholar] [CrossRef]
- Huang, G.; Wang, S.; Song, P.a.; Wu, C.; Chen, S.; Wang, X. Combination effect of carbon nanotubes with graphene on intumescent flame-retardant polypropylene nanocomposites. Compos. Part A Appl. Sci. Manuf. 2014, 59, 18–25. [Google Scholar] [CrossRef]
- Verdejo, R.; Bernal, M.M.; Romasanta, L.J.; Lopez-Manchado, M.A. Graphene filled polymer nanocomposites. J. Mater. Chem. 2011, 21, 3301–3310. [Google Scholar] [CrossRef]
- Lee, C.; Wei, X.; Kysar, J.W.; Hone, J. Measurement of the elastic properties and intrinsic strength of monolayer graphene. Science 2008, 321, 385–388. [Google Scholar] [CrossRef] [PubMed]
- El Mohajir, B.E.; Heymans, N. Changes in structural and mechanical behaviour of PVDF with processing and thermomechanical treatments. 1. Change in structure. Polymer 2001, 42, 5661–5667. [Google Scholar] [CrossRef]
- Lovinger, A.J. Crystallization of the β phase of poly(vinylidene fluoride) from the melt. Polymer 1981, 22, 412–413. [Google Scholar] [CrossRef]
- Dillon, D.R.; Tenneti, K.K.; Li, C.Y.; Ko, F.K.; Sics, I.; Hsiao, B.S. On the structure and morphology of polyvinylidene fluoride–nanoclay nanocomposites. Polymer 2006, 47, 1678–1688. [Google Scholar] [CrossRef]
- Wang, J.; Wu, J.; Xu, W.; Zhang, Q.; Fu, Q. Preparation of poly(vinylidene fluoride) films with excellent electric property, improved dielectric property and dominant polar crystalline forms by adding a quaternary phosphorus salt functionalized graphene. Compos. Sci. Technol. 2014, 91, 1–7. [Google Scholar] [CrossRef]
- Wu, C.M.; Chou, M.H.; Zeng, W.Y. Piezoelectric response of aligned electrospun polyvinylidene fluoride/carbon nanotube nanofibrous membranes. Nanomaterials 2018, 8, 420–453. [Google Scholar] [CrossRef] [PubMed]
- Cendrowski, K.; Kukulka, W.; Kedzierski, T.; Zhang, S.; Mijowska, E. Poly(vinylidene fluoride) and carbon derivative structures from eco-friendly MOF-5 for supercapacitor electrode preparation with improved electrochemical performance. Nanomaterials 2018, 8, 890–910. [Google Scholar] [CrossRef] [PubMed]
- Chiu, F.C. Comparisons of phase morphology and physical properties of PVDF nanocomposites filled with organoclay and/or multi-walled carbon nanotubes. Mater. Chem. Phys. 2014, 143, 681–692. [Google Scholar] [CrossRef]
- Liu, Y.L.; Li, Y.; Xu, J.T.; Fan, Z.Q. Cooperative effect of electrospinning and nanoclay on formation of polar crystalline phases in poly(vinylidene fluoride). ACS Appl. Mater. Inter. 2010, 2, 1759–1768. [Google Scholar] [CrossRef] [PubMed]
- Almasri, A.; Ounaies, Z.; Kim, Y.S.; Grunlan, J. Characterization of solution-processed double-walled carbon nanotube/poly(vinylidene fluoride) nanocomposites. Macromol. Mater. Eng. 2008, 293, 123–131. [Google Scholar] [CrossRef]
- Mago, G.; Fisher, F.T.; Kalyon, D.M. Deformation-induced crystallization and associated morphology development of carbon nanotube-PVDF nanocomposites. J. Nanosci. Nanotechnol. 2009, 9, 3330–3340. [Google Scholar] [CrossRef] [PubMed]
- Martins, J.N.; Bassani, T.S.; Barra, G.M.; Oliveira, R.V. Electrical and rheological percolation in poly(vinylidene fluoride)/multi-walled carbon nanotube nanocomposites. Polym. Int. 2011, 60, 430–435. [Google Scholar] [CrossRef]
- Kanagaraj, S.; Varanda, F.R.; Zhil’tsova, T.V.; Oliveira, M.S.A.; Simões, J.A.O. Mechanical properties of high density polyethylene/carbon nanotube composites. Compos. Sci. Technol. 2007, 67, 3071–3077. [Google Scholar] [CrossRef]
- Brandalise, R.N.; Zeni, M.; Martins, J.D.N.; Forte, M.M.C. Morphology, mechanical and dynamic mechanical properties of recycled high density polyethylene and poly(vinyl alcohol) blends. Polym. Bull. 2009, 62, 33–43. [Google Scholar] [CrossRef]
- Wang, C.; Wang, S.; Cheng, H.; Xian, Y.; Zhang, S. Mechanical properties and prediction for nanocalcium carbonate-treated bamboo fiber/high-density polyethylene composites. J. Mater. Sci. 2017, 52, 11482–11495. [Google Scholar] [CrossRef]
- Huang, C.Y.; Roan, M.L.; Kuo, M.C.; Lu, W.L. Effect of compatibiliser on the biodegradation and mechanical properties of high-content starch/low-density polyethylene blends. Polym. Degrad. Stab. 2005, 90, 95–105. [Google Scholar] [CrossRef]
- Rafeie, O.; Razavi Aghjeh, M.K.; Tavakoli, A.; Salami Kalajahi, M.; Jameie Oskooie, A. Conductive poly(vinylidene fluoride)/polyethylene/graphene blend-nanocomposites: Relationship between rheology, morphology, and electrical conductivity. J. Appl. Polym. Sci. 2018, 135, 46333–46346. [Google Scholar] [CrossRef]
- Deng, Y.; Song, X.; Ma, Z.; Zhang, X.; Shu, D.; Nan, J. Al2O3/PVDF-HFP-CMC/PE separator prepared using aqueous slurry and post-hot-pressing method for polymer lithium-ion batteries with enhanced safety. Electrochim. Acta 2016, 212, 416–425. [Google Scholar] [CrossRef]
- Shao, Y.; Yang, Z.X.; Deng, B.W.; Yin, B.; Yang, M.B. Tuning PVDF/PS/HDPE polymer blends to tri-continuous morphology by grafted copolymers as the compatibilizers. Polymer 2018, 140, 188–197. [Google Scholar] [CrossRef]
- Chiu, F.C. Poly(vinylidene fluoride)/polycarbonate blend-based nanocomposites with enhanced rigidity—Selective localization of carbon nanofillers and organoclay. Polym. Test. 2017, 62, 115–123. [Google Scholar] [CrossRef]
- Chiu, F.C.; Chen, Y.J. Evaluation of thermal, mechanical, and electrical properties of PVDF/GNP binary and PVDF/PMMA/GNP ternary nanocomposites. Compos. Part A Appl. Sci. Manuf. 2015, 68, 62–71. [Google Scholar] [CrossRef]
- Sumita, M.; Sakata, K.; Asai, S.; Miyasaka, K.; Nakagawa, H. Dispersion of fillers and the electrical conductivity of polymer blends filled with carbon black. Polym. Bullet. 1991, 25, 265–271. [Google Scholar] [CrossRef]
- Wu, S. Polymer Interface and Adhesion; Marcel Dekker Inc.: New York, NY, USA, 1982. [Google Scholar]
- Park, M.; Gu, G.Y.; Wang, Z.J.; Kwon, D.J.; Lawrence, K.; Vries, D. Interfacial durability and acoustical properties of transparent graphene nano platelets/poly(vinylidene fluoride) composite actuators. Thin Solid Films 2013, 539, 350–355. [Google Scholar]
- Yu, J.; Huang, X.; Wu, C.; Jiang, P. Permittivity, thermal conductivity and thermal stability of poly(vinylidene fluoride)/graphene nanocomposites. IEEE Trans. Dielectr. Electr. Insul. 2011, 18, 478–484. [Google Scholar]
- Kissinger, H.E. Variation of peak temperature with heating rate in differential thermal analysis. J. Res. Natl. Bur. Stand. 1956, 17, 217–221. [Google Scholar] [CrossRef]
- Behera, K.; Sivanjineyulu, V.; Chang, Y.H.; Chiu, F.C. Thermal properties, phase morphology and stability of biodegradable PLA/PBSL/HAp composites. Polym. Degrad. Stabil. 2018, 154, 248–260. [Google Scholar] [CrossRef]
- Behera, K.; Chang, Y.H.; Chiu, F.C.; Yang, J.C. Characterization of poly(lactic acid)s with reduced molecular weight fabricated through an autoclave process. Polym. Test. 2017, 60, 132–139. [Google Scholar] [CrossRef]
- Avrami, M. Kinetics of phase change. I general theory. J. Chem. Phys. 1939, 7, 1103–1112. [Google Scholar] [CrossRef]
- Martins, P.; Lopes, A.C.; Lanceros-Mendez, S. Electroactive phases of poly(vinylidene fluoride): Determination, processing and applications. Prog. Polym. Sci. 2014, 39, 683–706. [Google Scholar] [CrossRef]
- Priya, L.; Jog, J.P. Polymorphism in intercalated poly(vinylidene fluoride)/clay nanocomposites. J. Appl. Polym. Sci. 2003, 89, 2036–2040. [Google Scholar] [CrossRef]
- Kuila, T.; Bose, S.; Mishra, A.K.; Khanra, P.; Kim, N.H.; Lee, J.H. Effect of functionalized graphene on the physical properties of linear low density polyethylene nanocomposites. Polym. Test. 2012, 31, 31–38. [Google Scholar] [CrossRef]
- Yang, J.; Feng, C.; Dai, J.; Zhang, N.; Huang, T.; Wang, Y. Compatibilization of immiscible nylon 6/poly(vinylidene fluoride) blends using graphene oxides. Polym. Int. 2013, 62, 1085–1093. [Google Scholar] [CrossRef]
- Vasileiou, A.A.; Kontopoulou, M.; Docoslis, A. A noncovalent compatibilization approach to improve the filler dispersion and properties of polyethylene/graphene composites. ACS Appl. Mater. Inter. 2014, 6, 1916–1925. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | γ (mN m−1) | γd (mN m−1) | γp (mN m−1) | References |
|---|---|---|---|---|
| PVDF | 38.0 | 32.6 | 5.4 | [27] |
| HDPE | 25.9 | 25.9 | 0 | [33] |
| GNP | 52.6 | 47.7 | 4.9 | [34] |
| Samples | γPVDF-GNP | γHDPE-GNP | γPVDF-HDPE | ω (GNP) |
|---|---|---|---|---|
| Harmonic-mean Equation (2) | 2.90 | 11.35 | 6.17 | −1.37 |
| Geometric-mean Equation (3) | 1.45 | 8.20 | 5.78 | −1.17 |
| Samples | Properties | ||||||
|---|---|---|---|---|---|---|---|
| (Td10) a (°C) | (Td50) a (°C) | (Td10) b (°C) | (Td50) b (°C) | (Tp) c (°C) | (Tm) d (°C) | ∆Ec (kJ/mol) | |
| PVDF | 451 | 476 | 446 | 471 | 138.1 | 169.2 | −303 |
| HDPE | 422 | 461 | 376 | 411 | 118.2 | 135.0 | −153 |
| F7E3 | 447 | 480 | 421 | 464 | 138.2 | 167.7 | −326 |
| F7E3-1 | 457 | 481 | 427 | 475 | 140.6 | 167.9 | −330 |
| F7E3-2 | 458 | 484 | 435 | 476 | 140.1 | 168.4 | −363 |
| F7E3-3 | 463 | 491 | 449 | 489 | 140.7 | 168.5 | −374 |
| Samples | Properties | |||
|---|---|---|---|---|
| Young’s Modulus (σ) (MPa) | Flexural Modulus (σ) (MPa) | Impact Strength (σ) (J/m) | Electrical Resistivity (Ω-cm) | |
| PVDF | 1216 (90) | 927 (28) | 17.9 (2.10) | 1.03 × 1011 |
| HDPE | 665 (41) | 744 (36) | 10.5 (0.98) | 3.87 × 1011 |
| F7E3 | 575 (66) | 815 (29) | 6.4 (0.94) | 1.95 × 1011 |
| F7E3-1 | 672 (34) | 921 (18) | 7.8 (0.73) | 3.85 × 1010 |
| F7E3-2 | 697 (29) | 1010 (25) | 9.9 (0.45) | 2.32 × 109 |
| F7E3-3 | 654 (41) | 937 (34) | 8.5 (0.52) | 1.79 × 108 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Behera, K.; Yadav, M.; Chiu, F.-C.; Rhee, K.Y. Graphene Nanoplatelet-Reinforced Poly(vinylidene fluoride)/High Density Polyethylene Blend-Based Nanocomposites with Enhanced Thermal and Electrical Properties. Nanomaterials 2019, 9, 361. https://doi.org/10.3390/nano9030361
Behera K, Yadav M, Chiu F-C, Rhee KY. Graphene Nanoplatelet-Reinforced Poly(vinylidene fluoride)/High Density Polyethylene Blend-Based Nanocomposites with Enhanced Thermal and Electrical Properties. Nanomaterials. 2019; 9(3):361. https://doi.org/10.3390/nano9030361
Chicago/Turabian StyleBehera, Kartik, Mithilesh Yadav, Fang-Chyou Chiu, and Kyong Yop Rhee. 2019. "Graphene Nanoplatelet-Reinforced Poly(vinylidene fluoride)/High Density Polyethylene Blend-Based Nanocomposites with Enhanced Thermal and Electrical Properties" Nanomaterials 9, no. 3: 361. https://doi.org/10.3390/nano9030361
APA StyleBehera, K., Yadav, M., Chiu, F. -C., & Rhee, K. Y. (2019). Graphene Nanoplatelet-Reinforced Poly(vinylidene fluoride)/High Density Polyethylene Blend-Based Nanocomposites with Enhanced Thermal and Electrical Properties. Nanomaterials, 9(3), 361. https://doi.org/10.3390/nano9030361