Interaction in Li@Fullerenes and Li+@Fullerenes: First Principle Insights to Li-Based Endohedral Fullerenes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Model and Computational Methods
3. Results
3.1. Energy and Stability
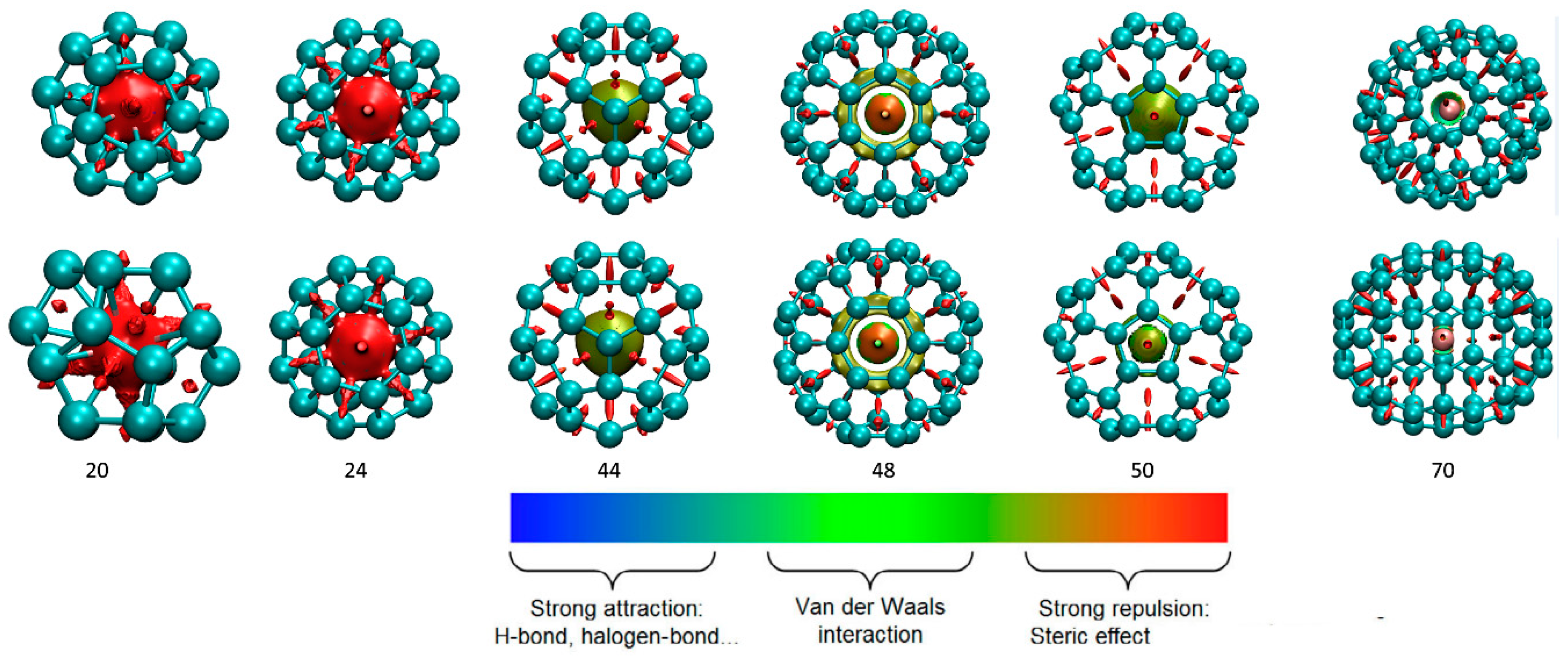
3.2. Interaction Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Yue, C.; Yu, Y.; Wu, Z.; Sun, S.; He, X.; Li, J. High stability induced by the tin/ti interlayer in three-dimensional si/ge nanorod arrays as anode in micro lithium ion battery. ACS Appl. Mater. Interfaces 2016, 8, 7806–7810. [Google Scholar] [CrossRef] [PubMed]
- Armand, M.B.; Whittingham, M.S.; Huggin, R.A. The iron cyanide bronzes. Mater. Res. Bull. 1972, 7, 101–108. [Google Scholar] [CrossRef]
- Li, W.; Dolocan, A.; Oh, P.; Celio, H.; Park, S.; Cho, J. Dynamic behaviour of interphases and its implication on high-energy-density cathode materials in lithium-ion batteries. Nat. Commun. 2017, 8, 14589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruce, P.; Scrosati, B.; Tarascon, J.M. Nanomaterials for rechargeable lithium batteries. Angew. Chem. Int. Ed. 2008, 47, 2930–2946. [Google Scholar] [CrossRef] [PubMed]
- Yamaki, J.I.; Tobishima, S.I.; Sakurai, Y.; Saito, K.I.; Hayashi, K. Safety evaluation of rechargeable cells with lithium metal anodes and amorphous V2O5 cathodes. J. Appl. Electrochem. 1998, 28, 135–140. [Google Scholar] [CrossRef]
- Abada, S.; Marlair, G.; Lecocq, A.; Petit, M.; Sauvant-Moynot, V.; Huet, F. Safety focused modeling of lithium-ion batteries: A review. J. Power Sour. 2016, 306, 178–192. [Google Scholar] [CrossRef] [Green Version]
- Croce, F.; Appetecchi, G.B.; Persi, L.; Scrosati, B. Nanocomposite polymer electrolytes for lithium batteries. Nature 1998, 394, 456. [Google Scholar] [CrossRef]
- Whittingham, M.S. Lithium Batteries and Cathode Materials. Chem. Rev. 2004, 104, 4271–4302. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Liu, J.; Gong, Y.; Wilkinson, D.P.; Zhang, J. Recent advances in all-solid-state rechargeable lithium batteries. Nano Energy 2017, 33, 363–386. [Google Scholar] [CrossRef]
- Li, J.; Ma, C.; Chi, M.; Liang, C.; Dudney, N.J. Solid electrolyte: The key for high-voltage lithium batteries. Adv. Energy Mater. 2015, 5. [Google Scholar] [CrossRef]
- Kroto, H.W.; Heath, J.R.; Obrien, S.C.; Curl, R.F.; Smalley, R.E. C60: Buckminsterfullerene. Nature 1985, 318, 162–163. [Google Scholar] [CrossRef]
- Huang, Y.; Bai, H. Theoretical study on the peanut-shaped dimers and nanotubes consisted of C50 cages. J. Nanosci. Nanotechnol. 2011, 11, 11104. [Google Scholar]
- Li, Z.; Liu, Z.; Sun, H.; Gao, C. Superstructured Assembly of Nanocarbons: Fullerenes, Nanotubes, and Graphene. Chem. Rev. 2015, 115, 7046–7117. [Google Scholar] [CrossRef]
- Bai, H.; Qiao, W.; Zhu, Y.; Huang, Y. Theoretical study on one-dimensional C50 polymers. Diam. Relat. Mater. 2012, 26, 20–24. [Google Scholar] [CrossRef]
- Fang, Y.; Bi, C.; Wang, D.; Huang, J. The functions of fullerenes in hybrid perovskite solar cells. ACS Energy Lett. 2017, 2, 782–794. [Google Scholar] [CrossRef]
- Hashikawa, Y.; Murata, M.; Wakamiya, A.; Murata, Y. Synthesis and Properties of Endohedral Aza[60] fullerenes: H2O@C59N and H2@C59N as Their Dimers and Monomers. J. Am. Chem. Soc. 2016, 138, 4096–4104. [Google Scholar] [CrossRef] [PubMed]
- Zlatko, B.; Vojtech, V.; Daniel, N.; Peter, M.F. Effects of symmetry breaking on the translation-rotation eigenstates of H2, HF, and H2O inside the fullerene C60. Faraday Discuss. 2018, 212, 547–567. [Google Scholar]
- Wang, L.; Ye, J.T.; Wang, H.Q.; Xie, H.M.; Qiu, Y.Q. Third-order nonlinear optical properties of endohedral fullerene (H2)2@C70 and (H2O)2@C70 accompanied by the prospective of novel (HF)2@C70. J. Phys. Chem. 2018, 122, 6835–6845. [Google Scholar] [CrossRef]
- EL-Barbary, A.A. Potential energy of H2 inside the C116 fullerene dimerization: An atomic analysis. J. Mol. Struct. 2016, 1112, 9–13. [Google Scholar] [CrossRef]
- Chen, C.S.; Kuo, T.S.; Yeh, W.Y. Encapsulation of formaldehyde and hydrogen cyanide in an open-cage fullerene. Chem. Eur. J. 2016, 22, 8773–8776. [Google Scholar] [CrossRef]
- Junghans, K.; Ghiassi, K.B.; Samoylova, N.A.; Deng, Q.; Rosenkranz, M.; Olmstead, M.; Balch, A.; Popov, A. Synthesis and isolation of the titanium–scandium endohedral fullerenes—Sc2TiC@Ih-C80, Sc2TiC@D5h-C80 and Sc2TiC2@Ih-C80: Metal size tuning of the tiiv/tiiii redox potentials. Chem. Eur. J. 2016, 22, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Churilov, G.N.; Popov, A.A.; Vnukova, N.G.; Dudnil, A.I.; Glushchenko, G.A.; Samoylova, N.A.; Dubinina, I.A.; Gulyaeva, U.E. A method and apparatus for high-throughput controlled synthesis of fullerenes and endohedral metal fullerenes. Tech. Phys. Lett. 2016, 42, 475–477. [Google Scholar] [CrossRef]
- Aoyagi, S.; Nishibori, E.; Sawa, H.; Sugimoto, K.; Takata, M.; Miyata, Y.; Kitaure, R.; Shinohara, H.; Okada, H.; Sakai, T.; et al. A layered ionic crystal of polar Li@C60 superatoms. Nat. Chem. 2010, 2, 678–683. [Google Scholar] [CrossRef] [PubMed]
- Pavanello, M.; Jalbout, A.F.; Trzaskowski, B.; Adamowicz, L. Fullerene as an electron buffer: Charge transfer in Li@C60. Chem. Phys. Lett. 2007, 442, 339–343. [Google Scholar] [CrossRef]
- Srivastava, A.K.; Pandey, S.K.; Misra, N. Prediction of superalkali@C60 endofullerenes, their enhanced stability and interesting properties. Chem. Phys. Lett. 2016, 655, 71–75. [Google Scholar] [CrossRef]
- Noguchi, Y.; Sugino, O.; Okada, H.; Matsuo, Y. First-principles investigation on structural and optical properties of M+@C60 (Where M = H, Li, Na, and K). J. Phys. Chem. 2013, 117, 15362–15368. [Google Scholar] [CrossRef]
- Etindele, A.J.; Maezono, R.; Melono, R.M.; Motapon, O. Influence of endohedral confinement of atoms on structural and dynamical properties of the C60 fullerene. Chem. Phys. Lett. 2017, 685, 395–400. [Google Scholar] [CrossRef]
- Debnath, T.; Saha, J.K.; Banu, T.; Ash, T.; Das, A.K. Structural and thermodynamic aspects of Lin@Cxendohedral metallofullerenes: A DFT approach. Theor. Chem. Acc. 2016, 135, 167. [Google Scholar] [CrossRef]
- Fowler, P.W.; Manolopoulos, D.E. An Atlas of Fullerenes; Clarendon Press: Oxford, UK, 1995. [Google Scholar]
- Bai, H. New solution method of pi-orbital axis vector and its applications in fullerenes and carbon nanotubes. Chin. J. Struct. Chem. 2013, 32, 695. [Google Scholar]
- Zhang, B.L.; Wang, C.Z.; Ho, K.M.; Xu, C.H.; Chan, C.T. The geometry of small fullerene cages: C20 to C70. J. Chem. Phys. 1992, 97, 5007–5011. [Google Scholar] [CrossRef]
- Han, S.S.; Van Duin, A.C.T.; Goddard, W.A.; Lee, H.M. Optimization and application of lithium parameters for the reactive force field, ReaxFF. J. Phys. Chem. 2005, 109, 4575–4582. [Google Scholar] [CrossRef] [PubMed]
- Van Duin, A.C.T.; Dasgupta, S.; Lorant, F.; Goddard, W.A. ReaxFF: A reactive force field for hydrocarbons. J. Phys. Chem. 2001, 105, 9396–9409. [Google Scholar] [CrossRef]
- Velde, G.T.; Bickelhaupt, F.M.; Baerends, E.J.; Guerra, C.F.; Gisbergen, S.J.A.V.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- Baerends, E.J.; Ziegler, T.; Autschbach, J.; Bashford, D.; Bérces, A.; Bickelhaupt, F.M.; Bo, C.; Boerrigter, P.M.; Cavallo, L.; Chong, D.P.; et al. ADF (2017), SCM, Theoretical Chemistry; Vrije Universiteit: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Zhao, Y.; Schultz, N.E.; Truhlar, D.G. Design of density functionals by combining the method of constraint satisfaction with parametrization for thermochemistry, thermochemical kinetics, and noncovalent interactions. J. Chem. Theory Comput. 2006, 2, 364–382. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Hansen, A.; Brandenburg, J.G.; Bannwarth, C. Dispersion-Corrected Mean-Field Electronic Structure Methods. Chem. Rev. 2016, 116, 5105. [Google Scholar] [CrossRef] [PubMed]
- Goerigk, L.; Hansen, A.; Bauer, C.; Ehrlich, S.; Najibi, A.; Grimme, S. A look at the density functional theory zoo with the advanced GMTKN55 database for general main group thermochemistry, kinetics and noncovalent interactions. Phys. Chem. Chem. Phys. 2017, 19, 32184. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheese, M.J.R.; Montgomery, J.A.; Vreven, T., Jr.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Johnson, E.R.; Keinan, S.; Mori–Sanchez, P.; Contreras–Garcıa, J.A.; Cohen, J.; Yang, W. Revealing noncovalent interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Hopffgarten, M.V.; Frenking, G. Energy decomposition analysis. Wire Comput. Mol. Sci. 2012, 2, 43–62. [Google Scholar] [CrossRef]
- Li, Y.; Bai, H.; Lin, F.; Huang, Y. Energetics and electronic structures of nitrogen chains encapsulated in zigzag carbon nanotube. Physica 2018, 103, 444–451. [Google Scholar]
- Li, Y.; Bai, H.; Li, L.; Huang, Y. Stabilities and electronic properties of nanowires made of single atomic sulfur chains encapsulated in zigzag carbon nanotubes. Nanotechnology 2018, 29, 415703. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Meher, B.R.; Eustache, D.; Wang, Y. Insight into the interaction between DNA bases and defective graphenes: Covalent or non-covalent. J. Mol. Graph. Model. 2014, 47, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Joseph, A.; Thomas, V.I.; Żyła, G.; Padmanabhan, A.S.; Mathew, S. Theoretical probing of weak anion−cation interactions in certain pyridinium-based ionic liquid ion pairs and the application of molecular electrostatic potential in their ionic crystal density determination: A comparative study using density functional approach. J. Phys. Chem. 2018, 122, 328–340. [Google Scholar]
- Gao, H.; Feng, W.; Li, X.; Li, N.; Du, Y.; Wu, Y. Insights into the non-covalent interaction between modified nucleobases and graphene nanoflake from first-principles. Physica 2019, 107, 73–79. [Google Scholar] [CrossRef]
- Maximillian, J.S.; Phipps, T.F.; Christofer, S.T.; Skylaris, C.K. Energy decomposition analysis approaches and their evaluation on prototypical protein–drug interaction patterns. Chem. Soc. Rev. 2015, 44, 3177. [Google Scholar]
- Lu, T. Multiwfn—A Multifunctional Wavefunction Analyze—Software Manual. Version 3.6 (dev); 2018; Available online: http://sobereva.com/multiwfn/misc/Multiwfn_3.6(dev).pdf (accessed on 18 April 2019).
- Fowler, P.W.; Manolopoulos, D.E. Magic numbers and stable structures for fullerenes, fullerides and fullerenium ions. Nature 1992, 355, 428. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bai, H.; Gao, H.; Feng, W.; Zhao, Y.; Wu, Y. Interaction in Li@Fullerenes and Li+@Fullerenes: First Principle Insights to Li-Based Endohedral Fullerenes. Nanomaterials 2019, 9, 630. https://doi.org/10.3390/nano9040630
Bai H, Gao H, Feng W, Zhao Y, Wu Y. Interaction in Li@Fullerenes and Li+@Fullerenes: First Principle Insights to Li-Based Endohedral Fullerenes. Nanomaterials. 2019; 9(4):630. https://doi.org/10.3390/nano9040630
Chicago/Turabian StyleBai, Hongcun, Hongfeng Gao, Wei Feng, Yaping Zhao, and Yuhua Wu. 2019. "Interaction in Li@Fullerenes and Li+@Fullerenes: First Principle Insights to Li-Based Endohedral Fullerenes" Nanomaterials 9, no. 4: 630. https://doi.org/10.3390/nano9040630
APA StyleBai, H., Gao, H., Feng, W., Zhao, Y., & Wu, Y. (2019). Interaction in Li@Fullerenes and Li+@Fullerenes: First Principle Insights to Li-Based Endohedral Fullerenes. Nanomaterials, 9(4), 630. https://doi.org/10.3390/nano9040630