Elucidating the Chemistry behind the Reduction of Graphene Oxide Using a Green Approach with Polydopamine



Abstract
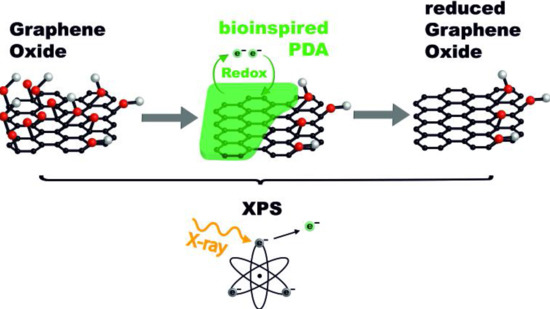
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of Graphene Oxide, Polydopamine-Reduced Graphene Oxide, and Reduced Graphene Oxide
2.3. Modeling and Characterization of GO, PDA-GO, and RGO
2.3.1. Ab-Initio Calculations
2.3.2. Raman Spectroscopy
2.3.3. X-Ray Photoelectron Spectroscopy (XPS)
2.3.4. Thermogravimetric Analysis (TGA)
2.3.5. Electrical Powder Conductivity
3. Results and Discussion
3.1. The Oxidation of Graphene Stacks
3.2. Raman Spectroscopy
3.3. X-Ray Photoelectron Spectroscopy (XPS)
3.3.1. XPS Spectra of Graphene Stacks and GO
3.3.2. XPS Spectra of PDA-GO
3.3.3. XPS Spectra of RGO
3.4. Thermogravimetric Analysis (TGA)
3.5. Electrical Powder Conductivity
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric field effect in atomically thin carbon films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [PubMed]
- Novoselov, K.S.; Jiang, D.; Schedin, F.; Booth, T.J.; Khotkevich, V.V.; Morozov, S.V.; Geim, A.K. Two-dimensional atomic crystals. Proc. Natl. Acad. Sci. USA 2005, 102, 10451–10453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.; Wei, X.; Li, Q.; Carpick, R.; Kysar, J.W.; Hone, J. Elastic and frictional properties of graphene. Phys. Status Solidi (b) 2009, 246, 2562–2567. [Google Scholar] [CrossRef]
- Novoselov, K.S.; Fal’ko, V.I.; Colombo, L.; Gellert, P.R.; Schwab, M.G.; Kim, K. A roadmap for graphene. Nature 2012, 490, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Murali, S.; Cai, W.; Li, X.; Suk, J.W.; Potts, J.R.; Ruoff, R.S. Graphene and graphene oxide: Synthesis, properties, and applications. Adv. Mater. 2010, 22, 3906–3924. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.-W.; Chai, S.-P.; Mohamed, A.R.; Hashim, U. Synthesis and characterization of graphene and carbon nanotubes: A review on the past and recent developments. J. Ind. Eng. Chem. 2014, 20, 1171–1185. [Google Scholar] [CrossRef]
- Warner, J.H.; Schäffel, F.; Bachmatiuk, A.; Rümmeli, M.H. Chapter 4—Methods for obtaining graphene. In Graphene; Warner, J.H., Schäffel, F., Bachmatiuk, A., Rümmeli, M.H., Eds.; Elsevier: Amsterdam, The Netherlands, 2013; pp. 129–228. [Google Scholar]
- Singh, V.; Joung, D.; Zhai, L.; Das, S.; Khondaker, S.I.; Seal, S. Graphene based materials: Past, present and future. Prog. Mater. Sci. 2011, 56, 1178–1271. [Google Scholar] [CrossRef]
- Liu, J.; Cui, L.; Losic, D. Graphene and graphene oxide as new nanocarriers for drug delivery applications. Acta Biomater. 2013, 9, 9243–9257. [Google Scholar] [CrossRef]
- Compton, O.C.; Nguyen, S.T. Graphene oxide, highly reduced graphene oxide, and graphene: Versatile building blocks for carbon-based materials. Small 2010, 6, 711–723. [Google Scholar] [CrossRef]
- Taniselass, S.; Md Arshad, M.K.; Gopinath, S.C.B. Current state of green reduction strategies: Solution-processed reduced graphene oxide for healthcare biodetection. Mater. Sci. Eng. C 2019, 96, 904–914. [Google Scholar] [CrossRef]
- Thakur, S.; Karak, N. Alternative methods and nature-based reagents for the reduction of graphene oxide: A review. Carbon 2015, 94, 224–242. [Google Scholar] [CrossRef]
- De Silva, K.K.H.; Huang, H.H.; Joshi, R.K.; Yoshimura, M. Chemical reduction of graphene oxide using green reductants. Carbon 2017, 119, 190–199. [Google Scholar] [CrossRef]
- De Marco, B.A.; Rechelo, B.S.; Tótoli, E.G.; Kogawa, A.C.; Salgado, H.R.N. Evolution of green chemistry and its multidimensional impacts: A review. Saudi Pharm. J. 2019, 27, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Liu, F.; Liu, Y.; Ma, N.; Wang, Z.; Zhang, X. Environment-friendly method to produce graphene that employs vitamin c and amino acid. Chem. Mater. 2010, 22, 2213–2218. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, H.; Shen, G.; Cheng, P.; Zhang, J.; Guo, S. Reduction of graphene oxide vial-ascorbic acid. Chem. Commun. 2010, 46, 1112–1114. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Merino, M.J.; Guardia, L.; Paredes, J.I.; Villar-Rodil, S.; Solís-Fernández, P.; Martínez-Alonso, A.; Tascón, J.M.D. Vitamin C is an ideal substitute for hydrazine in the reduction of graphene oxide suspensions. J. Phys. Chem. C 2010, 114, 6426–6432. [Google Scholar] [CrossRef]
- Zhu, C.; Guo, S.; Fang, Y.; Dong, S. Reducing sugar: New functional molecules for the green synthesis of graphene nanosheets. ACS Nano 2010, 4, 2429–2437. [Google Scholar] [CrossRef] [PubMed]
- Bose, S.; Kuila, T.; Mishra, A.K.; Kim, N.H.; Lee, J.H. Dual role of glycine as a chemical functionalizer and a reducing agent in the preparation of graphene: An environmentally friendly method. J. Mater. Chem. 2012, 22, 9696–9703. [Google Scholar] [CrossRef]
- Chen, D.; Li, L.; Guo, L. An environment-friendly preparation of reduced graphene oxide nanosheets via amino acid. Nanotechnology 2011, 22, 325601. [Google Scholar] [CrossRef]
- Ma, J.; Wang, X.; Liu, Y.; Wu, T.; Liu, Y.; Guo, Y.; Li, R.; Sun, X.; Wu, F.; Li, C.; et al. Reduction of graphene oxide with l-lysine to prepare reduced graphene oxide stabilized with polysaccharide polyelectrolyte. J. Mater. Chem. A 2013, 1, 2192–2201. [Google Scholar] [CrossRef]
- Bo, Z.; Shuai, X.; Mao, S.; Yang, H.; Qian, J.; Chen, J.; Yan, J.; Cen, K. Green preparation of reduced graphene oxide for sensing and energy storage applications. Sci. Rep. 2014, 4, 4684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, W.; Zhao, Z.; Hu, H.; Gogotsi, Y.; Qiu, J. Highly controllable and green reduction of graphene oxide to flexible graphene film with high strength. Mater. Res. Bull. 2013, 48, 4797–4803. [Google Scholar] [CrossRef]
- Zhang, Z.; Chen, H.; Xing, C.; Guo, M.; Xu, F.; Wang, X.; Gruber, H.J.; Zhang, B.; Tang, J. Sodium citrate: A universal reducing agent for reduction/decoration of graphene oxide with au nanoparticles. Nano Res. 2011, 4, 599–611. [Google Scholar] [CrossRef]
- Li, J.; Xiao, G.; Chen, C.; Li, R.; Yan, D. Superior dispersions of reduced graphene oxide synthesized by using gallic acid as a reductant and stabilizer. J. Mater. Chem. A 2013, 1, 1481–1487. [Google Scholar] [CrossRef]
- Kuila, T.; Bose, S.; Khanra, P.; Mishra, A.K.; Kim, N.H.; Lee, J.H. A green approach for the reduction of graphene oxide by wild carrot root. Carbon 2012, 50, 914–921. [Google Scholar] [CrossRef]
- Khanra, P.; Kuila, T.; Kim, N.H.; Bae, S.H.; Yu, D.-S.; Lee, J.H. Simultaneous bio-functionalization and reduction of graphene oxide by baker’s yeast. Chem. Eng. J. 2012, 183, 526–533. [Google Scholar] [CrossRef]
- Pham, T.A.; Kim, J.S.; Kim, J.S.; Jeong, Y.T. One-step reduction of graphene oxide with L-glutathione. Colloids Surf. A Physicochem. Eng. Asp. 2011, 384, 543–548. [Google Scholar] [CrossRef]
- Guo, C.; Book-Newell, B.; Irudayaraj, J. Protein-directed reduction of graphene oxide and intracellular imaging. Chem. Commun. 2011, 47, 12658–12660. [Google Scholar] [CrossRef] [PubMed]
- Esfandiar, A.; Akhavan, O.; Irajizad, A. Melatonin as a powerful bio-antioxidant for reduction of graphene oxide. J. Mater. Chem. 2011, 21, 10907–10914. [Google Scholar] [CrossRef]
- Lei, Z.; Lu, L.; Zhao, X.S. The electrocapacitive properties of graphene oxide reduced by urea. Energy Environ. Sci. 2012, 5, 6391–6399. [Google Scholar] [CrossRef]
- Lee, G.; Kim, B.S. Biological reduction of graphene oxide using plant leaf extracts. Biotechnol. Prog. 2014, 30, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Jana, M.; Saha, S.; Khanra, P.; Murmu, N.C.; Srivastava, S.K.; Kuila, T.; Lee, J.H. Bio-reduction of graphene oxide using drained water from soaked mung beans (Phaseolus aureus L.) and its application as energy storage electrode material. Mater. Sci. Eng. B 2014, 186, 33–40. [Google Scholar] [CrossRef]
- Wang, Y.; Shi, Z.; Yin, J. Facile synthesis of soluble graphene via a green reduction of graphene oxide in tea solution and its biocomposites. ACS Appl. Mater. Interfaces 2011, 3, 1127–1133. [Google Scholar] [CrossRef] [PubMed]
- Muthoosamy, K.; Bai, R.G.; Abubakar, I.B.; Sudheer, S.M.; Lim, H.N.; Loh, H.-S.; Huang, N.M.; Chia, C.H.; Manickam, S. Exceedingly biocompatible and thin-layered reduced graphene oxide nanosheets using an eco-friendly mushroom extract strategy. Int. J. Nanomed. 2015, 10, 1505–1519. [Google Scholar] [CrossRef]
- Xu, L.Q.; Yang, W.J.; Neoh, K.G.; Kang, E.T.; Fu, G.D. Dopamine-induced reduction and functionalization of graphene oxide nanosheets. Macromolecules 2010, 43, 8336–8339. [Google Scholar] [CrossRef]
- Lee, H.; Dellatore, S.M.; Miller, W.M.; Messersmith, P.B. Mussel-inspired surface chemistry for multifunctional coatings. Science 2007, 318, 426–430. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.Q.; Liu, Q.; Li, G.L.; Shi, J.B.; Liu, J.Y.; Wang, T.; Jiang, G.B. A mussel-inspired polydopamine coating as a versatile platform for the in situ synthesis of graphene-based nanocomposites. Nanoscale 2012, 4, 5864–5867. [Google Scholar] [CrossRef]
- Cong, H.P.; Wang, P.; Gong, M.; Yu, S.H. Facile synthesis of mesoporous nitrogen-doped graphene: An efficient methanol-tolerant cathodic catalyst for oxygen reduction reaction. Nano Energy 2014, 3, 55–63. [Google Scholar] [CrossRef]
- Wang, F.Y.; Sun, Q.Q.; Feng, B.; Xu, Z.A.; Zhang, J.Y.; Xu, J.; Lu, L.L.; Yu, H.J.; Wang, M.W.; Li, Y.P.; et al. Polydopamine-functionalized graphene oxide loaded with gold nanostars and doxorubicin for combined photothermal and chemotherapy of metastatic breast cancer. Adv. Healthc. Mater. 2016, 5, 2227–2236. [Google Scholar] [CrossRef]
- Zhang, X.Y.; Nan, X.; Shi, W.; Sun, Y.N.; Su, H.L.; He, Y.; Liu, X.; Zhang, Z.; Ge, D.T. Polydopamine-functionalized nanographene oxide: A versatile nanocarrier for chemotherapy and photothermal therapy. Nanotechnology 2017, 28. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, J.; Zhang, B.L.; Tang, J.L. Mussel-inspired functionalization of graphene for synthesizing Ag-polydopamine-graphene nanosheets as antibacterial materials. Nanoscale 2013, 5, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Deng, S.Y.; Li, D.L.; Shan, D.; He, W.; Zhang, X.J.; Shi, Y. Bioinspired polydopamine as the scaffold for the active AuNPs anchoring and the chemical simultaneously reduced graphene oxide: Characterization and the enhanced biosensing application. Biosens. Bioelectron. 2013, 49, 466–471. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.Y.; Xi, P.X.; Xie, G.Q.; Shi, Y.J.; Hou, F.P.; Huang, L.; Chen, F.J.; Zeng, Z.Z.; Shao, C.W.; Wang, J. Simultaneous reduction and surface functionalization of graphene oxide for hydroxyapatite mineralization. J. Phys. Chem. C 2012, 116, 3334–3341. [Google Scholar] [CrossRef]
- Da Silva, C.A.; Pötschke, P.; Simon, F.; Holzschuh, M.; Pionteck, J.; Heinrich; Wießner, S.; Zimmerer, C. Synthesis and characterization of graphene derivatives for application in magnetic high-field induction heating. AIP Conf. Proc. 2019, 2055, 130006. [Google Scholar] [CrossRef]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Briggs, D. Comprehensive Polymer Science. In Characterization of Surfaces; Chapter 24; Booth, C., Price, C., Briggs, D., Eds.; Pergamon: Oxford UK, 1989; Volume 1, pp. 543–559. [Google Scholar]
- Seah, M.P.; Dench, W.A. Quantitative electron spectroscopy of surfaces: A standard data base for electron inelastic mean free paths in solids. Surf. Interface Anal. 1979, 1, 2–11. [Google Scholar] [CrossRef]
- Paiva, M.C.; Simon, F.; Novais, R.M.; Ferreira, T.; Proença, M.F.; Xu, W.; Besenbacher, F. Controlled functionalization of carbon nanotubes by a solvent-free multicomponent approach. ACS Nano 2010, 4, 7379–7386. [Google Scholar] [CrossRef] [PubMed]
- Watts, J.F. High resolution XPS of organic polymers: The Scienta ESCA 300 database. G. Beamson and D. Briggs. 280pp., £65. John Wiley & Sons, Chichester, ISBN 0471 935921, (1992). Surf. Interface Anal. 1993, 20, 267. [Google Scholar] [CrossRef]
- Krause, B.; Boldt, R.; Häußler, L.; Pötschke, P. Ultralow percolation threshold in polyamide 6.6/MWCNT composites. Compos. Sci. Technol. 2015, 114, 119–125. [Google Scholar] [CrossRef]
- Lerf, A.; He, H.; Forster, M.; Klinowski, J. Structure of Graphite Oxide Revisited. J. Phys. Chem. B 1998, 102, 4477–4482. [Google Scholar] [CrossRef]
- He, H.; Klinowski, J.; Forster, M.; Lerf, A. A new structural model of graphite oxide. Chem. Phys. Lett. 1998, 287, 53–56. [Google Scholar] [CrossRef]
- Bellunato, A.; Arjmandi Tash, H.; Cesa, Y.; Schneider, G.F. Chemistry at the edge of graphene. ChemPhysChem 2016, 17, 785–801. [Google Scholar] [CrossRef] [PubMed]
- Grätz, S.; Beyer, D.; Tkachova, V.; Hellmann, S.; Berger, R.; Feng, X.; Borchardt, L. The mechanochemical scholl reaction—A solvent-free and versatile graphitization tool. Chem. Commun. 2018, 54, 5307–5310. [Google Scholar] [CrossRef] [PubMed]
- Malard, L.M.; Pimenta, M.A.; Dresselhaus, G.; Dresselhaus, M.S. Raman spectroscopy in graphene. Phys. Rep. 2009, 473, 51–87. [Google Scholar] [CrossRef]
- Ferrari, A.C. Raman spectroscopy of graphene and graphite: Disorder, electron–phonon coupling, doping and nonadiabatic effects. Solid State Commun. 2007, 143, 47–57. [Google Scholar] [CrossRef]
- Ferrari, A.C.; Meyer, J.C.; Scardaci, V.; Casiraghi, C.; Lazzeri, M.; Mauri, F.; Piscanec, S.; Jiang, D.; Novoselov, K.S.; Roth, S.; et al. Raman spectrum of graphene and graphene layers. Phys. Rev. Lett. 2006, 97, 187401. [Google Scholar] [CrossRef]
- Ferrari, A.C.; Basko, D.M. Raman spectroscopy as a versatile tool for studying the properties of graphene. Nat. Nanotechnol. 2013, 8, 235. [Google Scholar] [CrossRef]
- Song, J.; Dai, Z.; Li, J.; Tong, X.; Zhao, H. Polydopamine-decorated boron nitride as nano-reinforcing fillers for epoxy resin with enhanced thermomechanical and tribological properties. Mater. Res. Express 2018, 5, 075029. [Google Scholar] [CrossRef]
- Parviz, D.; Das, S.; Ahmed, H.S.T.; Irin, F.; Bhattacharia, S.; Green, M.J. Dispersions of non-covalently functionalized graphene with minimal stabilizer. Acs Nano 2012, 6, 8857–8867. [Google Scholar] [CrossRef]
- Tripathi, B.; Das, P.; Simon, F.; Stamm, M. Ultralow fouling membranes by surface modification with functional polydopamine. Eur. Polym. J. 2018, 99, 80–89. [Google Scholar] [CrossRef]
- Biesinger, M. Available online: http://www.xpsfitting.com/search/label/Adventitious (accessed on 11 June 2019).
- Yang, D.; Kong, X.X.; Ni, Y.F.; Ruan, M.N.; Huang, S.; Shao, P.Z.; Guo, W.L.; Zhang, L.Q. Improved mechanical and electrochemical properties of XNBR dielectric elastomer actuator by poly(dopamine) functionalized graphene nano-sheets. Polymers 2019, 11, 218. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Wang, J.; Zhang, H.; Zhang, T.; Zhang, B.; Cao, S.; Liu, J. Polydopamine-modified graphene oxide nanocomposite membrane for proton exchange membrane fuel cell under anhydrous conditions. J. Mater. Chem. A 2014, 2, 9548–9558. [Google Scholar] [CrossRef]
- Santha Kumar, A.R.S.; Piana, F.; Mičušík, M.; Pionteck, J.; Banerjee, S.; Voit, B. Preparation of graphite derivatives by selective reduction of graphite oxide and isocyanate functionalization. Mater. Chem. Phys. 2016, 182, 237–245. [Google Scholar] [CrossRef]
- Srivastava, S.K.; Pionteck, J. Recent advances in preparation, structure, properties and applications of graphite oxide. J. Nanosci. Nanotechnol. 2015, 15, 1984–2000. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time of Reaction (min) | Sample Description (PDA-GO_reduction Time in min) |
|---|---|
| 30 | PDA-GO_30 |
| 60 | PDA-GO_60 |
| 90 | PDA-GO_90 |
| 120 | PDA-GO_120 |
| 150 | PDA-GO_150 |
| 180 | PDA-GO_180 |
| 210 | PDA-GO_210 |
| Material | ID/IG | ID+PDA/IG+PDA |
|---|---|---|
| Graphite | 0.2 | — |
| GO | 0.5 | — |
| PDA-GO_30 | — | 1.2 |
| PDA-GO_90 | — | 1.2 |
| PDA-GO_180 | — | 1.2 |
| RGO_30 | — | 1.2 |
| RGO_90 | — | 1.2 |
| RGO_180 | — | 1.2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva, C.; Simon, F.; Friedel, P.; Pötschke, P.; Zimmerer, C. Elucidating the Chemistry behind the Reduction of Graphene Oxide Using a Green Approach with Polydopamine. Nanomaterials 2019, 9, 902. https://doi.org/10.3390/nano9060902
Silva C, Simon F, Friedel P, Pötschke P, Zimmerer C. Elucidating the Chemistry behind the Reduction of Graphene Oxide Using a Green Approach with Polydopamine. Nanomaterials. 2019; 9(6):902. https://doi.org/10.3390/nano9060902
Chicago/Turabian StyleSilva, Cláudia, Frank Simon, Peter Friedel, Petra Pötschke, and Cordelia Zimmerer. 2019. "Elucidating the Chemistry behind the Reduction of Graphene Oxide Using a Green Approach with Polydopamine" Nanomaterials 9, no. 6: 902. https://doi.org/10.3390/nano9060902
APA StyleSilva, C., Simon, F., Friedel, P., Pötschke, P., & Zimmerer, C. (2019). Elucidating the Chemistry behind the Reduction of Graphene Oxide Using a Green Approach with Polydopamine. Nanomaterials, 9(6), 902. https://doi.org/10.3390/nano9060902