Bioluminescent RIPoptosome Assay for FADD/RIPK1 Interaction Based on Split Luciferase Assay in a Human Neuroblastoma Cell Line SH-SY5Y


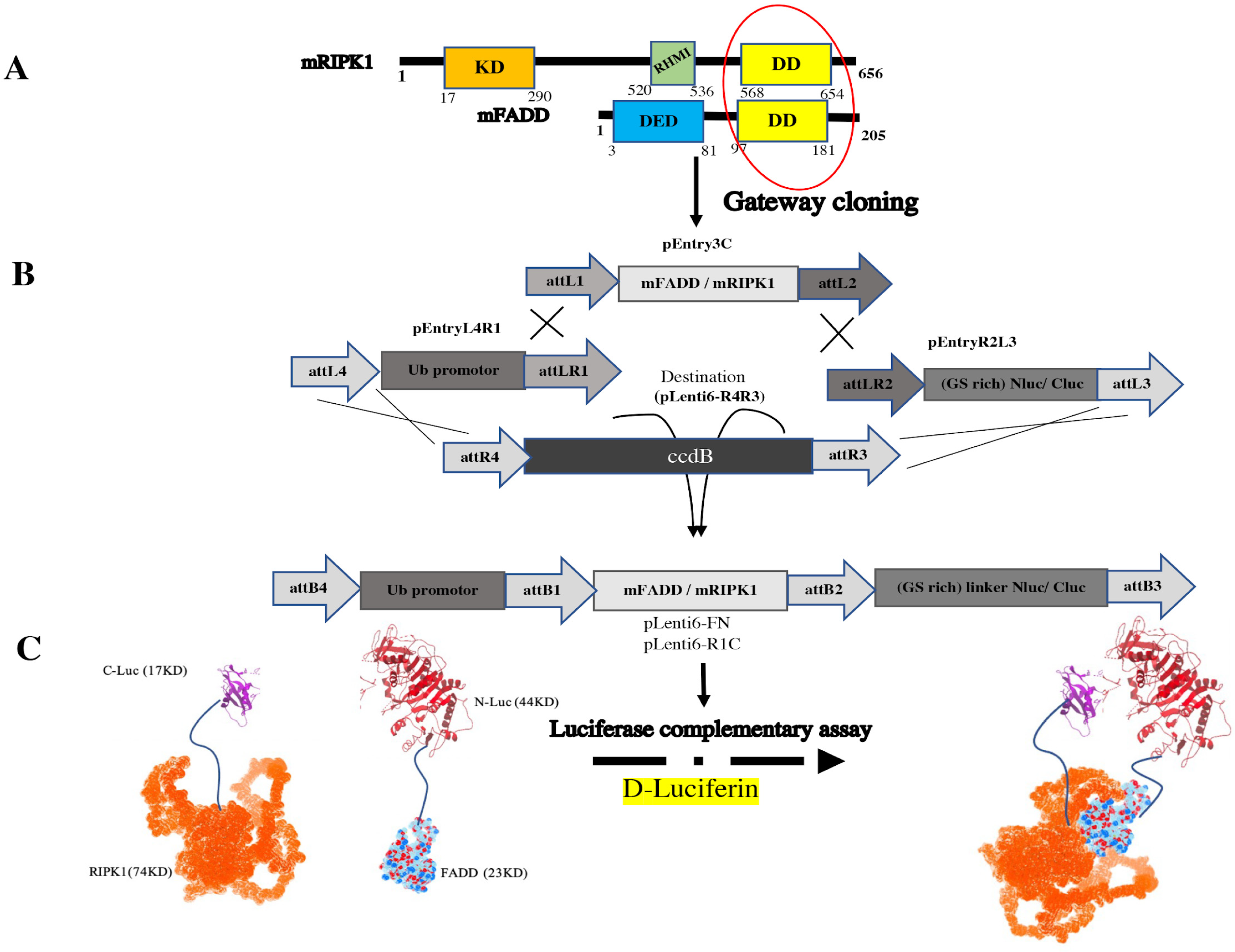{kind=link}
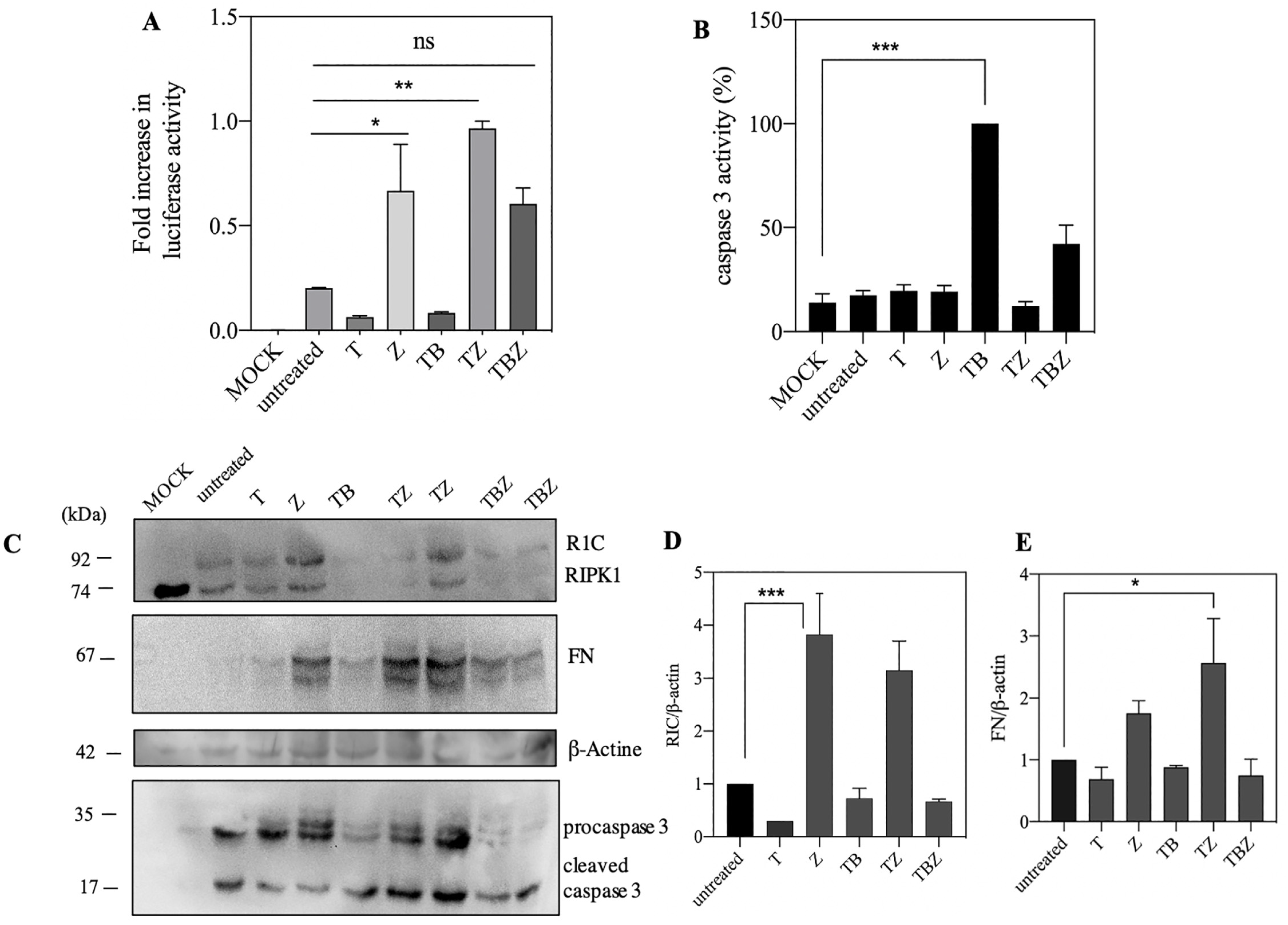{kind=link}
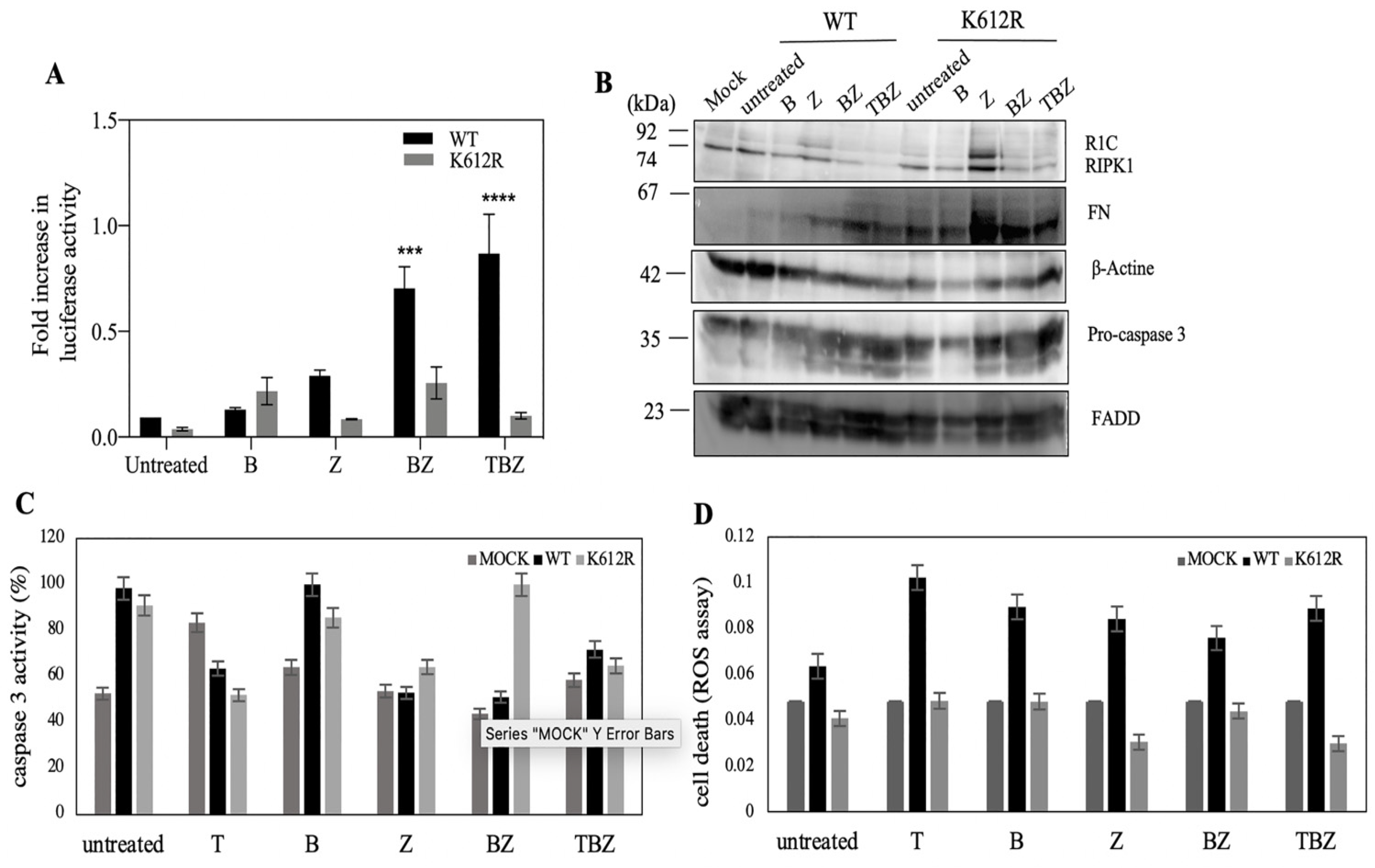{kind=link}
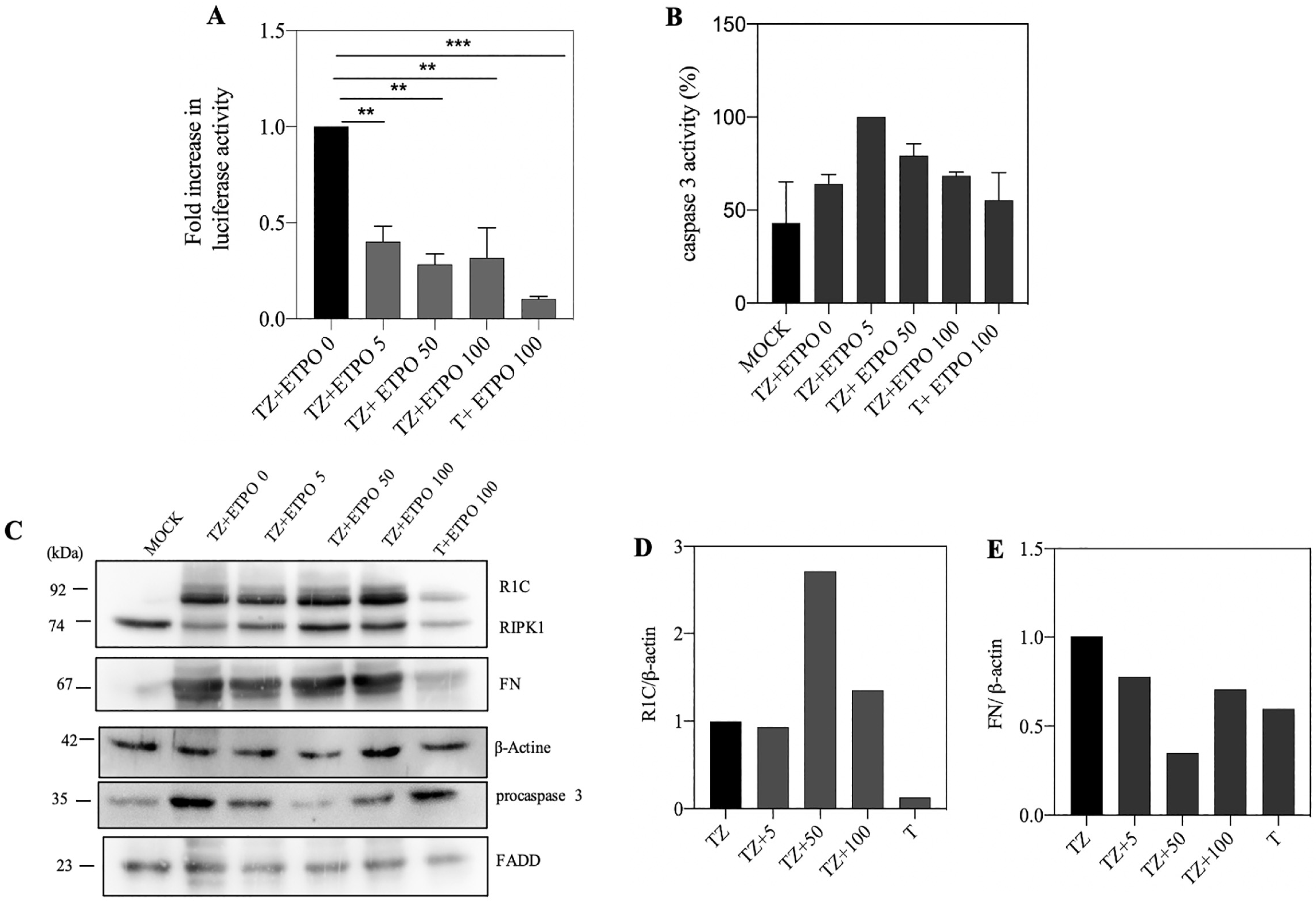{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and Cell Culture
2.2. Reporter Constructs and Site Directed Mutagenesis
2.3. Transient Transfection, Cellular Treatments and Extract Preparation
2.4. Western Blot Analysis
2.5. Luciferase Activity Measurements and Cell Death Assays
2.6. Caspases 3 Activity Measurement
2.7. Measurement of Reactive Oxygen Species
2.8. Statistical Analysis
3. Results
3.1. The pEntry and Final Constructs were Generated by Gateway Cloning
3.2. ZVAD.Fmk Trigger Luciferase Activity and Protein Expression in Caspase 8 Deficient SH-SY5Y Neuroblastoma Cells
3.3. K612R Mutation in DD of RIPK1 Reduce FADD/RIPK1 Interaction
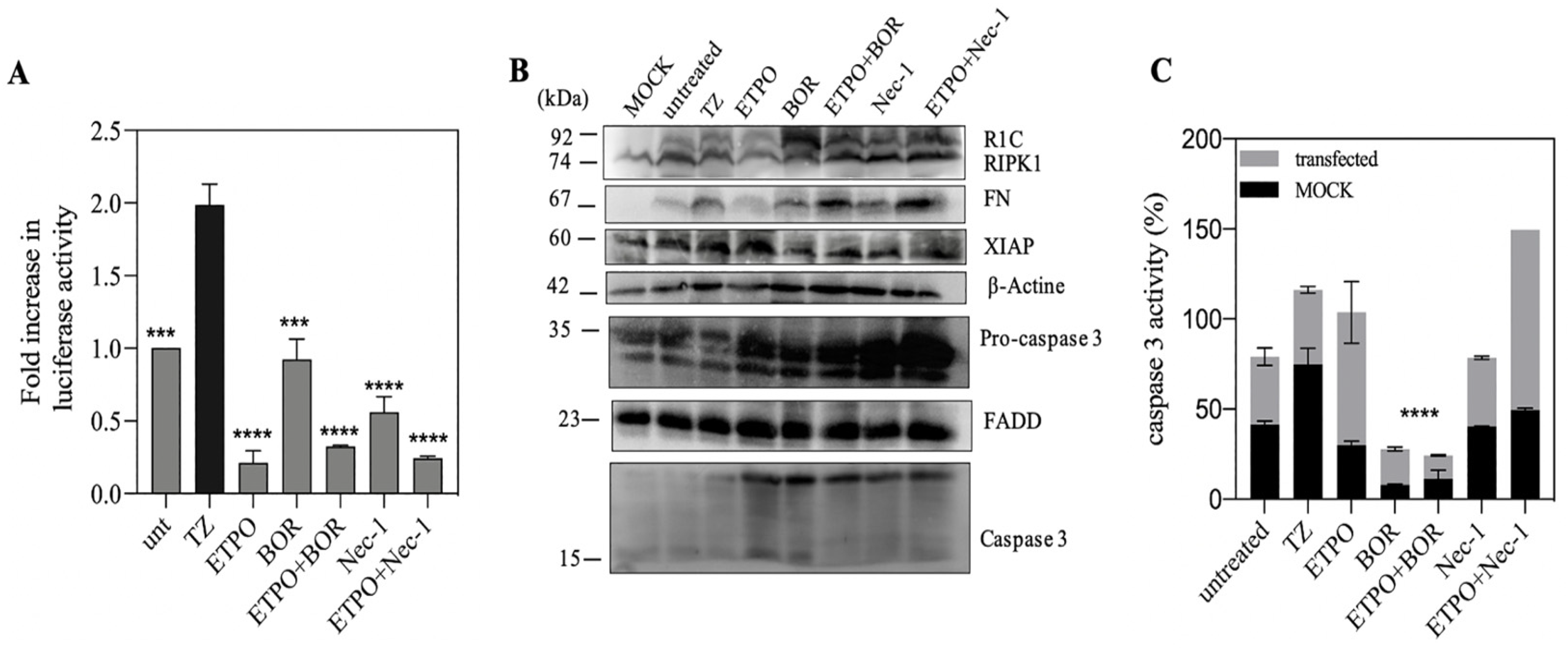
3.4. Genotoxic Drugs such as Etoposide Decrease FADD/RIPK1 Interaction
3.5. FN/R1C Interaction in Presence of as Bortezomib and Nec-1
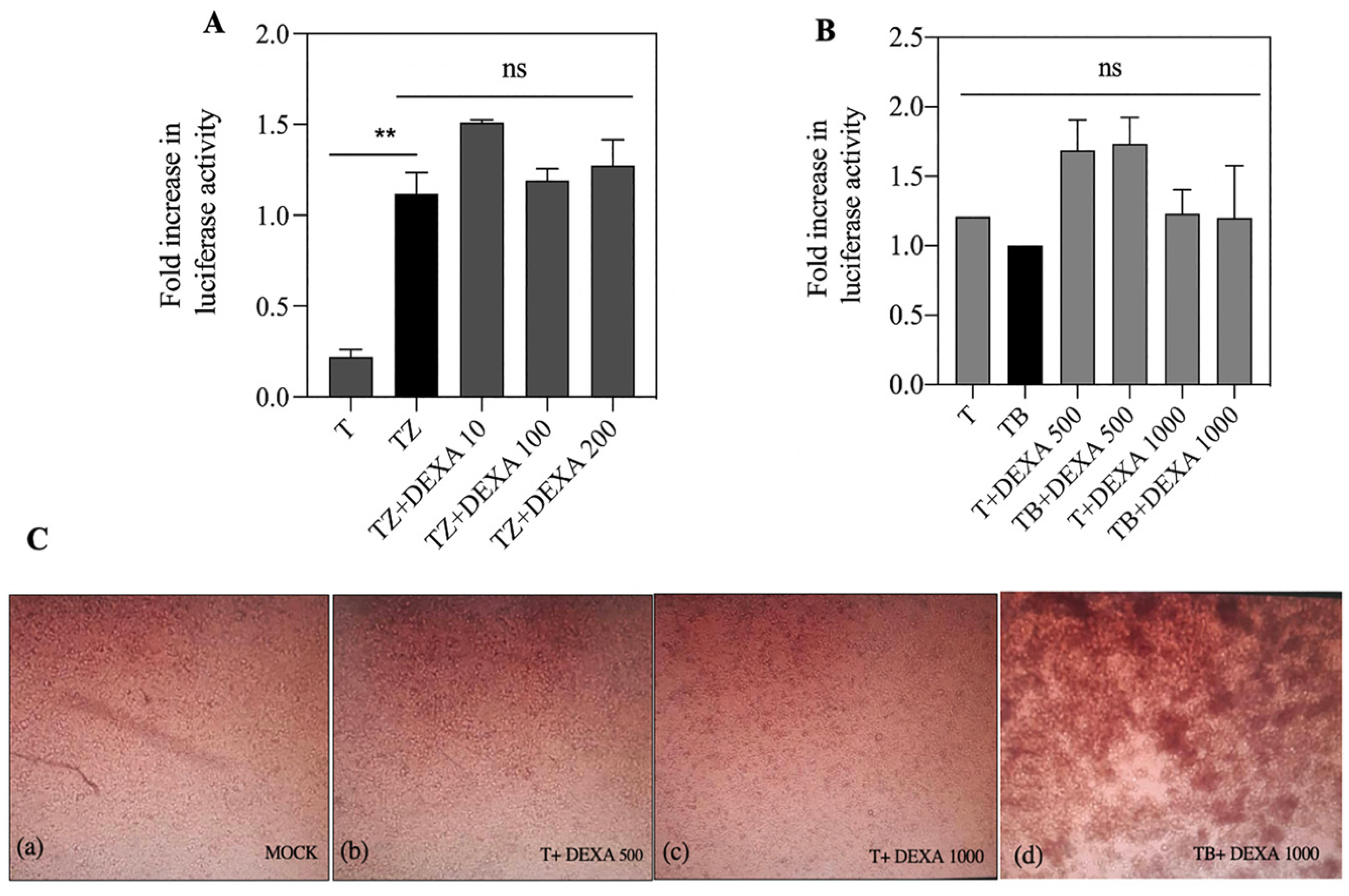
3.6. Dexamethasone Had no Effect on FN/R1C Interaction
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Gilad, Y.; Shiloh, R.; Ber, Y.; Bialik, S.; Kimchi, A. Discovering protein-protein interactions within the programmed cell death network using a protein-fragment complementation screen. Cell Rep. 2014, 8, 909–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Sun, J.; Gu, L.; Wang, S.; Yang, T.; Wei, T.; Shan, T.; Wang, H.; Wang, L. Programmed cell death: Complex regulatory networks in cardiovascular disease. Front. Cell Dev. Biol. 2021, 9, 794879. [Google Scholar] [CrossRef] [PubMed]
- Locquet, M.-A.; Ichim, G.; Bisaccia, J.; Dutour, A.; Lebecque, S.; Castets, M.; Weber, K. Caspase-8 deficiency induces a switch from TLR3 induced apoptosis to lysosomal cell death in neuroblastoma. Sci. Rep. 2021, 11, 10609. [Google Scholar] [CrossRef]
- Kepp, O.; Galluzzi, L.; Lipinski, M.; Yuan, J.; Kroemer, G. Cell death assays for drug discovery. Nat. Rev. Drug Discov. 2011, 10, 221–237. [Google Scholar] [CrossRef] [PubMed]
- Füllsack, S.; Rosenthal, A.; Wajant, H.; Siegmund, D. Redundant and receptor-specific activities of TRADD, RIPK1 and FADD in death receptor signaling. Cell Death Dis. 2019, 10, 122. [Google Scholar] [CrossRef] [Green Version]
- Holler, N.; Zaru, R.; Micheau, O.; Thome, M.; Attinger, A.; Valitutti, S.; Bodmer, J.-L.; Schneider, P.; Seed, B.; Tschopp, J. Fas triggers an alternative, caspase-8–independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 2000, 1, 489–495. [Google Scholar] [CrossRef]
- Ch’en, I.L.; Beisner, D.R.; Degterev, A.; Lynch, C.; Yuan, J.; Hoffmann, A.; Hedrick, S.M. Antigen-mediated T cell expansion regulated by parallel pathways of death. Proc. Natl. Acad. Sci. USA 2008, 105, 17463–17468. [Google Scholar] [CrossRef] [Green Version]
- Pasparakis, M.; Vandenabeele, P. Necroptosis and its role in inflammation. Nature 2015, 517, 311–320. [Google Scholar] [CrossRef]
- Holbrook, J.; Lara-Reyna, S.; Jarosz-Griffiths, H.; McDermott, M.F. Tumour necrosis factor signalling in health and disease. F1000Research 2019, 8, 111. [Google Scholar] [CrossRef] [Green Version]
- Tenev, T.; Bianchi, K.; Darding, M.; Broemer, M.; Langlais, C.; Wallberg, F.; Zachariou, A.; Lopez, J.; MacFarlane, M.; Cain, K. The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol. Cell 2011, 43, 432–448. [Google Scholar] [CrossRef]
- Feoktistova, M.; Geserick, P.; Kellert, B.; Dimitrova, D.P.; Langlais, C.; Hupe, M.; Cain, K.; MacFarlane, M.; Häcker, G.; Leverkus, M. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol. Cell 2011, 43, 449–463. [Google Scholar] [CrossRef] [Green Version]
- Neuper, T.; Weiss, R.; Horejs-Hoeck, J. Ripping the Ripoptosome: A novel path for blocking allergic inflammation? Cell. Mol. Immunol. 2022, 19, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Oberst, A.; Dillon, C.P.; Weinlich, R.; Salvesen, G.S. RIPK-dependent necrosis and its regulation by caspases: A mystery in five acts. Mol. Cell 2011, 44, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritsch, M.; Günther, S.D.; Schwarzer, R.; Albert, M.-C.; Schorn, F.; Werthenbach, J.P.; Schiffmann, L.M.; Stair, N.; Stocks, H.; Seeger, J.M. Caspase-8 is the molecular switch for apoptosis, necroptosis and pyroptosis. Nature 2019, 575, 683–687. [Google Scholar] [CrossRef] [PubMed]
- Ha, H.J.; Park, H.H. Identification and analysis of dominant negative mutants of RIP1 DD that disrupt RIPoptosome core formation. Mol. Biol. Rep. 2018, 45, 1715–1722. [Google Scholar] [CrossRef] [PubMed]
- Jang, T.-h.; Zheng, C.; Li, J.; Richards, C.; Hsiao, Y.-S.; Walz, T.; Wu, H.; Park, H.H. Structural study of the RIPoptosome core reveals a helical assembly for kinase recruitment. Biochemistry 2014, 53, 5424–5431. [Google Scholar] [CrossRef] [PubMed]
- Schilling, R.; Geserick, P.; Leverkus, M. Characterization of the ripoptosome and its components: Implications for anti-inflammatory and cancer therapy. Methods Enzymol. 2014, 545, 83–102. [Google Scholar]
- Zhang, S.; Tang, M.-b.; Luo, H.-y.; Shi, C.-h.; Xu, Y.-m. Necroptosis in neurodegenerative diseases: A potential therapeutic target. Cell Death Dis. 2017, 8, e2905. [Google Scholar] [CrossRef] [Green Version]
- Azad, T.; Tashakor, A.; Hosseinkhani, S. Split-luciferase complementary assay: Applications, recent developments, and future perspectives. Anal. Bioanal. Chem. 2014, 406, 5541–5560. [Google Scholar] [CrossRef]
- Bishnu, A.; Phadte, P.; Dhadve, A.; Sakpal, A.; Rekhi, B.; Ray, P. Molecular imaging of the kinetics of hyperactivated ERK1/2-mediated autophagy during acquirement of chemoresistance. Cell Death Dis. 2021, 12, 161. [Google Scholar] [CrossRef]
- Haga, S.; Kanno, A.; Ozawa, T.; Morita, N.; Asano, M.; Ozaki, M. Detection of necroptosis in ligand-mediated and hypoxia-induced injury of hepatocytes using a novel optic probe-detecting receptor-interacting protein (RIP) 1/RIP3 binding. Oncol. Res. 2018, 26, 503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isazadeh, M.; Amandadi, M.; Haghdoust, F.; Lotfollazadeh, S.; Orzáez, M.; Hosseinkhani, S. Split-luciferase complementary assay of NLRP3 PYD-PYD interaction indicates inflammasome formation during inflammation. Anal. Biochem. 2022, 638, 114510. [Google Scholar] [CrossRef] [PubMed]
- Mostafavi, M.; Ataei, F.; Hamidieh, A.A.; Hosseinkhani, S. Development of a bioluminescence assay for BIR2-caspase3 interaction through split luciferase complementary assay. Biochem. Eng. J. 2022, 186, 108584. [Google Scholar] [CrossRef]
- Sahebazzamani, F.; Hosseinkhani, S.; Eriksson, L.A.; Fearnhead, H.O. Apoptosome formation through disruption of the K192-D616 salt bridge in the Apaf-1 closed form. ACS Omega 2021, 6, 22551–22558. [Google Scholar] [CrossRef] [PubMed]
- Fan, F.; Wood, K.V. Bioluminescent assays for high-throughput screening. Assay Drug Dev. Technol. 2007, 5, 127–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moasses Ghafary, S.; Soriano-Teruel, P.M.; Lotfollahzadeh, S.; Sancho, M.; Serrano-Candelas, E.; Karami, F.; Barigye, S.J.; Fernández-Pérez, I.; Gozalbes, R.; Nikkhah, M. Identification of NLRP3PYD Homo-Oligomerization Inhibitors with Anti-Inflammatory Activity. Int. J. Mol. Sci. 2022, 23, 1651. [Google Scholar] [CrossRef]
- Hosseini, E.S.; Nikkhah, M.; Hamidieh, A.A.; Fearnhead, H.O.; Concordet, J.-P.; Hosseinkhani, S. The Lumiptosome, an engineered luminescent form of the apoptosome can report cell death by using the same Apaf-1 dependent pathway. J. Cell Sci. 2020, 133, jcs242636. [Google Scholar] [CrossRef]
- Noori, A.R.; Hosseini, E.S.; Nikkhah, M.; Hosseinkhani, S. Apoptosome formation upon overexpression of native and truncated Apaf-1 in cell-free and cell-based systems. Arch. Biochem. Biophys. 2018, 642, 46–51. [Google Scholar] [CrossRef]
- Oladzad, A.; Nikkhah, M.; Hosseinkhani, S. Optimization of experimental variables influencing apoptosome biosensor in HEK293T cells. Sensors 2020, 20, 1782. [Google Scholar] [CrossRef] [Green Version]
- Tashakor, A.; H-Dehkordi, M.; O’Connell, E.; Gomez Ganau, S.; Gozalbes, R.; Eriksson, L.A.; Hosseinkhani, S.; Fearnhead, H.O. A new split-luciferase complementation assay identifies pentachlorophenol as an inhibitor of apoptosome formation. FEBS Open Bio 2019, 9, 1194–1203. [Google Scholar] [CrossRef] [Green Version]
- Choosakoonkriang, S.; Lobo, B.A.; Koe, G.S.; Koe, J.G.; Middaugh, C.R. Biophysical characterization of PEI/DNA complexes. J. Pharm. Sci. 2003, 92, 1710–1722. [Google Scholar] [CrossRef] [PubMed]
- Kruger, N.J. The Bradford method for protein quantitation. In The Protein Protocols Handbook; Humana Press: Totowa, NJ, USA, 2009; pp. 17–24. [Google Scholar]
- Qin, J.Y.; Zhang, L.; Clift, K.L.; Hulur, I.; Xiang, A.P.; Ren, B.-Z.; Lahn, B.T. Systematic comparison of constitutive promoters and the doxycycline-inducible promoter. PLoS ONE 2010, 5, e10611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Zaro, J.L.; Shen, W.-C. Fusion protein linkers: Property, design and functionality. Adv. Drug Deliv. Rev. 2013, 65, 1357–1369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orning, P.; Lien, E. Multiple roles of caspase-8 in cell death, inflammation, and innate immunity. J. Leukoc. Biol. 2021, 109, 121–141. [Google Scholar] [CrossRef]
- Ribas, J.; Gomez-Arbones, X.; Boix, J. Caspase 8/10 are not mediating apoptosis in neuroblastoma cells treated with CDK inhibitory drugs. Eur. J. Pharmacol. 2005, 524, 49–52. [Google Scholar] [CrossRef] [Green Version]
- Vo, D.-K.H.; Urano, Y.; Takabe, W.; Saito, Y.; Noguchi, N. 24 (S)-Hydroxycholesterol induces RIPK1-dependent but MLKL-independent cell death in the absence of caspase-8. Steroids 2015, 99, 230–237. [Google Scholar] [CrossRef] [Green Version]
- Yamanaka, K.; Saito, Y.; Yamamori, T.; Urano, Y.; Noguchi, N. 24 (S)-hydroxycholesterol induces neuronal cell death through necroptosis, a form of programmed necrosis. J. Biol. Chem. 2011, 286, 24666–24673. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhang, M.; Huang, X.; Liang, W.; Li, G.; Lu, X.; Li, Y.; Pan, H.; Shi, L.; Zhu, H. Ubiquitination of RIPK1 regulates its activation mediated by TNFR1 and TLRs signaling in distinct manners. Nat. Commun. 2020, 11, 6364. [Google Scholar] [CrossRef]
- Yang, Y.; Fang, S.; Jensen, J.P.; Weissman, A.M.; Ashwell, J.D. Ubiquitin protein ligase activity of IAPs and their degradation in proteasomes in response to apoptotic stimuli. Science 2000, 288, 874–877. [Google Scholar] [CrossRef]
- Belz, K.; Schoeneberger, H.; Wehner, S.; Weigert, A.; Bönig, H.; Klingebiel, T.; Fichtner, I.; Fulda, S. Smac mimetic and glucocorticoids synergize to induce apoptosis in childhood ALL by promoting ripoptosome assembly. Blood J. Am. Soc. Hematol. 2014, 124, 240–250. [Google Scholar] [CrossRef] [Green Version]
- Eggert, A.; Grotzer, M.A.; Zuzak, T.J.; Wiewrodt, B.R.; Ho, R.; Ikegaki, N.; Brodeur, G.M. Resistance to tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis in neuroblastoma cells correlates with a loss of caspase-8 expression. Cancer Res. 2001, 61, 1314–1319. [Google Scholar] [PubMed]
- Simpson, D.S.; Gabrielyan, A.; Feltham, R. RIPK1 ubiquitination: Evidence, correlations and the undefined. Semin. Cell Dev. Biol. 2021, 109, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Amin, P.; Florez, M.; Najafov, A.; Pan, H.; Geng, J.; Ofengeim, D.; Dziedzic, S.A.; Wang, H.; Barrett, V.J.; Ito, Y. Regulation of a distinct activated RIPK1 intermediate bridging complex I and complex II in TNFα-mediated apoptosis. Proc. Natl. Acad. Sci. USA 2018, 115, E5944–E5953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandenabeele, P.; Vanden Berghe, T.; Festjens, N. Caspase inhibitors promote alternative cell death pathways. Sci. STKE 2006, 2006, pe44. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.-W.; Seo, J.-H.; Jeong, M.-H.; Lee, S.-S.; Song, J.-W. The roles of FADD in extrinsic apoptosis and necroptosis. BMB Rep. 2012, 45, 496–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laukens, B.; Jennewein, C.; Schenk, B.; Vanlangenakker, N.; Schier, A.; Cristofanon, S.; Zobel, K.; Deshayes, K.; Vucic, D.; Jeremias, I. Smac mimetic bypasses apoptosis resistance in FADD-or caspase-8-deficient cells by priming for tumor necrosis factor α-induced necroptosis. Neoplasia 2011, 13, 971-IN929. [Google Scholar] [CrossRef] [PubMed]
- Krelin, Y.; Zhang, L.; Kang, T.; Appel, E.; Kovalenko, A.; Wallach, D. Caspase-8 deficiency facilitates cellular transformation in vitro. Cell Death Differ. 2008, 15, 1350–1355. [Google Scholar] [CrossRef] [Green Version]
- Geserick, P.; Hupe, M.; Moulin, M.; Wong, W.W.-L.; Feoktistova, M.; Kellert, B.; Gollnick, H.; Silke, J.; Leverkus, M. Cellular IAPs inhibit a cryptic CD95-induced cell death by limiting RIP1 kinase recruitment. J. Cell Biol. 2009, 187, 1037–1054. [Google Scholar] [CrossRef] [Green Version]
- Imre, G.; Larisch, S.; Rajalingam, K. Ripoptosome: A novel IAP-regulated cell death-signalling platform. J. Mol. Cell Biol. 2011, 3, 324–326. [Google Scholar] [CrossRef] [Green Version]
- Maas, C.; Tromp, J.; Van Laar, J.; Thijssen, R.; Elias, J.; Malara, A.; Krippner-Heidenreich, A.; Silke, J.; Van Oers, M.H.; Eldering, E. CLL cells are resistant to smac mimetics because of an inability to form a ripoptosome complex. Cell Death Dis. 2013, 4, e782. [Google Scholar] [CrossRef] [Green Version]
- Zelic, M.; Pontarelli, F.; Woodworth, L.; Zhu, C.; Mahan, A.; Ren, Y.; LaMorte, M.; Gruber, R.; Keane, A.; Loring, P. RIPK1 activation mediates neuroinflammation and disease progression in multiple sclerosis. Cell Rep. 2021, 35, 109112. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghanavatian, P.; Salehi-Sedeh, H.; Ataei, F.; Hosseinkhani, S. Bioluminescent RIPoptosome Assay for FADD/RIPK1 Interaction Based on Split Luciferase Assay in a Human Neuroblastoma Cell Line SH-SY5Y. Biosensors 2023, 13, 297. https://doi.org/10.3390/bios13020297
Ghanavatian P, Salehi-Sedeh H, Ataei F, Hosseinkhani S. Bioluminescent RIPoptosome Assay for FADD/RIPK1 Interaction Based on Split Luciferase Assay in a Human Neuroblastoma Cell Line SH-SY5Y. Biosensors. 2023; 13(2):297. https://doi.org/10.3390/bios13020297
Chicago/Turabian StyleGhanavatian, Parisa, Hossein Salehi-Sedeh, Farangis Ataei, and Saman Hosseinkhani. 2023. "Bioluminescent RIPoptosome Assay for FADD/RIPK1 Interaction Based on Split Luciferase Assay in a Human Neuroblastoma Cell Line SH-SY5Y" Biosensors 13, no. 2: 297. https://doi.org/10.3390/bios13020297
APA StyleGhanavatian, P., Salehi-Sedeh, H., Ataei, F., & Hosseinkhani, S. (2023). Bioluminescent RIPoptosome Assay for FADD/RIPK1 Interaction Based on Split Luciferase Assay in a Human Neuroblastoma Cell Line SH-SY5Y. Biosensors, 13(2), 297. https://doi.org/10.3390/bios13020297