
Bacterial Targets of Antibiotics in Methicillin-Resistant Staphylococcus aureus



Abstract
:1. Introduction
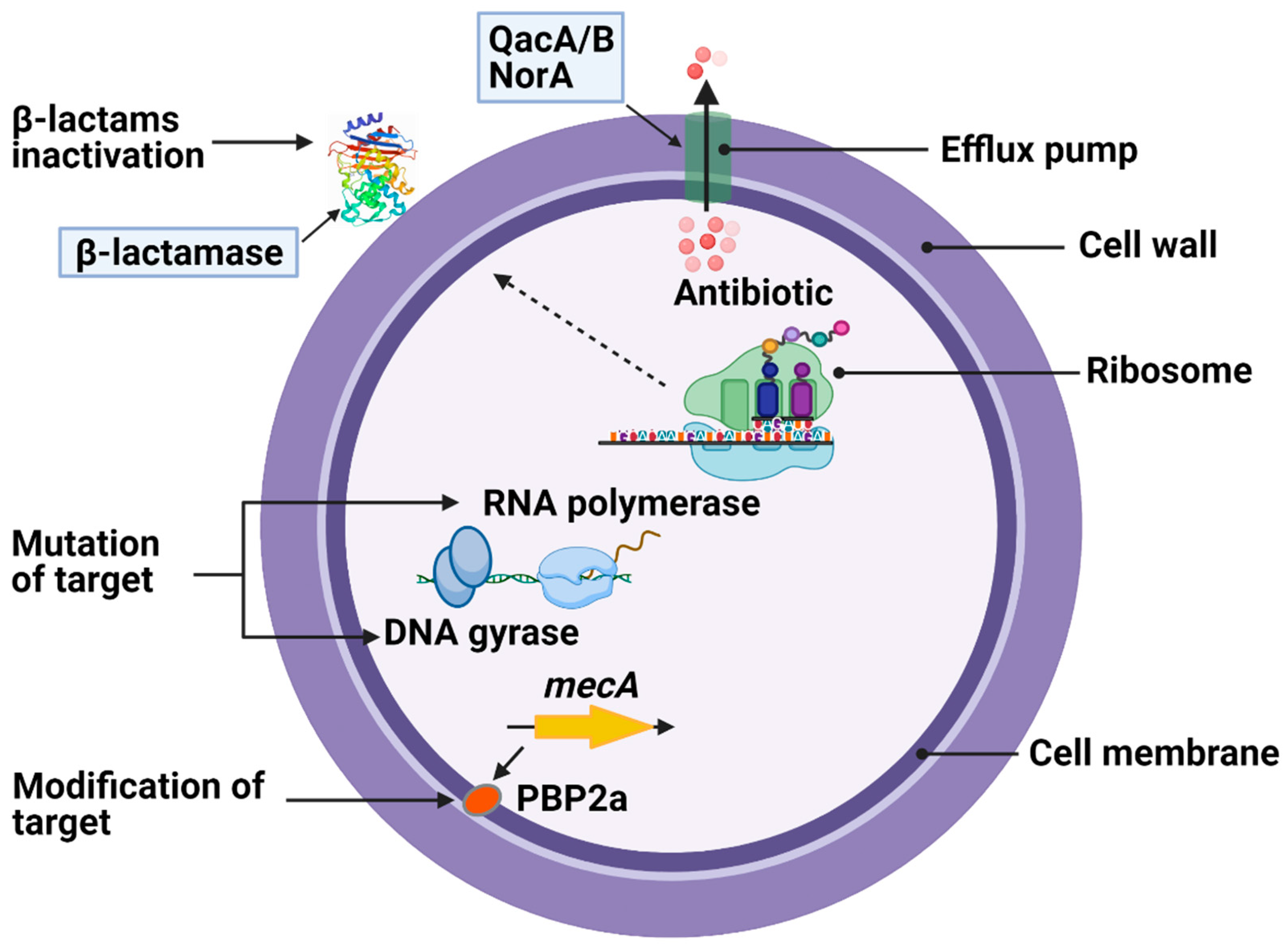
2. Antimicrobial Resistance in S. aureus
3. Treatment of MRSA Infections
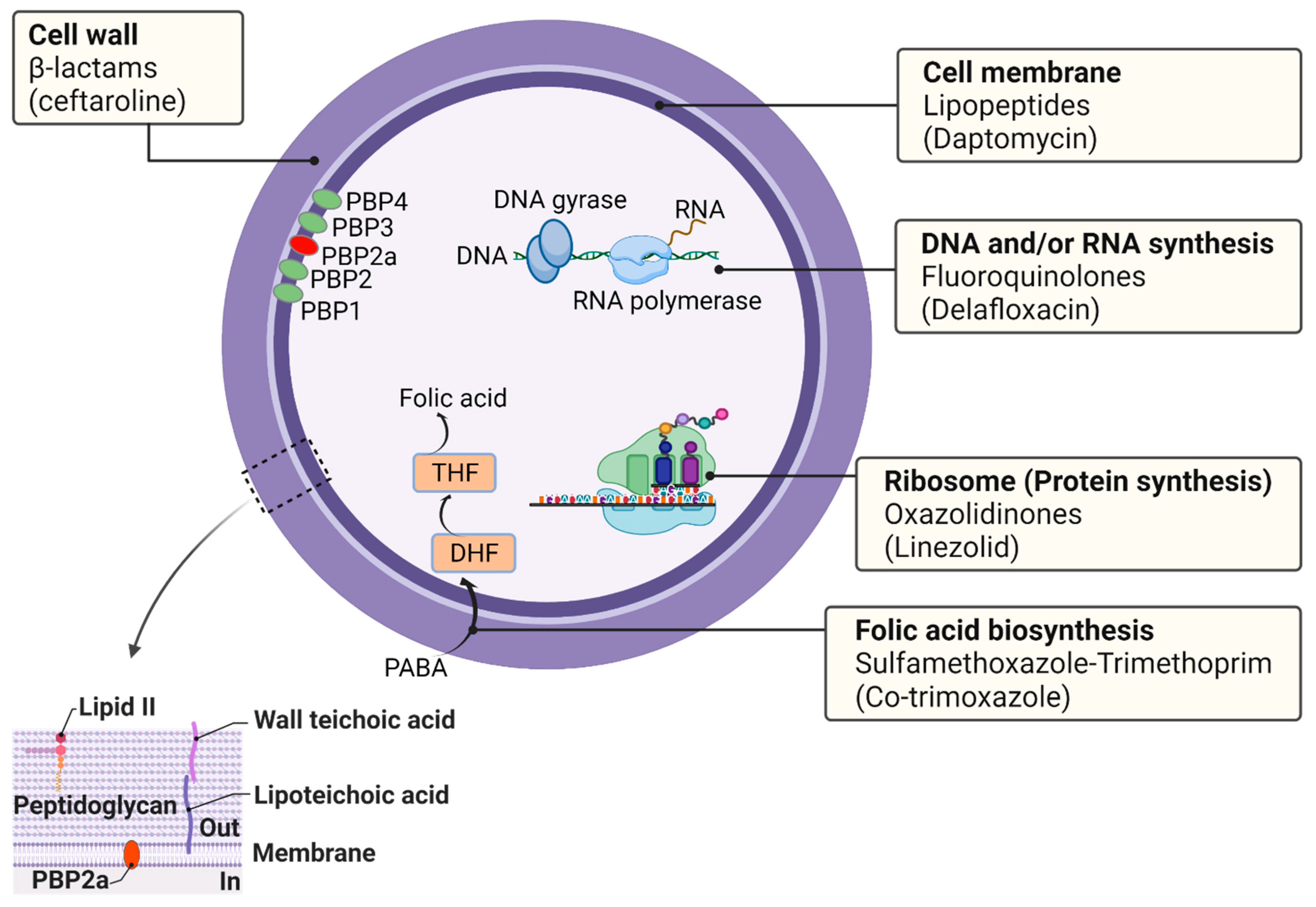
4. Antibiotic Targets in S. aureus
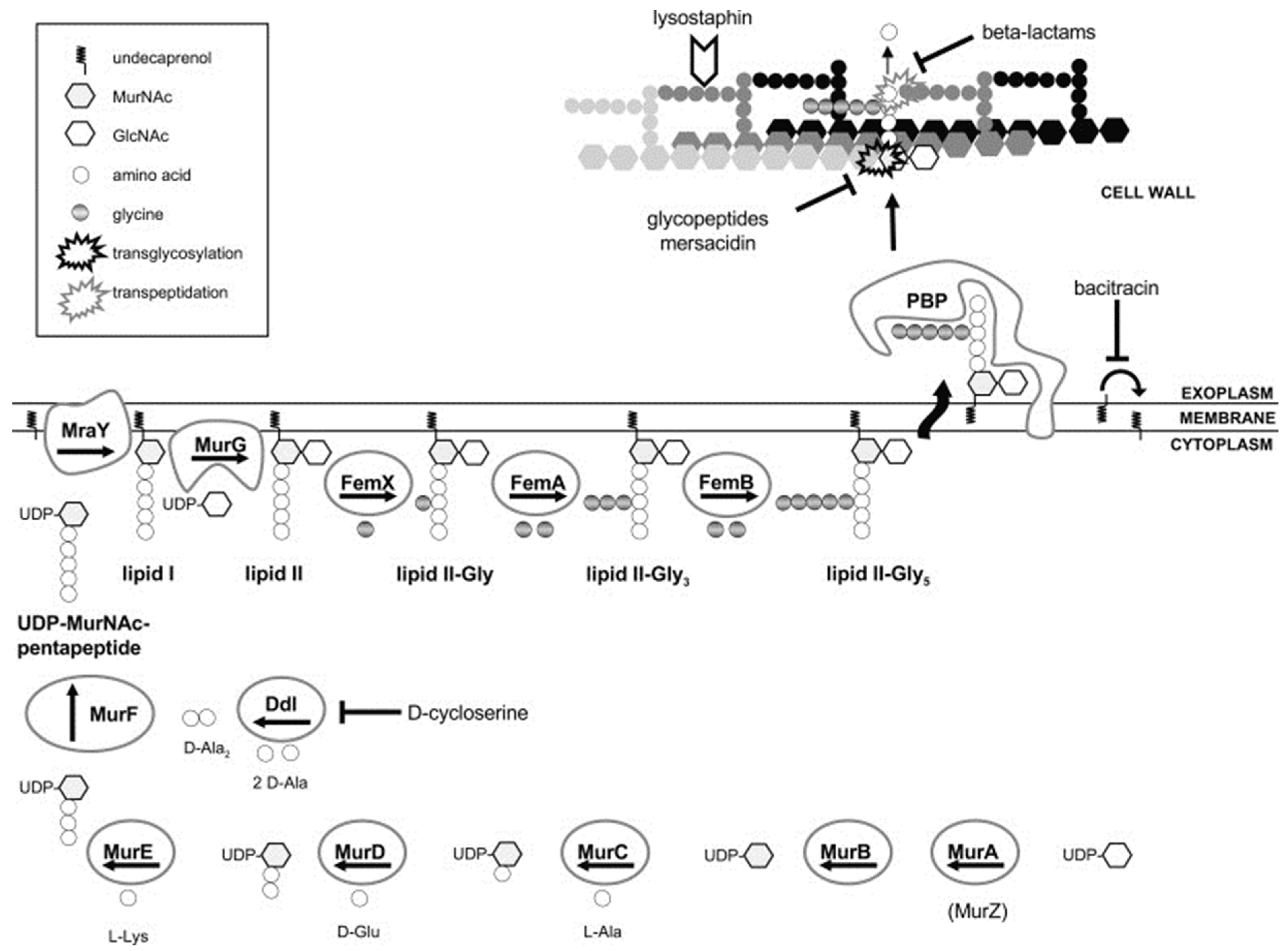
4.1. Cell Wall
4.2. Cell Membrane
4.3. DNA and/or RNA Synthesis
4.4. Ribosomes (Protein Synthesis)
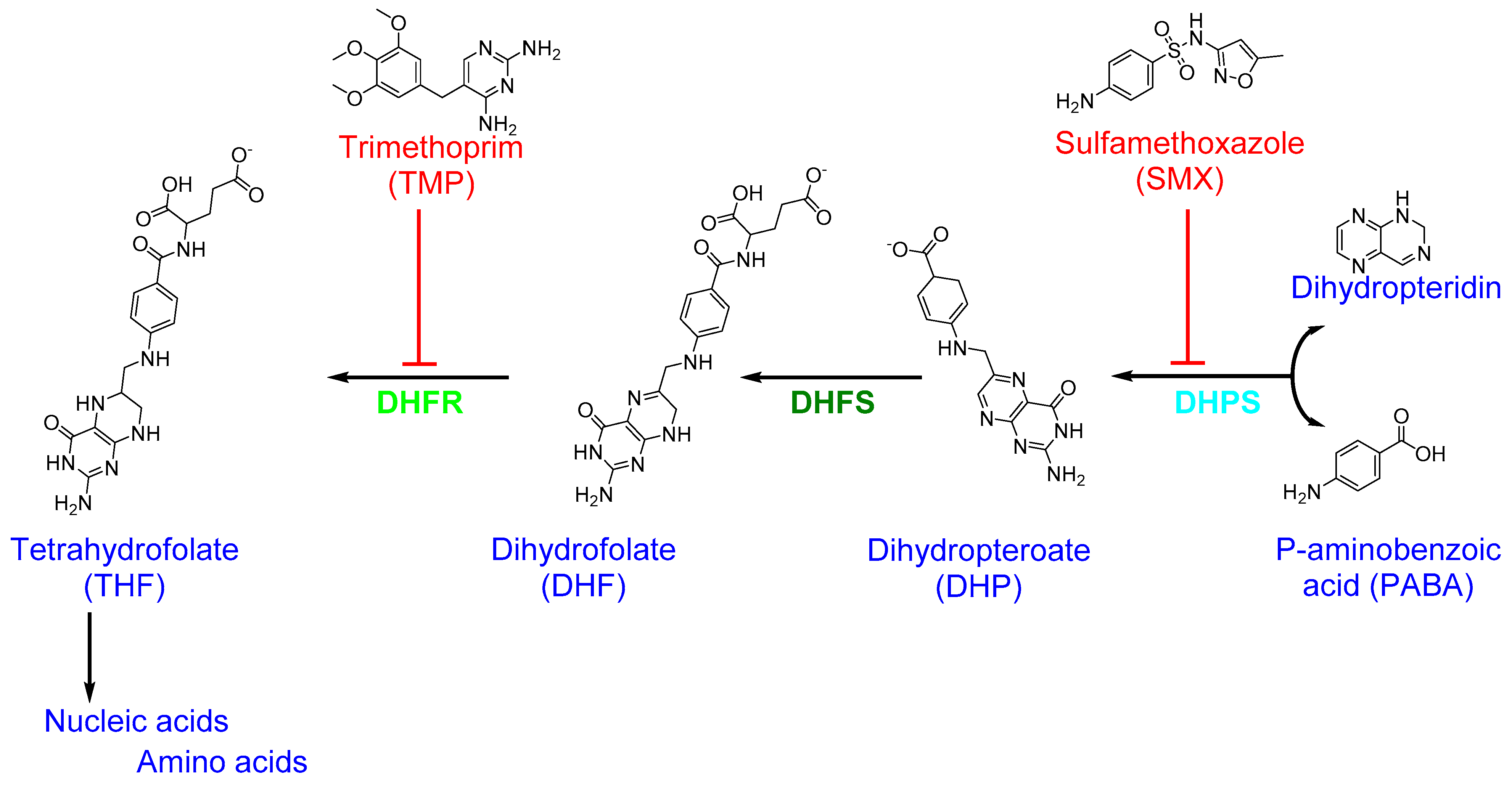
4.5. Folic Acid Biosynthesis (Folate Metabolism)
5. Other Promising Targets
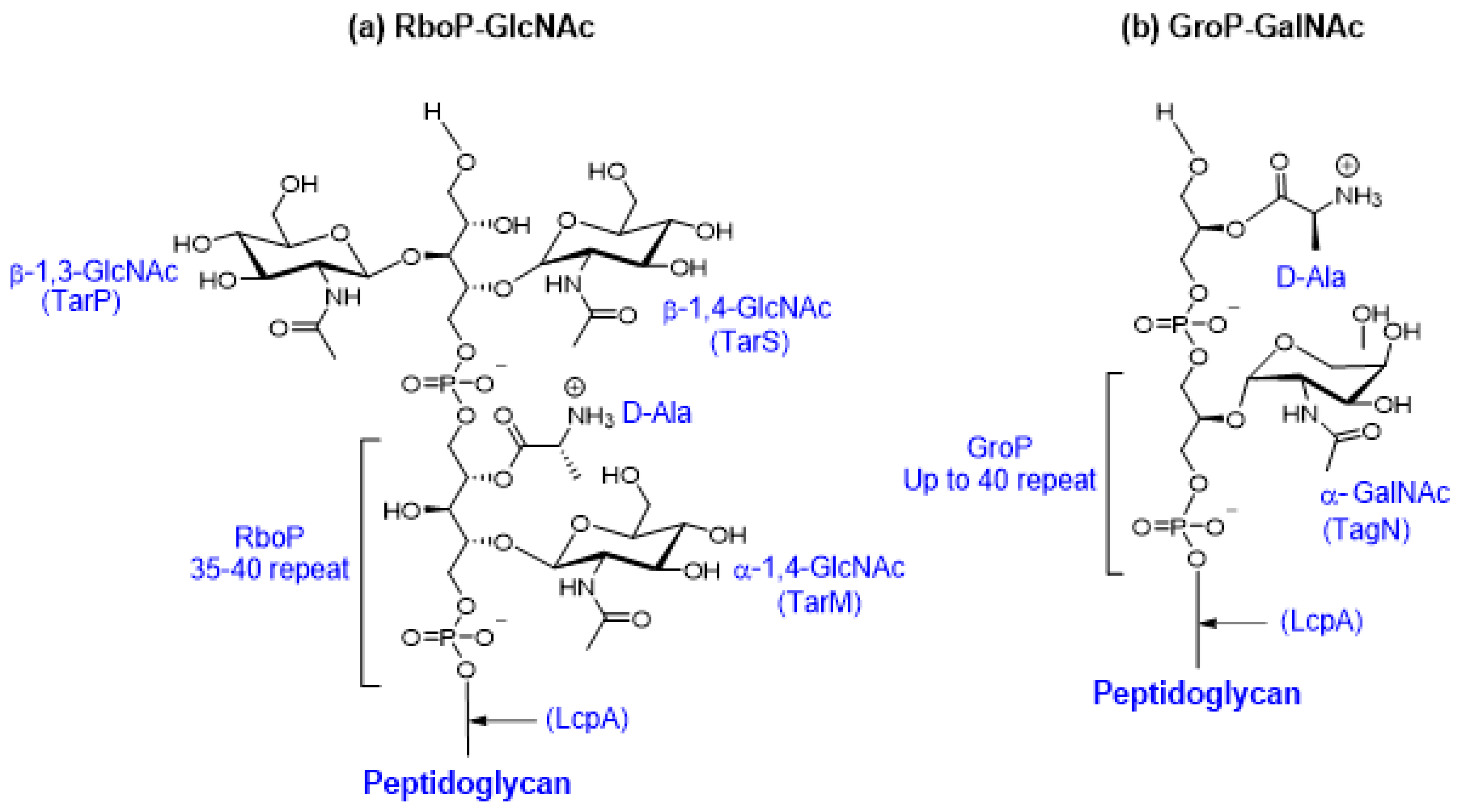
5.1. Teichoic Acid Synthesis
5.2. Aminoacyl-tRNA Synthetases
5.3. Lipid II Cycle
5.4. Auxiliary Factors in β-Lactam Resistance
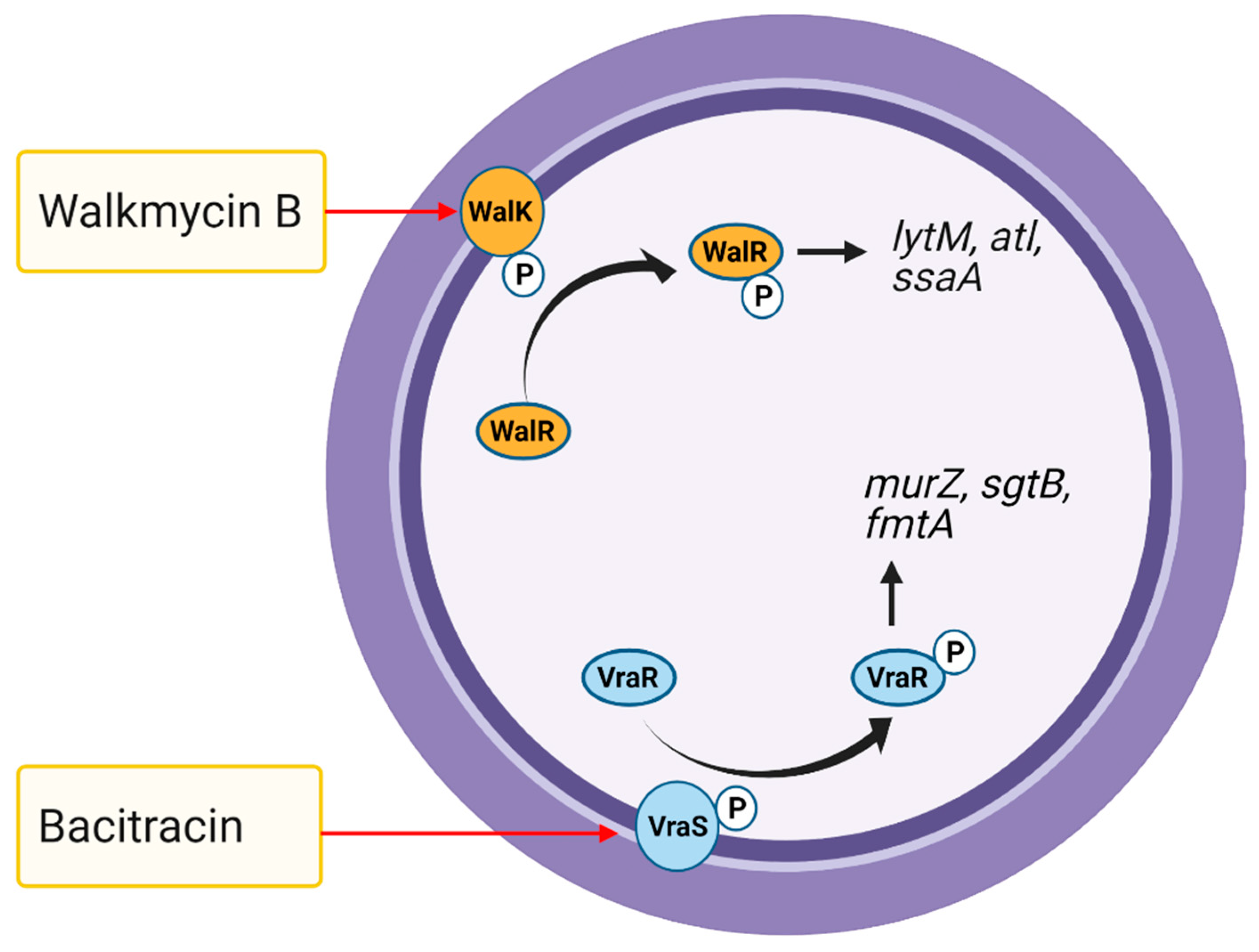
5.5. Two-Component Systems
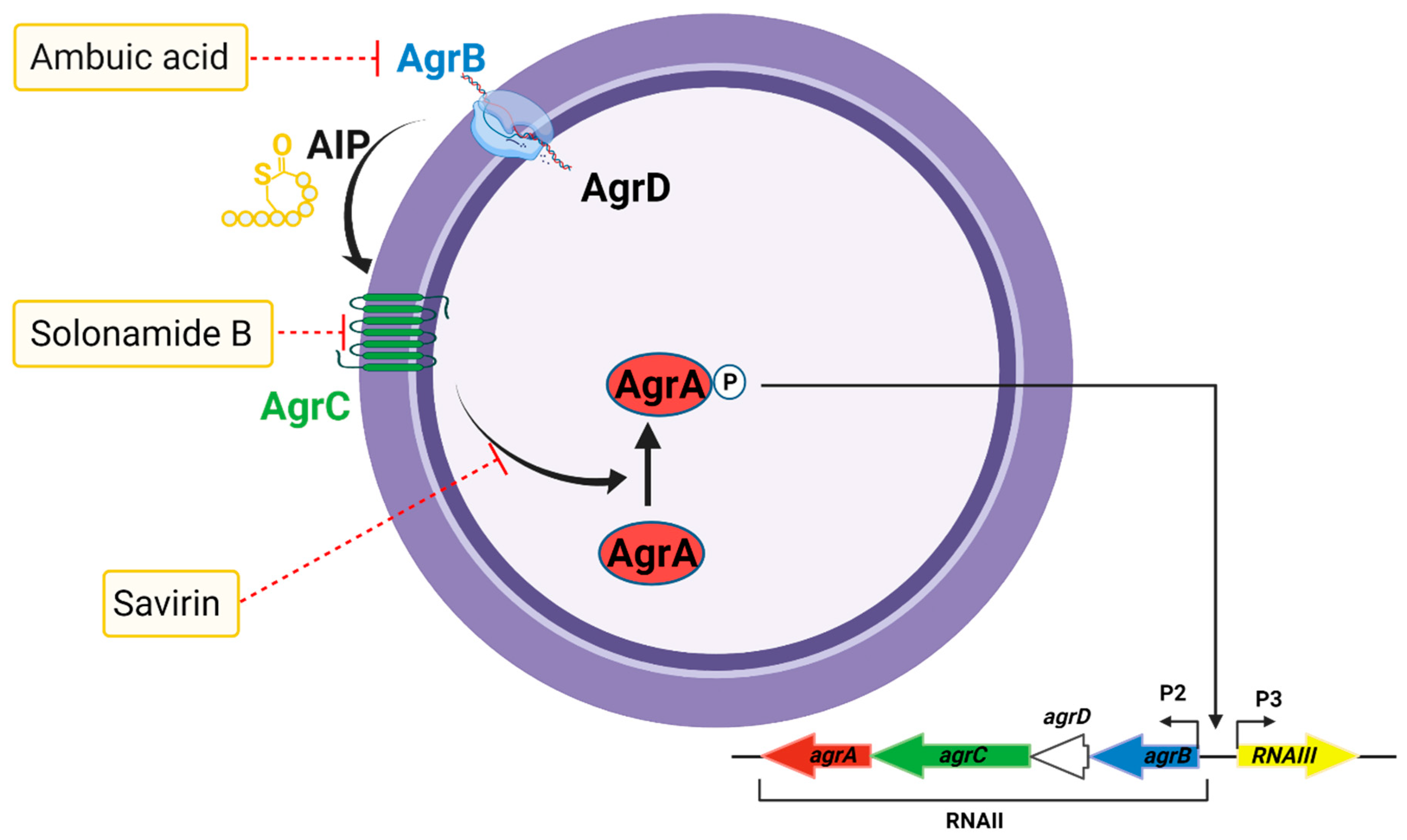
5.6. The Accessory Gene Regulator Quorum Sensing System
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Blair, J.M.A.; Webber, M.A.; Baylay, A.J.; Ogbolu, D.O.; Piddock, L.J.V. Molecular mechanisms of antibiotic resistance. Nat. Rev. Microbiol. 2015, 13, 42–51. [Google Scholar] [CrossRef]
- Boyle-Vavra, S.; Yin, S.; Challapalli, M.; Daum, R.S. Transcriptional Induction of the Penicillin-Binding Protein 2 Gene in Staphylococcus aureus by Cell Wall-Active Antibiotics Oxacillin and Vancomycin. Antimicrob. Agents Chemother. 2003, 47, 1028–1036. [Google Scholar] [CrossRef] [Green Version]
- Zeng, D.; Debabov, D.; Hartsell, T.L.; Cano, R.J.; Adams, S.; Schuyler, J.A.; McMillan, R.; Pace, J.L. Approved Glycopeptide Antibacterial Drugs: Mechanism of Action and Resistance. Cold Spring Harb. Perspect. Med. 2016, 6, a026989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swaney, S.M.; Aoki, H.; Ganoza, M.C.; Shinabarger, D.L. The oxazolidinone linezolid inhibits initiation of protein synthesis in bacteria. Antimicrob. Agents Chemother. 1998, 42, 3251–3255. [Google Scholar] [CrossRef] [Green Version]
- Kisgen, J.J.; Mansour, H.; Unger, N.R.; Childs, L.M. Tedizolid: A new oxazolidinone antimicrobial. Am. J. Health Pharm. 2014, 71, 621–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Champney, W.S.; Burdine, R. Macrolide antibiotics inhibit 50S ribosomal subunit assembly in Bacillus subtilis and Staphylococcus aureus. Antimicrob. Agents Chemother. 1995, 39, 2141–2144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drlica, K. Mechanism of fluoroquinolone action. Curr. Opin. Microbiol. 1999, 2, 504–508. [Google Scholar] [CrossRef]
- Drlica, K.; Malik, M.; Kerns, R.J.; Zhao, X. Quinolone-Mediated Bacterial Death. Antimicrob. Agents Chemother. 2008, 52, 385–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvori, C.; Frontali, L.; Leoni, L.; Tecce, G. Effect of Rifamycin on Protein Synthesis. Nature 1965, 207, 417–418. [Google Scholar] [CrossRef]
- Campbell, E.A.; Korzheva, N.; Mustaev, A.; Murakami, K.; Nair, S.; Goldfarb, A.; Darst, S.A. Structural Mechanism for Rifampicin Inhibition of Bacterial RNA Polymerase. Cell 2001, 104, 901–912. [Google Scholar] [CrossRef]
- Hitchings, G.H. Mechanism of Action of Trimethoprim-Sulfamethoxazole—I. J. Infect. Dis. 1973, 128, S433–S436. [Google Scholar] [CrossRef] [PubMed]
- Grim, S.A.; Rapp, R.P.; Martin, C.A.; Evans, M.E. Trimethoprim-Sulfamethoxazole as a Viable Treatment Option for Infections Caused by Methicillin-Resistant Staphylococcus aureus. Pharmacotherapy 2005, 25, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Rayner, C.; Munckhof, W.J. Antibiotics currently used in the treatment of infections caused by Staphylococcus aureus. Intern. Med. J. 2005, 35, S3–S16. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S.; de Lencastre, H.; Garau, J.; Kluytmans, J.; Malhotra-Kumar, S.; Peschel, A.; Harbarth, S. Methicillin-resistant Staphylococcus aureus. Nat. Rev. Dis. Prim. 2018, 4, 18033. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.A.; Sharma-Kuinkel, B.K.; Maskarinec, S.A.; Eichenberger, E.M.; Shah, P.P.; Carugati, M.; Holland, T.L.; Fowler, V.G. Methicillin-resistant Staphylococcus aureus: An overview of basic and clinical research. Nat. Rev. Microbiol. 2019, 17, 203–218. [Google Scholar] [CrossRef]
- Lowy, F.D. Staphylococcus aureus Infections. N. Engl. J. Med. 1998, 339, 520–532. [Google Scholar] [CrossRef] [PubMed]
- Tong, S.Y.C.; Davis, J.S.; Eichenberger, E.; Holland, T.L.; Fowler, V.G. Staphylococcus aureus infections: Epidemiology, pathophysiology, clinical manifestations, and management. Clin. Microbiol. Rev. 2015, 28, 603–661. [Google Scholar] [CrossRef] [Green Version]
- Wertheim, H.F.L.; Melles, D.C.; Vos, M.C.; van Leeuwen, W.; van Belkum, A.; Verbrugh, H.A.; Nouwen, J.L. The role of nasal carriage in Staphylococcus aureus infections. Lancet Infect. Dis. 2005, 5, 751–762. [Google Scholar] [CrossRef]
- Balasubramanian, D.; Harper, L.; Shopsin, B.; Torres, V.J. Staphylococcus aureus pathogenesis in diverse host environments. Pathog. Dis. 2017, 75, ftx005. [Google Scholar] [CrossRef] [Green Version]
- Kourtis, A.P.; Hatfield, K.; Baggs, J.; Mu, Y.; See, I.; Epson, E.; Nadle, J.; Kainer, M.A.; Dumyati, G.; Petit, S.; et al. Vital Signs: Epidemiology and Recent Trends in Methicillin-Resistant and in Methicillin-Susceptible Staphylococcus aureus Bloodstream Infections—United States. MMWR. Morb. Mortal. Wkly. Rep. 2019, 68, 214–219. [Google Scholar] [CrossRef] [Green Version]
- Tsuzuki, S.; Matsunaga, N.; Yahara, K.; Gu, Y.; Hayakawa, K.; Hirabayashi, A.; Kajihara, T.; Sugai, M.; Shibayama, K.; Ohmagari, N. National trend of blood-stream infection attributable deaths caused by Staphylococcus aureus and Escherichia coli in Japan. J. Infect. Chemother. 2020, 26, 367–371. [Google Scholar] [CrossRef]
- Boucher, H.W.; Talbot, G.H.; Bradley, J.S.; Edwards, J.E.; Gilbert, D.; Rice, L.B.; Scheld, M.; Spellberg, B.; Bartlett, J. Bad Bugs, No Drugs: No ESKAPE! An Update from the Infectious Diseases Society of America. Clin. Infect. Dis. 2009, 48, 1–12. [Google Scholar] [CrossRef] [Green Version]
- WHO. Antimicrobial Resistance: Global Report on Surveillance 2014. 2014. Available online: https://www.who.int/antimicrobial-resistance/publications/surveillancereport/en/ (accessed on 12 March 2021).
- Chambers, H.F.; DeLeo, F.R. Waves of resistance: Staphylococcus aureus in the antibiotic era. Nat. Rev. Microbiol. 2009, 7, 629–641. [Google Scholar] [CrossRef]
- Peacock, S.J.; Paterson, G.K. Mechanisms of Methicillin Resistance in Staphylococcus aureus. Annu. Rev. Biochem. 2015, 84, 577–601. [Google Scholar] [CrossRef] [PubMed]
- Vestergaard, M.; Frees, D.; Ingmer, H. Antibiotic Resistance and the MRSA Problem. In Gram-Positive Pathogens; ASM Press: Washington, DC, USA, 2019; pp. 747–765. [Google Scholar]
- Jensen, S.O.; Lyon, B.R. Genetics of antimicrobial resistance in Staphylococcus aureus. Future Microbiol. 2009, 4, 565–582. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Katayama, Y.; Hiramatsu, K. Cloning and Nucleotide Sequence Determination of the Entire mec DNA of Pre-Methicillin-Resistant Staphylococcus aureus N315. Antimicrob. Agents Chemother. 1999, 43, 1449–1458. [Google Scholar] [CrossRef] [Green Version]
- Jang, S. Multidrug efflux pumps in Staphylococcus aureus and their clinical implications. J. Microbiol. 2016, 54, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Foster, T.J. The remarkably multifunctional fibronectin binding proteins of Staphylococcus aureus. Eur. J. Clin. Microbiol. Infect. Dis. 2016, 35, 1923–1931. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.I.; Lee, Y.-M.; Park, K.-H.; Ryu, B.-H.; Hong, K.-W.; Kim, S.; Bae, I.-G.; Cho, O.-H. Clinical and Molecular Characteristics of qacA- and qacB-Positive Methicillin-Resistant Staphylococcus aureus Causing Bloodstream Infections. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef] [Green Version]
- Malachowa, N.; DeLeo, F.R. Mobile genetic elements of Staphylococcus aureus. Cell. Mol. Life Sci. 2010, 67, 3057–3071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, H.S.; Lee, H.; Roh, K.H.; Yum, J.H.; Yong, D.; Lee, K.; Chong, Y. Prevalence of Inducible Clindamycin Resistance in Staphylococcal Isolates at a Korean Tertiary Care Hospital. Yonsei Med. J. 2006, 47, 480. [Google Scholar] [CrossRef]
- Hartman, B.J.; Tomasz, A. Low-affinity penicillin-binding protein associated with beta-lactam resistance in Staphylococcus aureus. J. Bacteriol. 1984, 158, 513–516. [Google Scholar] [CrossRef] [Green Version]
- Fuda, C.C.S.; Fisher, J.F.; Mobashery, S. β-Lactam resistance in Staphylococcus aureus: The adaptive resistance of a plastic genome. Cell. Mol. Life Sci. 2005, 62, 2617–2633. [Google Scholar] [CrossRef] [PubMed]
- Chambers, H.F.; Sachdeva, M. Binding of β-Lactam Antibiotics to Penicillin-Binding Proteins in Methicillin-Resistant Staphylococcus aureus. J. Infect. Dis. 1990, 161, 1170–1176. [Google Scholar] [CrossRef] [PubMed]
- Kirby, W.M.M. Extraction of a highly potent penicillin inactivator from penicillin resistant staphylococci. Science 1944, 99, 452–453. [Google Scholar] [CrossRef]
- Fisher, J.F.; Mobashery, S. β-Lactams against the Fortress of the Gram-Positive Staphylococcus aureus Bacterium. Chem. Rev. 2020. [Google Scholar] [CrossRef]
- Sauvage, E.; Kerff, F.; Terrak, M.; Ayala, J.A.; Charlier, P. The penicillin-binding proteins: Structure and role in peptidoglycan biosynthesis. FEMS Microbiol. Rev. 2008, 32, 234–258. [Google Scholar] [CrossRef] [Green Version]
- Shalaby, M.-A.W.; Dokla, E.M.E.; Serya, R.A.T.; Abouzid, K.A.M. Penicillin binding protein 2a: An overview and a medicinal chemistry perspective. Eur. J. Med. Chem. 2020, 199, 112312. [Google Scholar] [CrossRef]
- de Lencastre, H.; Tomasz, A. Reassessment of the number of auxiliary genes essential for expression of high-level methicillin resistance in Staphylococcus aureus. Antimicrob. Agents Chemother. 1994, 38, 2590–2598. [Google Scholar] [CrossRef] [Green Version]
- Berger-Bächi, B.; Barberis-Maino, L.; Strässle, A.; Kayser, F.H. FemA, a host-mediated factor essential for methicillin resistance in Staphylococcus aureus: Molecular cloning and characterization. Mol. Gen. Genet. MGG 1989, 219, 263–269. [Google Scholar] [CrossRef]
- Katayama, Y.; Ito, T.; Hiramatsu, K. A New Class of Genetic Element, Staphylococcus Cassette Chromosome mec, Encodes Methicillin Resistance in Staphylococcus aureus. Antimicrob. Agents Chemother. 2000, 44, 1549–1555. [Google Scholar] [CrossRef] [Green Version]
- Matsuhashi, M.; Song, M.D.; Ishino, F.; Wachi, M.; Doi, M.; Inoue, M.; Ubukata, K.; Yamashita, N.; Konno, M. Molecular cloning of the gene of a penicillin-binding protein supposed to cause high resistance to beta-lactam antibiotics in Staphylococcus aureus. J. Bacteriol. 1986, 167, 975–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakhundi, S.; Zhang, K. Methicillin-Resistant Staphylococcus aureus: Molecular Characterization, Evolution, and Epidemiology. Clin. Microbiol. Rev. 2018, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, M.Z.; Daum, R.S. Community-associated methicillin-resistant Staphylococcus aureus: Epidemiology and clinical consequences of an emerging epidemic. Clin. Microbiol. Rev. 2010, 23, 616–687. [Google Scholar]
- Kim, C.; Milheiriço, C.; Gardete, S.; Holmes, M.A.; Holden, M.T.G.; de Lencastre, H.; Tomasz, A. Properties of a Novel PBP2A Protein Homolog from Staphylococcus aureus Strain LGA251 and Its Contribution to the β-Lactam-resistant Phenotype*. J. Biol. Chem. 2012, 287, 36854–36863. [Google Scholar] [CrossRef] [Green Version]
- Zhao, G.; Meier, T.I.; Hoskins, J.; Jaskunas, S.R. Penicillin-Binding Protein 2a of Streptococcus pneumoniae: Expression in Escherichia coli and Purification and Refolding of Inclusion Bodies into a Soluble and Enzymatically Active Enzyme. Protein Expr. Purif. 1999, 16, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Sanbongi, Y.; Ida, T.; Ishikawa, M.; Osaki, Y.; Kataoka, H.; Suzuki, T.; Kondo, K.; Ohsawa, F.; Yonezawa, M. Complete Sequences of Six Penicillin-Binding Protein Genes from 40 Streptococcus pneumoniae Clinical Isolates Collected in Japan. Antimicrob. Agents Chemother. 2004, 48, 2244–2250. [Google Scholar] [CrossRef] [Green Version]
- Zhao, G.; Meier, T.I.; Hoskins, J.; McAllister, K.A. Identification and Characterization of the Penicillin-Binding Protein 2a of Streptococcus pneumoniae and Its Possible Role in Resistance to β-Lactam Antibiotics. Antimicrob. Agents Chemother. 2000, 44, 1745–1748. [Google Scholar] [CrossRef] [Green Version]
- Grebe, T.; Hakenbeck, R. Penicillin-binding proteins 2b and 2x of Streptococcus pneumoniae are primary resistance determinants for different classes of beta-lactam antibiotics. Antimicrob. Agents Chemother. 1996, 40, 829–834. [Google Scholar] [CrossRef] [Green Version]
- Lowy, F.D. Antimicrobial resistance: The example of Staphylococcus aureus. J. Clin. Investig. 2003, 111, 1265–1273. [Google Scholar] [CrossRef]
- Maddux, M.S. Effects of ß-Lactamase-Mediated Antimicrobial Resistance: The Role of ß-Lactamase Inhibitors. Pharmacother. J. Hum. Pharmacol. Drug Ther. 1991, 11, 40S–50S. [Google Scholar]
- Tooke, C.L.; Hinchliffe, P.; Bragginton, E.C.; Colenso, C.K.; Hirvonen, V.H.A.; Takebayashi, Y.; Spencer, J. β-Lactamases and β-Lactamase Inhibitors in the 21st Century. J. Mol. Biol. 2019, 431, 3472–3500. [Google Scholar] [CrossRef] [PubMed]
- Babic, M.; Hujer, A.; Bonomo, R. What’s new in antibiotic resistance? Focus on beta-lactamases. Drug Resist. Updat. 2006, 9, 142–156. [Google Scholar] [CrossRef]
- Bush, K.; Bradford, P.A. Epidemiology of β-lactamase-producing pathogens. Clin. Microbiol. Rev. 2020, 33. [Google Scholar] [CrossRef]
- Bush, K. Past and present perspectives on β-lactamases. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef] [Green Version]
- Meini, S.; Tascini, C.; Cei, M.; Sozio, E.; Rossolini, G.M. AmpC β-lactamase-producing Enterobacterales: What a clinician should know. Infection 2019, 47, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Kot, B.; Wierzchowska, K.; Piechota, M.; Grużewska, A. Antimicrobial Resistance Patterns in Methicillin-Resistant Staphylococcus aureus from Patients Hospitalized during 2015–2017 in Hospitals in Poland. Med. Princ. Pract. 2020, 29, 61–68. [Google Scholar] [CrossRef]
- Rossato, A.M.; Primon-Barros, M.; Rocha, L.d.L.; Reiter, K.C.; Dias, C.A.G.; D’Azevedo, P.A. Resistance profile to antimicrobials agents in methicillin-resistant Staphylococcus aureus isolated from hospitals in South Brazil between 2014–2019. Rev. Soc. Bras. Med. Trop. 2020, 53, e20200431. [Google Scholar] [CrossRef] [PubMed]
- Weigel, L.M. Genetic Analysis of a High-Level Vancomycin-Resistant Isolate of Staphylococcus aureus. Science 2003, 302, 1569–1571. [Google Scholar] [CrossRef]
- Chang, S.; Sievert, D.M.; Hageman, J.C.; Boulton, M.L.; Tenover, F.C.; Downes, F.P.; Shah, S.; Rudrik, J.T.; Pupp, G.R.; Brown, W.J.; et al. Infection with Vancomycin-Resistant Staphylococcus aureus Containing the vanA Resistance Gene. N. Engl. J. Med. 2003, 348, 1342–1347. [Google Scholar] [CrossRef]
- Mwangi, M.M.; Wu, S.W.; Zhou, Y.; Sieradzki, K.; de Lencastre, H.; Richardson, P.; Bruce, D.; Rubin, E.; Myers, E.; Siggia, E.D.; et al. Tracking the in vivo evolution of multidrug resistance in Staphylococcus aureus by whole-genome sequencing. Proc. Natl. Acad. Sci. USA 2007, 104, 9451–9456. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-J.; Huang, Y.-C.; Chiu, C.-H. Multiple pathways of cross-resistance to glycopeptides and daptomycin in persistent MRSA bacteraemia. J. Antimicrob. Chemother. 2015, 70, 2965–2972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thitiananpakorn, K.; Aiba, Y.; Tan, X.-E.; Watanabe, S.; Kiga, K.; Sato’o, Y.; Boonsiri, T.; Li, F.-Y.; Sasahara, T.; Taki, Y.; et al. Association of mprF mutations with cross-resistance to daptomycin and vancomycin in methicillin-resistant Staphylococcus aureus (MRSA). Sci. Rep. 2020, 10, 16107. [Google Scholar] [CrossRef] [PubMed]
- Bayer, A.S.; Mishra, N.N.; Cheung, A.L.; Rubio, A.; Yang, S.-J. Dysregulation of mprF and dltABCD expression among daptomycin-non-susceptible MRSA clinical isolates. J. Antimicrob. Chemother. 2016, 71, 2100–2104. [Google Scholar] [CrossRef] [Green Version]
- Dubrac, S.; Bisicchia, P.; Devine, K.M.; Msadek, T. A matter of life and death: Cell wall homeostasis and the WalKR (YycGF) essential signal transduction pathway. Mol. Microbiol. 2008, 70, 1307–1322. [Google Scholar] [CrossRef] [PubMed]
- Jones, T.; Yeaman, M.R.; Sakoulas, G.; Yang, S.-J.; Proctor, R.A.; Sahl, H.-G.; Schrenzel, J.; Xiong, Y.Q.; Bayer, A.S. Failures in Clinical Treatment of Staphylococcus aureus Infection with Daptomycin Are Associated with Alterations in Surface Charge, Membrane Phospholipid Asymmetry, and Drug Binding. Antimicrob. Agents Chemother. 2008, 52, 269–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.-J.; Nast, C.C.; Mishra, N.N.; Yeaman, M.R.; Fey, P.D.; Bayer, A.S. Cell Wall Thickening Is Not a Universal Accompaniment of the Daptomycin Nonsusceptibility Phenotype in Staphylococcus aureus: Evidence for Multiple Resistance Mechanisms. Antimicrob. Agents Chemother. 2010, 54, 3079–3085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, N.; Cascella, M. Beta Lactam Antibiotics. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Arbeit, R.D.; Maki, D.; Tally, F.P.; Campanaro, E.; Eisenstein, B.I. The Safety and Efficacy of Daptomycin for the Treatment of Complicated Skin and Skin-Structure Infections. Clin. Infect. Dis. 2004, 38, 1673–1681. [Google Scholar] [CrossRef]
- Shorr, A.F.; Kunkel, M.J.; Kollef, M. Linezolid versus vancomycin for Staphylococcus aureus bacteraemia: Pooled analysis of randomized studies. J. Antimicrob. Chemother. 2005, 56, 923–929. [Google Scholar] [CrossRef]
- Weigelt, J.; Itani, K.; Stevens, D.; Lau, W.; Dryden, M.; Knirsch, C. Linezolid versus Vancomycin in Treatment of Complicated Skin and Soft Tissue Infections. Antimicrob. Agents Chemother. 2005, 49, 2260–2266. [Google Scholar] [CrossRef] [Green Version]
- Fowler, V.G.; Boucher, H.W.; Corey, G.R.; Abrutyn, E.; Karchmer, A.W.; Rupp, M.E.; Levine, D.P.; Chambers, H.F.; Tally, F.P.; Vigliani, G.A.; et al. Daptomycin versus Standard Therapy for Bacteremia and Endocarditis Caused by Staphylococcus aureus. N. Engl. J. Med. 2006, 355, 653–665. [Google Scholar] [CrossRef] [Green Version]
- Goto, R.; Inose, R.; Kusama, Y.; Kawabe, A.; Ishii, S.; Ebisui, A.; Ishikane, M.; Yagi, T.; Ohmagari, N.; Muraki, Y. Trends of the Use of Anti-methicillin-Resistant Staphylococcus aureus Agents in Japan Based on Sales Data from 2006 to 2015. Biol. Pharm. Bull. 2020, 43, 1906–1910. [Google Scholar] [CrossRef]
- Pfizer Summary of Product Characteristics: Zinforo 600 mg Powder for Concentrate for Solution for Infusion. Available online: https://www.ema.europa.eu/en/documents/product-information/zinforo-epar-product-information_en.pdf (accessed on 20 January 2021).
- Allergan Teflaro® (Ceftaroline Fosamil) for Injection, for Intravenous Use Prescribing Information. Available online: https://media.allergan.com/actavis/actavis/media/allergan-pdf-documents/product-prescribing/Teflaro-USPI-09_2019-2.pdf (accessed on 20 January 2021).
- Tyers, M.; Wright, G.D. Drug combinations: A strategy to extend the life of antibiotics in the 21st century. Nat. Rev. Microbiol. 2019, 17, 141–155. [Google Scholar] [CrossRef]
- Salam, A.M.; Quave, C.L. Targeting Virulence in Staphylococcus aureus by Chemical Inhibition of the Accessory Gene Regulator System In Vivo. mSphere 2018, 3, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, R.C.; Popat, R.; Diggle, S.P.; Brown, S.P. Targeting virulence: Can we make evolution-proof drugs? Nat. Rev. Microbiol. 2014, 12, 300–308. [Google Scholar] [CrossRef] [PubMed]
- Moller, A.G.; Winston, K.; Ji, S.; Wang, J.; Hargita Davis, M.N.; Solís-Lemus, C.R.; Read, T.D. Genes Influencing Phage Host Range in Staphylococcus aureus on a Species-Wide Scale. mSphere 2021, 6. [Google Scholar] [CrossRef] [PubMed]
- Nobrega, F.L.; Costa, A.R.; Kluskens, L.D.; Azeredo, J. Revisiting phage therapy: New applications for old resources. Trends Microbiol. 2015, 23, 185–191. [Google Scholar] [CrossRef] [Green Version]
- Gilmer, D.B.; Schmitz, J.E.; Euler, C.W.; Fischetti, V.A. Novel Bacteriophage Lysin with Broad Lytic Activity Protects against Mixed Infection by Streptococcus pyogenes and Methicillin-Resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2013, 57, 2743–2750. [Google Scholar] [CrossRef] [Green Version]
- Lood, R.; Molina, H.; Fischetti, V.A. Determining bacteriophage endopeptidase activity using either fluorophore-quencher labeled peptides combined with liquid chromatography-mass spectrometry (LC-MS) or Förster resonance energy transfer (FRET) assays. PLoS ONE 2017, 12, e0173919. [Google Scholar] [CrossRef]
- Wright, G.D. Q&A: Antibiotic resistance: Where does it come from and what can we do about it? BMC Biol. 2010, 8, 123. [Google Scholar]
- Frère, J.M.; Page, M.G.P. Penicillin-binding proteins: Evergreen drug targets. Curr. Opin. Pharmacol. 2014, 18, 112–119. [Google Scholar] [CrossRef]
- Biedenbach, D.J.; Alm, R.A.; Lahiri, S.D.; Reiszner, E.; Hoban, D.J.; Sahm, D.F.; Bouchillon, S.K.; Ambler, J.E. In vitro activity of ceftaroline against Staphylococcus aureus isolated in 2012 from Asia-Pacific countries as part of the AWARE surveillance program. Antimicrob. Agents Chemother. 2016, 60, 343–347. [Google Scholar] [CrossRef] [Green Version]
- Kosowska-Shick, K.; McGhee, P.L.; Appelbaum, P.C. Affinity of Ceftaroline and Other -Lactams for Penicillin-Binding Proteins from Staphylococcus aureus and Streptococcus pneumoniae. Antimicrob. Agents Chemother. 2010, 54, 1670–1677. [Google Scholar] [CrossRef] [Green Version]
- Moisan, H.; Pruneau, M.; Malouin, F. Binding of ceftaroline to penicillin-binding proteins of Staphylococcus aureus and Streptococcus pneumoniae. J. Antimicrob. Chemother. 2010, 65, 713–716. [Google Scholar] [CrossRef]
- Laudano, J.B. Ceftaroline fosamil: A new broad-spectrum cephalosporin. J. Antimicrob. Chemother. 2011, 66, iii11–iii18. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, P.E. Structure, biochemistry and mechanism of action of glycopeptide antibiotics. Eur. J. Clin. Microbiol. Infect. Dis. 1989, 8, 943–950. [Google Scholar] [CrossRef] [PubMed]
- Parenti, F. Structure and mechanism of action of teicoplanin. J. Hosp. Infect. 1986, 7, 79–83. [Google Scholar] [CrossRef]
- Grein, F.; Müller, A.; Scherer, K.M.; Liu, X.; Ludwig, K.C.; Klöckner, A.; Strach, M.; Sahl, H.; Kubitscheck, U.; Schneider, T. Ca2+-Daptomycin targets cell wall biosynthesis by forming a tripartite complex with undecaprenyl-coupled intermediates and membrane lipids. Nat. Commun. 2020, 11, 1455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, S.D.; Palmer, M. The action mechanism of daptomycin. Bioorg. Med. Chem. 2016, 24, 6253–6268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayer, A.S.; Schneider, T.; Sahl, H.G. Mechanisms of daptomycin resistance in Staphylococcus aureus: Role of the cell membrane and cell wall. Ann. N. Y. Acad. Sci. 2013, 1277, 139–158. [Google Scholar] [CrossRef] [Green Version]
- Muraih, J.K.; Pearson, A.; Silverman, J.; Palmer, M. Oligomerization of daptomycin on membranes. Biochim. Biophys. Acta—Biomembr. 2011, 1808, 1154–1160. [Google Scholar] [CrossRef] [Green Version]
- Silverman, J.A.; Perlmutter, N.G.; Shapiro, H.M. Correlation of Daptomycin Bactericidal Activity and Membrane Depolarization in Staphylococcus aureus. Antimicrob. Agents Chemother. 2003, 47, 2538–2544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mascio, C.T.M.; Alder, J.D.; Silverman, J.A. Bactericidal Action of Daptomycin against Stationary-Phase and Nondividing Staphylococcus aureus Cells. Antimicrob. Agents Chemother. 2007, 51, 4255–4260. [Google Scholar] [CrossRef] [Green Version]
- Allington, D. Quinupristin/dalfopristin: A therapeutic review. Clin. Ther. 2001, 23, 24–44. [Google Scholar] [CrossRef]
- Long, J.K. Agents for the treatment of multidrug-resistant gram-positive endocarditis. Curr. Infect. Dis. Rep. 2005, 7, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Schluenzen, F.; Tocilj, A.; Zarivach, R.; Harms, J.; Gluehmann, M.; Janell, D.; Bashan, A.; Bartels, H.; Agmon, I.; Franceschi, F.; et al. Structure of Functionally Activated Small Ribosomal Subunit at 3.3 Å Resolution. Cell 2000, 102, 615–623. [Google Scholar] [CrossRef] [Green Version]
- Carter, A.P.; Clemons, W.M.; Brodersen, D.E.; Morgan-Warren, R.J.; Wimberly, B.T.; Ramakrishnan, V. Functional insights from the structure of the 30S ribosomal subunit and its interactions with antibiotics. Nature 2000, 407, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Magnet, S.; Blanchard, J.S. Molecular Insights into Aminoglycoside Action and Resistance. Chem. Rev. 2005, 105, 477–498. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T. Arbekacin: Another novel agent for treating infections due to methicillin-resistant Staphylococcus aureus and multidrug-resistant Gram-negative pathogens. Clin. Pharmacol. Adv. Appl. 2014, 6, 139. [Google Scholar] [CrossRef] [Green Version]
- Zhanel, G.G.; Love, R.; Adam, H.; Golden, A.; Zelenitsky, S.; Schweizer, F.; Gorityala, B.; Lagacé-Wiens, P.R.S.; Rubinstein, E.; Walkty, A.; et al. Tedizolid: A Novel Oxazolidinone with Potent Activity Against Multidrug-Resistant Gram-Positive Pathogens. Drugs 2015, 75, 253–270. [Google Scholar] [CrossRef]
- Ross, J.I.; Eady, E.A.; Cove, J.H.; Cunliffe, W.J. 16S rRNA Mutation Associated with Tetracycline Resistance in a Gram-Positive Bacterium. Antimicrob. Agents Chemother. 1998, 42, 1702–1705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geigenmuller, U.; Nierhaus, K.H. Tetracycline can inhibit tRNA binding to the ribosomal P site as well as to the A site. Eur. J. Biochem. 1986, 161, 723–726. [Google Scholar] [CrossRef] [PubMed]
- Chernyshev, A.; Fleischmann, T.; Kohen, A. Thymidyl biosynthesis enzymes as antibiotic targets. Appl. Microbiol. Biotechnol. 2007, 74, 282–289. [Google Scholar] [CrossRef]
- Hampele, I.C.; D’Arcy, A.; Dale, G.E.; Kostrewa, D.; Nielsen, J.; Oefner, C.; Page, M.G.P.; Schönfeld, H.-J.; Stüber, D.; Then, R.L. Structure and function of the dihydropteroate synthase from staphylococcus aureus. J. Mol. Biol. 1997, 268, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Kompis, I.M.; Islam, K.; Then, R.L. DNA and RNA Synthesis: Antifolates. Chem. Rev. 2005, 105, 593–620. [Google Scholar] [CrossRef] [PubMed]
- Sköld, O. Sulfonamide resistance: Mechanisms and trends. Drug Resist. Updat. 2000, 3, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Woods, D.D. The Relation of p-aminobenzoic Acid to the Mechanism of the Action of Sulphanilamide; Wiley-Blackwell: Hoboken, NJ, USA, 1940; Volume 21. [Google Scholar]
- Proctor, R.A. Role of Folate Antagonists in the Treatment of Methicillin-Resistant Staphylococcus aureus Infection. Clin. Infect. Dis. 2008, 46, 584–593. [Google Scholar]
- Koch, A.L. Bacterial Wall as Target for Attack. Clin. Microbiol. Rev. 2003, 16, 673–687. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Sun, S.X. Morphology, Growth, and Size Limit of Bacterial Cells. Phys. Rev. Lett. 2010, 105, 028101. [Google Scholar] [CrossRef] [Green Version]
- Cabeen, M.T.; Jacobs-Wagner, C. Bacterial cell shape. Nat. Rev. Microbiol. 2005, 3, 601–610. [Google Scholar] [CrossRef]
- Walter, A.; Mayer, C. Peptidoglycan Structure, Biosynthesis, and Dynamics During Bacterial Growth. In Extracellular Sugar-Based Biopolymers Matrices; Springer: Cham, Switzerland, 2019; pp. 237–299. [Google Scholar]
- Pasquina-Lemonche, L.; Burns, J.; Turner, R.D.; Kumar, S.; Tank, R.; Mullin, N.; Wilson, J.S.; Chakrabarti, B.; Bullough, P.A.; Foster, S.J.; et al. The architecture of the Gram-positive bacterial cell wall. Nature 2020, 582, 294–297. [Google Scholar] [CrossRef]
- Schleifer, K.H.; Kandler, O. Peptidoglycan types of bacterial cell walls and their taxonomic implications. Bacteriol. Rev. 1972, 36, 407–477. [Google Scholar] [CrossRef]
- Boneca, I.G.; Huang, Z.-H.; Gage, D.A.; Tomasz, A. Characterization of Staphylococcus aureus Cell Wall Glycan Strands, Evidence for a New β-N-Acetylglucosaminidase Activity. J. Biol. Chem. 2000, 275, 9910–9918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egan, A.J.F.; Errington, J.; Vollmer, W. Regulation of peptidoglycan synthesis and remodelling. Nat. Rev. Microbiol. 2020, 18, 446–460. [Google Scholar] [CrossRef] [PubMed]
- Vollmer, W.; Seligman, S.J. Architecture of peptidoglycan: More data and more models. Trends Microbiol. 2010, 18, 59–66. [Google Scholar] [CrossRef]
- Silhavy, T.J.; Kahne, D.; Walker, S. The Bacterial Cell Envelope. Cold Spring Harb. Perspect. Biol. 2010, 2, a000414. [Google Scholar] [CrossRef]
- Turner, R.D.; Vollmer, W.; Foster, S.J. Different walls for rods and balls: The diversity of peptidoglycan. Mol. Microbiol. 2014, 91, 862–874. [Google Scholar] [CrossRef] [Green Version]
- Matias, V.R.F.; Beveridge, T.J. Native cell wall organization shown by cryo-electron microscopy confirms the existence of a periplasmic space in Staphylococcus aureus. J. Bacteriol. 2006, 188, 1011–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Jonge, B.L.; Chang, Y.S.; Gage, D.; Tomasz, A. Peptidoglycan composition of a highly methicillin-resistant Staphylococcus aureus strain. The role of penicillin binding protein 2A. J. Biol. Chem. 1992, 267, 11248–11254. [Google Scholar] [CrossRef]
- Naclerio, G.A.; Sintim, H.O. Multiple ways to kill bacteria via inhibiting novel cell wall or membrane targets. Future Med. Chem. 2020, 12, 1253–1279. [Google Scholar] [CrossRef]
- Meroueh, S.O.; Bencze, K.Z.; Hesek, D.; Lee, M.; Fisher, J.F.; Stemmler, T.L.; Mobashery, S. Three-dimensional structure of the bacterial cell wall peptidoglycan. Proc. Natl. Acad. Sci. USA 2006, 103, 4404–4409. [Google Scholar] [CrossRef] [Green Version]
- Fishovitz, J.; Hermoso, J.A.; Chang, M.; Mobashery, S. Penicillin-binding protein 2a of methicillin-resistant Staphylococcus aureus. IUBMB Life 2014, 66, 572–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bugg, T.D.H. Bacterial Peptidoglycan Biosynthesis and Its Inhibition. In Comprehensive Natural Products Chemistry; Elsevier: Amsterdam, The Netherlands, 1999; pp. 241–294. [Google Scholar]
- Otero, L.H.; Rojas-Altuve, A.; Llarrull, L.I.; Carrasco-Lopez, C.; Kumarasiri, M.; Lastochkin, E.; Fishovitz, J.; Dawley, M.; Hesek, D.; Lee, M.; et al. How allosteric control of Staphylococcus aureus penicillin binding protein 2a enables methicillin resistance and physiological function. Proc. Natl. Acad. Sci. USA 2013, 110, 16808–16813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCallum, N.; Berger-Bächi, B.; Senn, M.M. Regulation of antibiotic resistance in Staphylococcus aureus. Int. J. Med. Microbiol. 2010, 300, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Weidenmaier, C.; Peschel, A. Teichoic acids and related cell-wall glycopolymers in Gram-positive physiology and host interactions. Nat. Rev. Microbiol. 2008, 6, 276–287. [Google Scholar] [CrossRef]
- Epand, R.M.; Walker, C.; Epand, R.F.; Magarvey, N.A. Molecular mechanisms of membrane targeting antibiotics. Biochim. Biophys. Acta—Biomembr. 2016, 1858, 980–987. [Google Scholar] [CrossRef]
- Halder, S.; Yadav, K.K.; Sarkar, R.; Mukherjee, S.; Saha, P.; Haldar, S.; Karmakar, S.; Sen, T. Alteration of Zeta potential and membrane permeability in bacteria: A study with cationic agents. Springerplus 2015, 4, 672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, W.; Zou, G.; Hari, T.P.A.; Wilt, I.K.; Zhu, W.; Galle, N.; Faizi, H.A.; Hendricks, G.L.; Tori, K.; Pan, W.; et al. A selective membrane-targeting repurposed antibiotic with activity against persistent methicillin-resistant Staphylococcus aureus. Proc. Natl. Acad. Sci. USA 2019, 116, 16529–16534. [Google Scholar] [CrossRef] [Green Version]
- Luther, A.; Bisang, C.; Obrecht, D. Advances in macrocyclic peptide-based antibiotics. Bioorg. Med. Chem. 2018, 26, 2850–2858. [Google Scholar] [CrossRef]
- Upert, G.; Luther, A.; Obrecht, D.; Ermert, P. Emerging peptide antibiotics with therapeutic potential. Med. Drug Discov. 2021, 9, 100078. [Google Scholar] [CrossRef]
- Hurdle, J.G.; O’Neill, A.J.; Chopra, I.; Lee, R.E. Targeting bacterial membrane function: An underexploited mechanism for treating persistent infections. Nat. Rev. Microbiol. 2011, 9, 62–75. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, V.; Feio, M.J.; Bastos, M. Role of lipids in the interaction of antimicrobial peptides with membranes. Prog. Lipid Res. 2012, 51, 149–177. [Google Scholar] [CrossRef] [PubMed]
- Hooper, D.C. Mechanisms of fluoroquinolone resistance. Drug Resist. Updat. 1999, 2, 38–55. [Google Scholar] [CrossRef] [PubMed]
- Poehlsgaard, J.; Douthwaite, S. The bacterial ribosome as a target for antibiotics. Nat. Rev. Microbiol. 2005, 3, 870–881. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.N. The A–Z of bacterial translation inhibitors. Crit. Rev. Biochem. Mol. Biol. 2009, 44, 393–433. [Google Scholar] [CrossRef]
- Steitz, T.A. A structural understanding of the dynamic ribosome machine. Nat. Rev. Mol. Cell Biol. 2008, 9, 242–253. [Google Scholar] [CrossRef]
- Wilson, D.N.; Nierhaus, K.H. The Ribosome through the Looking Glass. Angew. Chemie Int. Ed. 2003, 42, 3464–3486. [Google Scholar] [CrossRef]
- Ramakrishnan, V. Ribosome Structure and the Mechanism of Translation. Cell 2002, 108, 557–572. [Google Scholar] [CrossRef] [Green Version]
- Chellat, M.F.; Raguž, L.; Riedl, R. Targeting Antibiotic Resistance. Angew. Chem. Int. Ed. 2016, 55, 6600–6626. [Google Scholar] [CrossRef] [PubMed]
- Champney, W.S. Antibiotics targeting bacterial ribosomal subunit biogenesis. J. Antimicrob. Chemother. 2020, 75, 787–806. [Google Scholar] [CrossRef]
- Greulich, P.; Doležal, J.; Scott, M.; Evans, M.R.; Allen, R.J. Predicting the dynamics of bacterial growth inhibition by ribosome-targeting antibiotics. Phys. Biol. 2017, 14, 065005. [Google Scholar] [CrossRef] [PubMed]
- McCoy, L.S.; Xie, Y.; Tor, Y. Antibiotics that target protein synthesis. Wiley Interdiscip. Rev. RNA 2011, 2, 209–232. [Google Scholar] [CrossRef]
- Wilson, D.N. Ribosome-targeting antibiotics and mechanisms of bacterial resistance. Nat. Rev. Microbiol. 2014, 12, 35–48. [Google Scholar] [CrossRef]
- Stokes, J.M.; Brown, E.D. Chemical modulators of ribosome biogenesis as biological probes. Nat. Chem. Biol. 2015, 11, 924–932. [Google Scholar] [CrossRef] [PubMed]
- Stokes, J.M.; Davis, J.H.; Mangat, C.S.; Williamson, J.R.; Brown, E.D. Discovery of a small molecule that inhibits bacterial ribosome biogenesis. Elife 2014, 3, e03574. [Google Scholar] [CrossRef] [PubMed]
- Becker, B.; Cooper, M.A. Aminoglycoside Antibiotics in the 21st Century. ACS Chem. Biol. 2013, 8, 105–115. [Google Scholar] [CrossRef]
- Davies, J.; Davis, B.D. Misreading of Ribonucleic Acid Code Words Induced by Aminoglycoside Antibiotics. J. Biol. Chem. 1968, 243, 3312–3316. [Google Scholar] [CrossRef]
- Ida, T.; Okamoto, R.; Nonoyama, M.; Irinoda, K.; Kurazono, M.; Inoue, M. Antagonism between Aminoglycosides and β-Lactams in a Methicillin-Resistant Staphylococcus aureus Isolate Involves Induction of an Aminoglycoside-Modifying Enzyme. Antimicrob. Agents Chemother. 2002, 46, 1516–1521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordeiro, J.C.R.; Reis, A.O.; Miranda, E.A.; Sader, H.S. In vitro antimicrobial activity of the aminoglycoside arbekacin tested against oxacillin-resistant Staphylococcus aureus isolated in Brazilian hospitals. Braz. J. Infect. Dis. 2001, 5, 130–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shigemura, K.; Osawa, K.; Mukai, A.; Yoshida, H.; Fujisawa, M.; Arakawa, S. Anti-MRSA drug use and antibiotic susceptibilities of MRSA at a university hospital in Japan from 2007 to 2011. J. Antibiot. 2013, 66, 273–276. [Google Scholar] [CrossRef]
- Bermingham, A.; Derrick, J.P. The folic acid biosynthesis pathway in bacteria: Evaluation of potential for antibacterial drug discovery. BioEssays 2002, 24, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Bertacine Dias, M.V.; Santos, J.C.; Libreros-Zúñiga, G.A.; Ribeiro, J.A.; Chavez-Pacheco, S.M. Folate biosynthesis pathway: Mechanisms and insights into drug design for infectious diseases. Future Med. Chem. 2018, 10, 935–959. [Google Scholar] [CrossRef]
- Fernández-Villa, D.; Aguilar, M.R.; Rojo, L. Folic Acid Antagonists: Antimicrobial and Immunomodulating Mechanisms and Applications. Int. J. Mol. Sci. 2019, 20, 4996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schweitzer, B.I.; Dicker, A.P.; Bertino, J.R. Dihydrofolate reductase as a therapeutic target. FASEB J. 1990, 4, 2441–2452. [Google Scholar] [CrossRef] [PubMed]
- Dale, G.E.; Broger, C.; D’Arcy, A.; Hartman, P.G.; DeHoogt, R.; Jolidon, S.; Kompis, I.; Labhardt, A.M.; Langen, H.; Locher, H.; et al. A single amino acid substitution in Staphylococcus aureus dihydrofolate reductase determines trimethoprim resistance 1 1 Edited by T.Richmond. J. Mol. Biol. 1997, 266, 23–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouch, D.A.; Messerotti, L.J.; Loo, L.S.L.; Jackson, C.A.; Skurray, R.A. Trimethoprim resistance transposon Tn4003 from Staphylococcus aureus encodes genes for a dihydrofolate reductase and thymidylate synthetase flanked by three copies of IS257. Mol. Microbiol. 1989, 3, 161–175. [Google Scholar] [CrossRef]
- Kadlec, K.; Schwarz, S. Novel ABC Transporter Gene, vga(C), Located on a Multiresistance Plasmid from a Porcine Methicillin-Resistant Staphylococcus aureus ST398 Strain. Antimicrob. Agents Chemother. 2009, 53, 3589–3591. [Google Scholar] [CrossRef] [Green Version]
- Perreten, V.; Kadlec, K.; Schwarz, S.; Gronlund Andersson, U.; Finn, M.; Greko, C.; Moodley, A.; Kania, S.A.; Frank, L.A.; Bemis, D.A.; et al. Clonal spread of methicillin-resistant Staphylococcus pseudintermedius in Europe and North America: An international multicentre study. J. Antimicrob. Chemother. 2010, 65, 1145–1154. [Google Scholar] [CrossRef] [Green Version]
- Nurjadi, D.; Schäfer, J.; Friedrich-Jänicke, B.; Mueller, A.; Neumayr, A.; Calvo-Cano, A.; Goorhuis, A.; Molhoek, N.; Lagler, H.; Kantele, A.; et al. Predominance of dfrG as determinant of trimethoprim resistance in imported Staphylococcus aureus. Clin. Microbiol. Infect. 2015, 21, 1095.e5–1095.e9. [Google Scholar] [CrossRef] [Green Version]
- Paul, M.; Bishara, J.; Yahav, D.; Goldberg, E.; Neuberger, A.; Ghanem-Zoubi, N.; Dickstein, Y.; Nseir, W.; Dan, M.; Leibovici, L. Trimethoprim-sulfamethoxazole versus vancomycin for severe infections caused by meticillin resistant Staphylococcus aureus: Randomised controlled trial. BMJ 2015, 350, h2219. [Google Scholar] [CrossRef] [Green Version]
- Elwell, L.P.; Wilson, H.R.; Knick, V.B.; Keith, B.R. In vitro and in vivo efficacy of the combination trimethoprim-sulfamethoxazole against clinical isolates of methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 1986, 29, 1092–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frei, C.R.; Miller, M.L.; Lewis, J.S.; Lawson, K.A.; Hunter, J.M.; Oramasionwu, C.U.; Talbert, R.L. Trimethoprim-Sulfamethoxazole or Clindamycin for Community-Associated MRSA (CA-MRSA) Skin Infections. J. Am. Board Fam. Med. 2010, 23, 714–719. [Google Scholar] [CrossRef] [PubMed]
- Kalkut, G. Sulfonamides and Trimethoprim. Cancer Investig. 1998, 16, 612–615. [Google Scholar] [CrossRef] [PubMed]
- Khamash, D.F.; Voskertchian, A.; Tamma, P.D.; Akinboyo, I.C.; Carroll, K.C.; Milstone, A.M. Increasing Clindamycin and Trimethoprim-Sulfamethoxazole Resistance in Pediatric Staphylococcus aureus Infections. J. Pediatric Infect. Dis. Soc. 2019, 8, 351–353. [Google Scholar] [CrossRef]
- Acree, M.E.; Morgan, E.; David, M.Z. S. aureus infections in chicago, 2006–2014: Increase in CA MSSA and decrease in MRSA incidence. Infect. Control. Hosp. Epidemiol. 2017, 38, 1226–1234. [Google Scholar] [CrossRef]
- Harris, T.; Bowen, A.; Holt, D.; Sarovich, D.; Stevens, K.; Currie, B.; Howden, B.; Carapetis, J.; Giffard, P.; Tong, S. Investigation of trimethoprim/sulfamethoxazole resistance in an emerging sequence type 5 methicillin-resistant Staphylococcus aureus clone reveals discrepant resistance reporting. Clin. Microbiol. Infect. 2018, 24, 1027–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, G.; Kohler, T.; Peschel, A. The wall teichoic acid and lipoteichoic acid polymers of Staphylococcus aureus. Int. J. Med. Microbiol. 2010, 300, 148–154. [Google Scholar] [CrossRef]
- Deininger, S.; Stadelmaier, A.; von Aulock, S.; Morath, S.; Schmidt, R.R.; Hartung, T. Definition of Structural Prerequisites for Lipoteichoic Acid-Inducible Cytokine Induction by Synthetic Derivatives. J. Immunol. 2003, 170, 4134–4138. [Google Scholar] [CrossRef] [Green Version]
- Percy, M.G.; Gründling, A. Lipoteichoic Acid Synthesis and Function in Gram-Positive Bacteria. Annu. Rev. Microbiol. 2014, 68, 81–100. [Google Scholar] [CrossRef] [Green Version]
- Brown, S.; Santa Maria, J.P.; Walker, S. Wall Teichoic Acids of Gram-Positive Bacteria. Annu. Rev. Microbiol. 2013, 67, 313–336. [Google Scholar] [CrossRef] [Green Version]
- Neuhaus, F.C.; Baddiley, J. A Continuum of Anionic Charge: Structures and Functions of d-Alanyl-Teichoic Acids in Gram-Positive Bacteria. Microbiol. Mol. Biol. Rev. 2003, 67, 686–723. [Google Scholar] [CrossRef] [Green Version]
- Peschel, A.; Otto, M.; Jack, R.W.; Kalbacher, H.; Jung, G.; Götz, F. Inactivation of the dlt Operon inStaphylococcus aureus Confers Sensitivity to Defensins, Protegrins, and Other Antimicrobial Peptides. J. Biol. Chem. 1999, 274, 8405–8410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peschel, A.; Vuong, C.; Otto, M.; Götz, F. The d-Alanine residues of Staphylococcus aureus teichoic acids alter the susceptibility to vancomycin and the activity of autolytic enzymes. Antimicrob. Agents Chemother. 2000, 44, 2845–2847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atilano, M.L.; Pereira, P.M.; Yates, J.; Reed, P.; Veiga, H.; Pinho, M.G.; Filipe, S.R. Teichoic acids are temporal and spatial regulators of peptidoglycan cross-linking in Staphylococcus aureus. Proc. Natl. Acad. Sci. USA 2010, 107, 18991–18996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovács, M.; Halfmann, A.; Fedtke, I.; Heintz, M.; Peschel, A.; Vollmer, W.; Hakenbeck, R.; Brückner, R. A Functional dlt Operon, Encoding Proteins Required for Incorporation of d-Alanine in Teichoic Acids in Gram-Positive Bacteria, Confers Resistance to Cationic Antimicrobial Peptides in Streptococcus pneumoniae. J. Bacteriol. 2006, 188, 5797–5805. [Google Scholar] [CrossRef] [Green Version]
- Mechler, L.; Bonetti, E.-J.; Reichert, S.; Flötenmeyer, M.; Schrenzel, J.; Bertram, R.; François, P.; Götz, F. Daptomycin Tolerance in the Staphylococcus aureus pitA6 Mutant Is Due to Upregulation of the dlt Operon. Antimicrob. Agents Chemother. 2016, 60, 2684–2691. [Google Scholar] [CrossRef] [Green Version]
- van Dalen, R.; Peschel, A.; van Sorge, N.M. Wall Teichoic Acid in Staphylococcus aureus Host Interaction. Trends Microbiol. 2020, 28, 985–998. [Google Scholar] [CrossRef]
- Kho, K.; Meredith, T.C. Salt-Induced Stress Stimulates a Lipoteichoic Acid-Specific Three-Component Glycosylation System in Staphylococcus aureus. J. Bacteriol. 2018, 200, e00017-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winstel, V.; Sanchez-Carballo, P.; Holst, O.; Xia, G.; Peschel, A. Biosynthesis of the Unique Wall Teichoic Acid of Staphylococcus aureus Lineage ST395. MBio 2014, 5, e000869-14. [Google Scholar] [CrossRef] [Green Version]
- Winstel, V.; Liang, C.; Sanchez-Carballo, P.; Steglich, M.; Munar, M.; Bröker, B.M.; Penadés, J.R.; Nübel, U.; Holst, O.; Dandekar, T.; et al. Wall teichoic acid structure governs horizontal gene transfer between major bacterial pathogens. Nat. Commun. 2013, 4, 2345. [Google Scholar] [CrossRef]
- Swoboda, J.G.; Campbell, J.; Meredith, T.C.; Walker, S. Wall Teichoic Acid Function, Biosynthesis, and Inhibition. ChemBioChem 2009, 11, 35–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simanski, M.; Gläser, R.; Köten, B.; Meyer-Hoffert, U.; Wanner, S.; Weidenmaier, C.; Peschel, A.; Harder, J. Staphylococcus aureus subverts cutaneous defense by d -alanylation of teichoic acids. Exp. Dermatol. 2013, 22, 294–296. [Google Scholar] [CrossRef]
- Ernst, C.M.; Staubitz, P.; Mishra, N.N.; Yang, S.-J.; Hornig, G.; Kalbacher, H.; Bayer, A.S.; Kraus, D.; Peschel, A. The Bacterial Defensin Resistance Protein MprF Consists of Separable Domains for Lipid Lysinylation and Antimicrobial Peptide Repulsion. PLoS Pathog. 2009, 5, e1000660. [Google Scholar] [CrossRef] [Green Version]
- Schlag, M.; Biswas, R.; Krismer, B.; Kohler, T.; Zoll, S.; Yu, W.; Schwarz, H.; Peschel, A.; Götz, F. Role of staphylococcal wall teichoic acid in targeting the major autolysin Atl. Mol. Microbiol. 2010, 75, 864–873. [Google Scholar] [CrossRef] [PubMed]
- Santa Maria, J.P.; Sadaka, A.; Moussa, S.H.; Brown, S.; Zhang, Y.J.; Rubin, E.J.; Gilmore, M.S.; Walker, S. Compound-gene interaction mapping reveals distinct roles for Staphylococcus aureus teichoic acids. Proc. Natl. Acad. Sci. USA 2014, 111, 12510–12515. [Google Scholar] [CrossRef] [Green Version]
- Campbell, J.; Singh, A.K.; Santa Maria, J.P.; Kim, Y.; Brown, S.; Swoboda, J.G.; Mylonakis, E.; Wilkinson, B.J.; Walker, S. Synthetic Lethal Compound Combinations Reveal a Fundamental Connection between Wall Teichoic Acid and Peptidoglycan Biosyntheses in Staphylococcus aureus. ACS Chem. Biol. 2011, 6, 106–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farha, M.A.; Leung, A.; Sewell, E.W.; D’Elia, M.A.; Allison, S.E.; Ejim, L.; Pereira, P.M.; Pinho, M.G.; Wright, G.D.; Brown, E.D. Inhibition of WTA Synthesis Blocks the Cooperative Action of PBPs and Sensitizes MRSA to β-Lactams. ACS Chem. Biol. 2013, 8, 226–233. [Google Scholar] [CrossRef]
- Campbell, J.; Singh, A.K.; Swoboda, J.G.; Gilmore, M.S.; Wilkinson, B.J.; Walker, S. An Antibiotic That Inhibits a Late Step in Wall Teichoic Acid Biosynthesis Induces the Cell Wall Stress Stimulon in Staphylococcus aureus. Antimicrob. Agents Chemother. 2012, 56, 1810–1820. [Google Scholar] [CrossRef] [Green Version]
- Schirner, K.; Marles-Wright, J.; Lewis, R.J.; Errington, J. Distinct and essential morphogenic functions for wall- and lipo-teichoic acids in Bacillus subtilis. EMBO J. 2009, 28, 830–842. [Google Scholar] [CrossRef] [Green Version]
- Oku, Y.; Kurokawa, K.; Matsuo, M.; Yamada, S.; Lee, B.-L.; Sekimizu, K. Pleiotropic Roles of Polyglycerolphosphate Synthase of Lipoteichoic Acid in Growth of Staphylococcus aureus Cells. J. Bacteriol. 2009, 191, 141–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, J.; Reynolds, N.; Ibba, M. Aminoacyl-tRNA Synthesis and Translational Quality Control. Annu. Rev. Microbiol. 2009, 63, 61–78. [Google Scholar] [CrossRef]
- Ribas de Pouplana, L.; Schimmel, P. Aminoacyl-tRNA synthetases: Potential markers of genetic code development. Trends Biochem. Sci. 2001, 26, 591–596. [Google Scholar] [CrossRef]
- Ibba, M.; Söll, D. Aminoacyl-tRNA Synthesis. Annu. Rev. Biochem. 2000, 69, 617–650. [Google Scholar] [CrossRef]
- Eriani, G.; Delarue, M.; Poch, O.; Gangloff, J.; Moras, D. Partition of tRNA synthetases into two classes based on mutually exclusive sets of sequence motifs. Nature 1990, 347, 203–206. [Google Scholar] [CrossRef]
- Cassels, R.; Oliva, B.; Knowles, D. Occurrence of the regulatory nucleotides ppGpp and pppGpp following induction of the stringent response in staphylococci. J. Bacteriol. 1995, 177, 5161–5165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, Y.I.; Aravind, L.; Grishin, N.V.; Koonin, E.V. Evolution of Aminoacyl-tRNA synthetases-analysis of unique domain architectures and phylogenetic trees reveals a complex history of horizontal gene transfer events. Genome Res. 1999, 9, 689–710. [Google Scholar]
- Cusack, S. Aminoacyl-tRNA synthetases. Curr. Opin. Struct. Biol. 1997, 7, 881–889. [Google Scholar] [CrossRef]
- Elbaramawi, S.S.; Ibrahim, S.M.; Lashine, E.-S.M.; El-Sadek, M.E.; Mantzourani, E.; Simons, C. Exploring the binding sites of Staphylococcus aureus phenylalanine tRNA synthetase: A homology model approach. J. Mol. Graph. Model. 2017, 73, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, R.; Boon, R.J.; Griffin, K.E.; Masters, P.J.; Slocombe, B.; White, A.R. Antibacterial activity of mupirocin (pseudomonic acid), a new antibiotic for topical use. Antimicrob. Agents Chemother. 1985, 27, 495–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurdle, J.G.; O’Neill, A.J.; Chopra, I. Prospects for Aminoacyl-tRNA Synthetase Inhibitors as New Antimicrobial Agents. Antimicrob. Agents Chemother. 2005, 49, 4821–4833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breukink, E.; de Kruijff, B. Lipid II as a target for antibiotics. Nat. Rev. Drug Discov. 2006, 5, 321–323. [Google Scholar] [CrossRef] [PubMed]
- Schneider, T.; Sahl, H.-G. An oldie but a goodie—Cell wall biosynthesis as antibiotic target pathway. Int. J. Med. Microbiol. 2010, 300, 161–169. [Google Scholar] [CrossRef]
- van Heijenoort, J. Lipid Intermediates in the Biosynthesis of Bacterial Peptidoglycan. Microbiol. Mol. Biol. Rev. 2007, 71, 620–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikkelsen, K.; Sirisarn, W.; Alharbi, O.; Alharbi, M.; Liu, H.; Nøhr-Meldgaard, K.; Mayer, K.; Vestergaard, M.; Gallagher, L.A.; Derrick, J.P.; et al. The Novel Membrane-Associated Auxiliary Factors AuxA and AuxB Modulate β-lactam Resistance in MRSA by stabilizing Lipoteichoic Acids. Int. J. Antimicrob. Agents 2021, 57, 106283. [Google Scholar] [CrossRef]
- Jousselin, A.; Manzano, C.; Biette, A.; Reed, P.; Pinho, M.G.; Rosato, A.E.; Kelley, W.L.; Renzoni, A. The Staphylococcus aureus Chaperone PrsA Is a New Auxiliary Factor of Oxacillin Resistance Affecting Penicillin-Binding Protein 2A. Antimicrob. Agents Chemother. 2016, 60, 1656–1666. [Google Scholar] [CrossRef] [Green Version]
- Gardete, S.; Ludovice, A.M.; Sobral, R.G.; Filipe, S.R.; de Lencastre, H.; Tomasz, A. Role of murE in the Expression of β-Lactam Antibiotic Resistance in Staphylococcus aureus. J. Bacteriol. 2004, 186, 1705–1713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Memmi, G.; Filipe, S.R.; Pinho, M.G.; Fu, Z.; Cheung, A. Staphylococcus aureus PBP4 Is Essential for β-Lactam Resistance in Community-Acquired Methicillin-Resistant Strains. Antimicrob. Agents Chemother. 2008, 52, 3955–3966. [Google Scholar] [CrossRef] [Green Version]
- Rohrer, S.; Ehlert, K.; Tschierske, M.; Labischinski, H.; Berger-Bachi, B. The essential Staphylococcus aureus gene fmhB is involved in the first step of peptidoglycan pentaglycine interpeptide formation. Proc. Natl. Acad. Sci. USA 1999, 96, 9351–9356. [Google Scholar] [CrossRef] [Green Version]
- Brown, S.; Xia, G.; Luhachack, L.G.; Campbell, J.; Meredith, T.C.; Chen, C.; Winstel, V.; Gekeler, C.; Irazoqui, J.E.; Peschel, A.; et al. Methicillin resistance in Staphylococcus aureus requires glycosylated wall teichoic acids. Proc. Natl. Acad. Sci. USA 2012, 109, 18909–18914. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Jarantow, L.W.; Wang, H.; Sillaots, S.; Cheng, H.; Meredith, T.C.; Thompson, J.; Roemer, T. Antagonism of Chemical Genetic Interaction Networks Resensitize MRSA to β-Lactam Antibiotics. Chem. Biol. 2011, 18, 1379–1389. [Google Scholar] [CrossRef] [Green Version]
- Gardete, S.; Wu, S.W.; Gill, S.; Tomasz, A. Role of VraSR in Antibiotic Resistance and Antibiotic-Induced Stress Response in Staphylococcus aureus. Antimicrob. Agents Chemother. 2006, 50, 3424–3434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, A.H.; Stock, A.M. Histidine kinases and response regulator proteins in two-component signaling systems. Trends Biochem. Sci. 2001, 26, 369–376. [Google Scholar] [CrossRef]
- Szurmant, H.; White, R.A.; Hoch, J.A. Sensor complexes regulating two-component signal transduction. Curr. Opin. Struct. Biol. 2007, 17, 706–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuroda, M.; Ohta, T.; Uchiyama, I.; Baba, T.; Yuzawa, H.; Kobayashi, I.; Cui, L.; Oguchi, A.; Aoki, K.; Nagai, Y.; et al. Whole genome sequencing of meticillin-resistant Staphylococcus aureus. Lancet 2001, 357, 1225–1240. [Google Scholar] [CrossRef]
- Haag, A.F.; Bagnoli, F. The Role of Two-Component Signal Transduction Systems in Staphylococcus aureus Virulence Regulation. In Current Topics in Microbiology and Immunology; Springer: Berlin/Heidelberg, Germany, 2015; Volume 409, pp. 145–198. [Google Scholar]
- Taglialegna, A.; Varela, M.C.; Rosato, R.R.; Rosato, A.E. VraSR and Virulence Trait Modulation during Daptomycin Resistance in Methicillin-Resistant Staphylococcus aureus Infection. mSphere 2019, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belcheva, A.; Verma, V.; Golemi-Kotra, D. DNA-Binding Activity of the Vancomycin Resistance Associated Regulator Protein VraR and the Role of Phosphorylation in Transcriptional Regulation of the vraSR Operon. Biochemistry 2009, 48, 5592–5601. [Google Scholar] [CrossRef] [PubMed]
- Flannagan, R.S.; Kuiack, R.C.; McGavin, M.J.; Heinrichs, D.E. Staphylococcus aureus Uses the GraXRS Regulatory System to Sense and Adapt to the Acidified Phagolysosome in Macrophages. mBio 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rapun-Araiz, B.; Haag, A.F.; De Cesare, V.; Gil, C.; Dorado-Morales, P.; Penades, J.R.; Lasa, I. Systematic Reconstruction of the Complete Two-Component Sensorial Network in Staphylococcus aureus. mSystems 2020, 5. [Google Scholar] [CrossRef]
- Novick, R.P.; Ross, H.F.; Projan, S.J.; Kornblum, J.; Kreiswirth, B.; Moghazeh, S. Synthesis of staphylococcal virulence factors is controlled by a regulatory RNA molecule. EMBO J. 1993, 12, 3967–3975. [Google Scholar] [CrossRef]
- Liang, X.; Yu, C.; Sun, J.; Liu, H.; Landwehr, C.; Holmes, D.; Ji, Y. Inactivation of a Two-Component Signal Transduction System, SaeRS, Eliminates Adherence and Attenuates Virulence of Staphylococcus aureus. Infect. Immun. 2006, 74, 4655–4665. [Google Scholar] [CrossRef] [Green Version]
- Giraudo, A.T.; Calzolari, A.; Cataldi, A.A.; Bogni, C.; Nagel, R. The sae locus of Staphylococcus aureus encodes a two-component regulatory system. FEMS Microbiol. Lett. 1999, 177, 15–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, P.K.; Li, T.; Sun, D.; Biek, D.P.; Schmid, M.B. Role in Cell Permeability of an Essential Two-Component System in Staphylococcus aureus. J. Bacteriol. 1999, 181, 3666–3673. [Google Scholar] [CrossRef] [Green Version]
- Fournier, B.; Hooper, D.C. A new two-component regulatory system involved in adhesion, autolysis, and extracellular proteolytic activity of Staphylococcus aureus. J. Bacteriol. 2000, 182, 3955–3964. [Google Scholar] [CrossRef] [Green Version]
- Brunskill, E.W.; Bayles, K.W. Identification of LytSR-regulated genes from Staphylococcus aureus. J. Bacteriol. 1996, 178, 5810–5812. [Google Scholar] [CrossRef] [Green Version]
- Throup, J.P.; Zappacosta, F.; Lunsford, R.D.; Annan, R.S.; Carr, S.A.; Lonsdale, J.T.; Bryant, A.P.; McDevitt, D.; Rosenberg, M.; Burnham, M.K.R. The srhSR Gene Pair from Staphylococcus aureus: Genomic and Proteomic Approaches to the Identification and Characterization of Gene Function. Biochemistry 2001, 40, 10392–10401. [Google Scholar] [CrossRef] [PubMed]
- Yarwood, J.M.; McCormick, J.K.; Schlievert, P.M. Identification of a Novel Two-Component Regulatory System That Acts in Global Regulation of Virulence Factors ofStaphylococcus aureus. J. Bacteriol. 2001, 183, 1113–1123. [Google Scholar] [CrossRef] [Green Version]
- Schlag, S.; Fuchs, S.; Nerz, C.; Gaupp, R.; Engelmann, S.; Liebeke, M.; Lalk, M.; Hecker, M.; Götz, F. Characterization of the Oxygen-Responsive NreABC Regulon of Staphylococcus aureus. J. Bacteriol. 2008, 190, 7847–7858. [Google Scholar] [CrossRef] [Green Version]
- Xue, T.; You, Y.; Hong, D.; Sun, H.; Sun, B. The Staphylococcus aureus KdpDE Two-Component System Couples Extracellular K + Sensing and Agr Signaling to Infection Programming. Infect. Immun. 2011, 79, 2154–2167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Xue, T.; Shang, F.; Sun, H.; Sun, B. Staphylococcus aureus AI-2 Quorum Sensing Associates with the KdpDE Two-Component System to Regulate Capsular Polysaccharide Synthesis and Virulence. Infect. Immun. 2010, 78, 3506–3515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, V.J.; Stauff, D.L.; Pishchany, G.; Bezbradica, J.S.; Gordy, L.E.; Iturregui, J.; Anderson, K.L.; Dunman, P.M.; Joyce, S.; Skaar, E.P. A Staphylococcus aureus Regulatory System that Responds to Host Heme and Modulates Virulence. Cell Host Microbe 2007, 1, 109–119. [Google Scholar] [CrossRef] [Green Version]
- Kelliher, J.L.; Radin, J.N.; Kehl-Fie, T.E. PhoPR Contributes to Staphylococcus aureus Growth during Phosphate Starvation and Pathogenesis in an Environment-Specific Manner. Infect. Immun. 2018, 86. [Google Scholar] [CrossRef] [Green Version]
- Baldry, M.; Nielsen, A.; Bojer, M.S.; Zhao, Y.; Friberg, C.; Ifrah, D.; Glasser Heede, N.; Larsen, T.O.; Frøkiær, H.; Frees, D.; et al. Norlichexanthone Reduces Virulence Gene Expression and Biofilm Formation in Staphylococcus aureus. PLoS ONE 2016, 11, e0168305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prieto, J.M.; Rapún-Araiz, B.; Gil, C.; Penadés, J.R.; Lasa, I.; Latasa, C. Inhibiting the two-component system GraXRS with verteporfin to combat Staphylococcus aureus infections. Sci. Rep. 2020, 10, 17939. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, M.; García, B.; Valle, J.; Rapún, B.; Ruiz De Los Mozos, I.; Solano, C.; Martí, M.; Penadés, J.R.; Toledo-Arana, A.; Lasa, I. Sensory deprivation in Staphylococcus aureus. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Yin, S.; Daum, R.S.; Boyle-Vavra, S. VraSR Two-Component Regulatory System and Its Role in Induction of pbp2 and vraSR Expression by Cell Wall Antimicrobials in Staphylococcus aureus. Antimicrob. Agents Chemother. 2006, 50, 336–343. [Google Scholar] [CrossRef] [Green Version]
- Kuroda, M.; Kuroda, H.; Oshima, T.; Takeuchi, F.; Mori, H.; Hiramatsu, K. Two-component system VraSR positively modulates the regulation of cell-wall biosynthesis pathway in Staphylococcus aureus. Mol. Microbiol. 2004, 49, 807–821. [Google Scholar] [CrossRef]
- Watanabe, T.; Okada, A.; Gotoh, Y.; Utsumi, R. Inhibitors Targeting Two-Component Signal Transduction. In Bacterial Signal Transduction: Networks and Drug Targets; Springer: New York, NY, USA, 2008; Volume 631, pp. 229–236. ISBN 9780387788845. [Google Scholar]
- Macielag, M.J.; Demers, J.P.; Fraga-Spano, S.A.; Hlasta, D.J.; Johnson, S.G.; Kanojia, R.M.; Russell, R.K.; Sui, Z.; Weidner-Wells, M.A.; Werblood, H.; et al. Substituted Salicylanilides as Inhibitors of Two-Component Regulatory Systems in Bacteria. J. Med. Chem. 1998, 41, 2939–2945. [Google Scholar] [CrossRef] [PubMed]
- Okada, A.; Igarashi, M.; Okajima, T.; Kinoshita, N.; Umekita, M.; Sawa, R.; Inoue, K.; Watanabe, T.; Doi, A.; Martin, A.; et al. Walkmycin B targets WalK (YycG), a histidine kinase essential for bacterial cell growth. J. Antibiot. 2010, 63, 89–94. [Google Scholar] [CrossRef]
- Gotoh, Y.; Doi, A.; Furuta, E.; Dubrac, S.; Ishizaki, Y.; Okada, M.; Igarashi, M.; Misawa, N.; Yoshikawa, H.; Okajima, T.; et al. Novel antibacterial compounds specifically targeting the essential WalR response regulator. J. Antibiot. 2010, 63, 127–134. [Google Scholar] [CrossRef] [Green Version]
- Hiron, A.; Falord, M.; Valle, J.; Débarbouillé, M.; Msadek, T. Bacitracin and nisin resistance in Staphylococcus aureus: A novel pathway involving the BraS/BraR two-component system (SA2417/SA2418) and both the BraD/BraE and VraD/VraE ABC transporters. Mol. Microbiol. 2011, 81, 602–622. [Google Scholar] [CrossRef]
- Le, K.Y.; Otto, M. Quorum-sensing regulation in staphylococci-an overview. Front. Microbiol. 2015, 6. [Google Scholar] [CrossRef] [Green Version]
- Otto, M. Quorum-sensing control in Staphylococci—A target for antimicrobial drug therapy? FEMS Microbiol. Lett. 2004, 241, 135–141. [Google Scholar] [CrossRef]
- Novick, R.P. Autoinduction and signal transduction in the regulation of staphylococcal virulence. Mol. Microbiol. 2003, 48, 1429–1449. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, S.T.; Bassler, B.L. Bacterial Quorum Sensing: Its Role in Virulence and Possibilities for Its Control. Cold Spring Harb. Perspect. Med. 2012, 2, a012427. [Google Scholar] [CrossRef]
- Lyon, G.J.; Muir, T.W. Chemical Signaling among Bacteria and Its Inhibition. Chem. Biol. 2003, 10, 1007–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.-C.; Liu, F.; Zhu, K.; Shen, J. Natural Products That Target Virulence Factors in Antibiotic-Resistant Staphylococcus aureus. J. Agric. Food Chem. 2019, 67, 13195–13211. [Google Scholar] [CrossRef]
- Clatworthy, A.E.; Pierson, E.; Hung, D.T. Targeting virulence: A new paradigm for antimicrobial therapy. Nat. Chem. Biol. 2007, 3, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Scutera, S.; Zucca, M.; Savoia, D. Novel approaches for the design and discovery of quorum-sensing inhibitors. Expert Opin. Drug Discov. 2014, 9, 353–366. [Google Scholar] [CrossRef]
- Khan, B.A.; Yeh, A.J.; Cheung, G.Y.; Otto, M. Investigational therapies targeting quorum-sensing for the treatment of Staphylococcus aureus infections. Expert Opin. Investig. Drugs 2015, 24, 689–704. [Google Scholar] [CrossRef]
- Piewngam, P.; Chiou, J.; Chatterjee, P.; Otto, M. Alternative approaches to treat bacterial infections: Targeting quorum-sensing. Expert Rev. Anti-Infect. Ther. 2020, 18, 499–510. [Google Scholar] [CrossRef]
- Yarwood, J.M.; Bartels, D.J.; Volper, E.M.; Greenberg, E.P. Quorum Sensing in Staphylococcus aureus Biofilms. J. Bacteriol. 2004, 186, 1838–1850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lina, G.; Jarraud, S.; Ji, G.; Greenland, T.; Pedraza, A.; Etienne, J.; Novick, R.P.; Vandenesch, F. Transmembrane topology and histidine protein kinase activity of AgrC, the agr signal receptor in Staphylococcus aureus. Mol. Microbiol. 1998, 28, 655–662. [Google Scholar] [CrossRef]
- Thoendel, M.; Kavanaugh, J.S.; Flack, C.E.; Horswill, A.R. Peptide Signaling in the Staphylococci. Chem. Rev. 2011, 111, 117–151. [Google Scholar] [CrossRef] [Green Version]
- Wright, J.S.; Jin, R.; Novick, R.P. Transient interference with staphylococcal quorum sensing blocks abscess formation. Proc. Natl. Acad. Sci. USA 2005, 102, 1691–1696. [Google Scholar] [CrossRef] [Green Version]
- Montgomery, C.P.; Boyle-Vavra, S.; Daum, R.S. Importance of the Global Regulators Agr and SaeRS in the Pathogenesis of CA-MRSA USA300 Infection. PLoS ONE 2010, 5, e15177. [Google Scholar] [CrossRef] [Green Version]
- Cheung, G.Y.C.; Wang, R.; Khan, B.A.; Sturdevant, D.E.; Otto, M. Role of the Accessory Gene Regulator agr in Community-Associated Methicillin-Resistant Staphylococcus aureus Pathogenesis. Infect. Immun. 2011, 79, 1927–1935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heyer, G.; Saba, S.; Adamo, R.; Rush, W.; Soong, G.; Cheung, A.; Prince, A. Staphylococcus aureusagr and sarA Functions Are Required for Invasive Infection but Not Inflammatory Responses in the Lung. Infect. Immun. 2002, 70, 127–133. [Google Scholar] [CrossRef] [Green Version]
- Mansson, M.; Nielsen, A.; Kjærulff, L.; Gotfredsen, C.H.; Wietz, M.; Ingmer, H.; Gram, L.; Larsen, T.O. Inhibition of Virulence Gene Expression in Staphylococcus aureus by Novel Depsipeptides from a Marine Photobacterium. Mar. Drugs 2011, 9, 2537–2552. [Google Scholar] [CrossRef] [Green Version]
- Baldry, M.; Nakamura, Y.; Nakagawa, S.; Frees, D.; Matsue, H.; Núñez, G.; Ingmer, H. Application of an agr-Specific Antivirulence Compound as Therapy for Staphylococcus aureus-Induced Inflammatory Skin Disease. J. Infect. Dis. 2018, 218, 1009–1013. [Google Scholar] [CrossRef]
- Nakayama, J.; Uemura, Y.; Nishiguchi, K.; Yoshimura, N.; Igarashi, Y.; Sonomoto, K. Ambuic Acid Inhibits the Biosynthesis of Cyclic Peptide Quormones in Gram-Positive Bacteria. Antimicrob. Agents Chemother. 2009, 53, 580–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todd, D.A.; Parlet, C.P.; Crosby, H.A.; Malone, C.L.; Heilmann, K.P.; Horswill, A.R.; Cech, N.B. Signal Biosynthesis Inhibition with Ambuic Acid as a Strategy to Target Antibiotic-Resistant Infections. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sully, E.K.; Malachowa, N.; Elmore, B.O.; Alexander, S.M.; Femling, J.K.; Gray, B.M.; DeLeo, F.R.; Otto, M.; Cheung, A.L.; Edwards, B.S.; et al. Selective Chemical Inhibition of agr Quorum Sensing in Staphylococcus aureus Promotes Host Defense with Minimal Impact on Resistance. PLoS Pathog. 2014, 10, e1004174. [Google Scholar] [CrossRef] [PubMed]
- Vuong, C.; Saenz, H.L.; Götz, F.; Otto, M. Impact of the agr Quorum-Sensing System on Adherence to Polystyrene in Staphylococcus aureus. J. Infect. Dis. 2000, 182, 1688–1693. [Google Scholar] [CrossRef] [Green Version]
- Nishitani, K.; Sutipornpalangkul, W.; de Mesy Bentley, K.L.; Varrone, J.J.; Bello-Irizarry, S.N.; Ito, H.; Matsuda, S.; Kates, S.L.; Daiss, J.L.; Schwarz, E.M. Quantifying the natural history of biofilm formation in vivo during the establishment of chronic implant-associated Staphylococcus aureus osteomyelitis in mice to identify critical pathogen and host factors. J. Orthop. Res. 2015, 33, 1311–1319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, L.; Le, K.Y.; Khan, B.A.; Nguyen, T.H.; Hunt, R.L.; Bae, J.S.; Kabat, J.; Zheng, Y.; Cheung, G.Y.C.; Li, M.; et al. Resistance to leukocytes ties benefits of quorum sensing dysfunctionality to biofilm infection. Nat. Microbiol. 2019, 4, 1114–1119. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Class and Agent | Primary Target | Potential Use |
|---|---|---|
| Glycopeptide-β-lactam hybrid | ||
| Cefilavancin | Peptidoglycan chain elongation + PBP | ABSSSI |
| Triazaacenaphthylene | ||
| Gepotidacin | Type IIA topoisomerase | UTI |
| Benzofuran naphthyridine | ||
| Afabicin | FabI | SSSIs, Bone and joint infections |
| Defensin mimetic | ||
| Brilacidin | Cell membrane | ABSSSI |
| Fluoroquinolone | ||
| Finafloxacin EMROK | Type II topoisomerase Type II topoisomerase | ABSSSI, UTI ABSSSI, HA pneumonia |
| Macrolide | ||
| Nafithromycin | 50S ribosome subunit | CA pneumonia |
| Quinolone | ||
| Taigexyn | Type II topoisomerase | ABSSSI, CA pneumonia |
| Rifamycin-quinolone hybrid | ||
| TNP-2092 | RNA polymerase, DNA gyrase, DNA topoisomerase IV | ABSSSI, BSI |
| Aminoglycoside | ||
| Apramycin | 30S ribosome subunit | BSI, Complicated UTI, HA pneumonia |
| Benzyl pyridinone | ||
| CG-549 | FabI | ABSSSI |
| Oxazolidinone | ||
| Delpazolid Contezolid/contezolid acefosamil | 50S ribosome subunit 50S ribosome subunit | Gram-positive infections (Specific use unclear) ABSSSI |
| Tetracycline | ||
| KBP-7072 TP-271 | 30S ribosome subunit 30S ribosome subunit | CA and HA pneumonia CA pneumonia |
| Benzamide | ||
| TXA709/ TXA707 | FtsZ (Cell wall division) | ABSSSI |
| Cephalosporin + Diazabicyclooctane | ||
| WCK 5222 (Cefepime + Zidebactam) | PBP + β-lactamase | Complicated UTI, HA pneumonia |
| Cephalosporin + Cyclic boronate | ||
| Cefepime + Taniborbactam | PBP + β-lactamase | Complicated UTI |
| Antibiotic Class and Agent | Primary Target (Specific Target) | Net Effect | Mechanism of Action |
|---|---|---|---|
| β-lactams Oxacillin | Cell wall synthesis (PBPs) | Peptidoglycan damage Destruction of cell membranes | Oxacillin covalently binds to PBPs, thereby inhibiting the transpeptidase activity of PBP required for bacterial cell wall synthesis [2,86]. This decreases the integrity of the bacterial cell wall and, ultimately, cell death through autolysis. |
| Cephalosporins Ceftaroline | Cell wall synthesis (PBP2a transpeptidase) | Conformational changes in PBPs | Ceftaroline is a novel β-lactam broad-spectrum cephalosporin that binds to PBPs, including PBP2a, in MRSA with high affinity, thereby inhibiting cell wall synthesis [87,88]. The 1,3-thiazole ring attaches to the 3-position of the cephalosporin nucleus, while the oxime group in the C7 acyl moiety confers enhanced lethality against MRSA [89,90]. |
| Glycopeptides Vancomycin Teicoplanin | Cell wall synthesis (MurNac-pentapeptide, Transglycosylase) | Peptidoglycan damage Destruction of cell membranes | Vancomycin forms hydrogen bonds with the D-Ala-D-Ala termini moieties of the peptidoglycan precursor lipid II, leading to a conformational alteration that prevents incorporation of NAM/NAG peptide subunits into the growing peptidoglycan chain and consequent transpeptidation [91,92]. This alters bacterial cell membrane integrity and increases permeability, leading to bacterial death [3]. |
| Lipopeptides Daptomycin | Cell wall synthesis Cell membrane (Note: the precise mechanism of action has not been established, and a specific molecular target has not been identified) | Destruction of cell membranes | Daptomycin-Ca2+ complex targets cell wall biosynthesis in S. aureus by forming a tripartite complex with undecaprenyl-coupled intermediates and membrane lipids [93]. Daptomycin-Ca2+ complex oligomerizes to form micelles [94], which penetrate the cell wall and insert into the cytoplasmic membrane by binding to phosphatidylglycerol head groups [95,96]. This causes membrane depolarization, permeabilization, K+ ions leakage, and rapid cell death [97,98]. |
| Streptogramins quinupristin/dalfopristin | Protein synthesis (50S ribosome subunit) | Inhibition of protein synthesis | Dalfopristin binds to 23S ribosomal RNA (rRNA) in the 50S ribosome subunit, causing confirmational change, which increases the binding of quinupristin and results in inhibition of peptidyl transfer [99,100]. Quinupristin binds to a nearby site on the 50S ribosome, preventing elongation of polypeptide and causes incomplete chain release. (Note: Each antibiotic alone is bacteriostatic, while their combination shows bactericidal activity) |
| Aminoglycosides Arbekacin | Protein synthesis (30S ribosome subunit) | Inhibition of protein synthesis | Arbekacin binds to four nucleotides of 16S rRNA and single amino acid of protein S12, thereby interfering with the decoding center of the bacterial 30S ribosome subunit [101]. This leads to inaccurate induction and inhibition of translation, preventing protein synthesis [102,103,104]. |
| Oxazolidinones Linezolid Tedizolid | Protein synthesis (70S ribosome by linezolid 50S ribosome by tedizolid) | Inhibition of protein synthesis | Linezolid inhibits the initiation of ternary complex formation between N-formylmethionyl-tRNA (tRNAfMet), mRNA, and the 70S ribosome, resulting in the inhibition of bacterial protein synthesis [4]. Tedizolid binds to 23S rRNA of the 50S ribosome subunit, thereby preventing the formation of the 70S ribosomal initial complex, resulting in the inhibition of bacterial protein synthesis [5,105]. |
| Macrolides Azithromycin | Protein synthesis (50S ribosome subunit) | Inhibition of protein synthesis | Azithromycin interacts with bacterial 23S rRNA on the 50S ribosome subunit and inhibits translation by targeting aminoacyl-tRNA, peptidyl-tRNA, or the peptide exit tunnel [6]. |
| Tetracyclines Tetracycline | Protein synthesis (30S ribosome subunit) | Inhibition of protein synthesis | Tetracycline interacts with the bacterial 30S ribosome subunit, preventing the binding of aminoacyl-tRNA (aa-tRNA) to the A site, resulting in inhibition of bacterial protein synthesis [106,107]. |
| Rifamycins Rifampicin (rifampin) | Nucleic acid (RNA) synthesis (RNA polymerase) | Inhibition of protein synthesis Destruction of cell membranes | Rifampicin inhibits bacterial DNA-dependent RNA polymerase, resulting in the suppression of RNA synthesis and bacterial cell death [9,10]. Rifampicin binds in a pocket of the RNA polymerase β subunit within the DNA/RNA channel, preventing transcription by blocking elongation of the 5′ end of the RNA transcript, thus inhibiting protein synthesis [10]. (Note: Rifampicin retains bactericidal activity against non-growing bacterial cultures) |
| Fluoroquinolones Delafloxacin | Nucleic acid (DNA) synthesis (DNA gyrase and DNA topoisomerase IV) | Damage to DNA replication Destruction of chromosome | Delafloxacin targets two bacterial enzymes: DNA topoisomerase II (DNA gyrase) and DNA topoisomerase IV. Control of supercoiling within treated cells is lost, resulting in impaired DNA replication [7]. Generally, topoisomerase IV is the preferred target in Gram-positive bacteria, whereas DNA gyrase is the preferred target in Gram-negative bacteria [8]. |
| Sulfonamides Sulfadiazine | Folic acid biosynthesis (Dihydropteroate synthase) | Inhibition of nucleic acid (DNA) synthesis Inhibition of cell division | Sulfadiazine acts as a competitive inhibitor of dihydropteroate synthase [108], an enzyme that reduces p-aminobenzoic acid (PABA) to form dihydropteroate in the folic acid biosynthesis pathway [109,110], resulting in a slow-acting bacteriostatic effect [111]. |
| Sulfamethoxazole-Trimethoprim Co-trimoxazole | Folic acid biosynthesis (Dihydropteroate synthase by Sulfamethoxazole (SMX) and dihydrofolate reductase by trimethoprim (TMP)) | Inhibition of nucleic acid (DNA) synthesis Inhibition of protein synthesis | SMX inhibits dihydropteroate synthase, leading to the inhibition of folic acid biosynthesis, while TMP binds and inhibits dihydrofolate reductase, preventing the conversion of dihydrofolic acid to tetrahydrofolate [11,112]. The SMX–TMP combination acts synergistically to block two consecutive steps in nucleic acid and protein biosynthesis [113]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lade, H.; Kim, J.-S. Bacterial Targets of Antibiotics in Methicillin-Resistant Staphylococcus aureus. Antibiotics 2021, 10, 398. https://doi.org/10.3390/antibiotics10040398
Lade H, Kim J-S. Bacterial Targets of Antibiotics in Methicillin-Resistant Staphylococcus aureus. Antibiotics. 2021; 10(4):398. https://doi.org/10.3390/antibiotics10040398
Chicago/Turabian StyleLade, Harshad, and Jae-Seok Kim. 2021. "Bacterial Targets of Antibiotics in Methicillin-Resistant Staphylococcus aureus" Antibiotics 10, no. 4: 398. https://doi.org/10.3390/antibiotics10040398
APA StyleLade, H., & Kim, J. -S. (2021). Bacterial Targets of Antibiotics in Methicillin-Resistant Staphylococcus aureus. Antibiotics, 10(4), 398. https://doi.org/10.3390/antibiotics10040398