
A Fine-Tuned Lipophilicity/Hydrophilicity Ratio Governs Antibacterial Potency and Selectivity of Bifurcated Halogen Bond-Forming NBTIs


 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results and Discussion
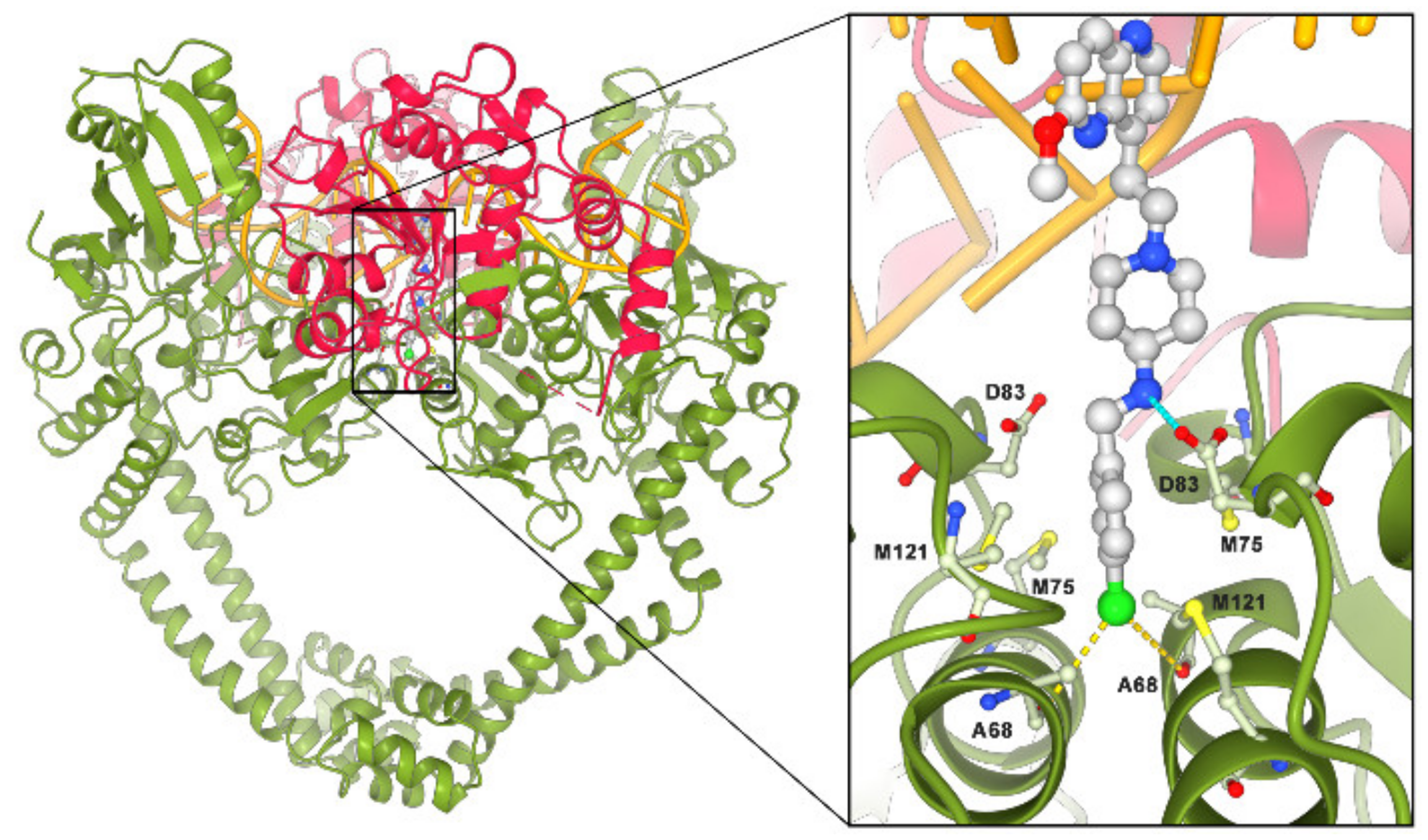
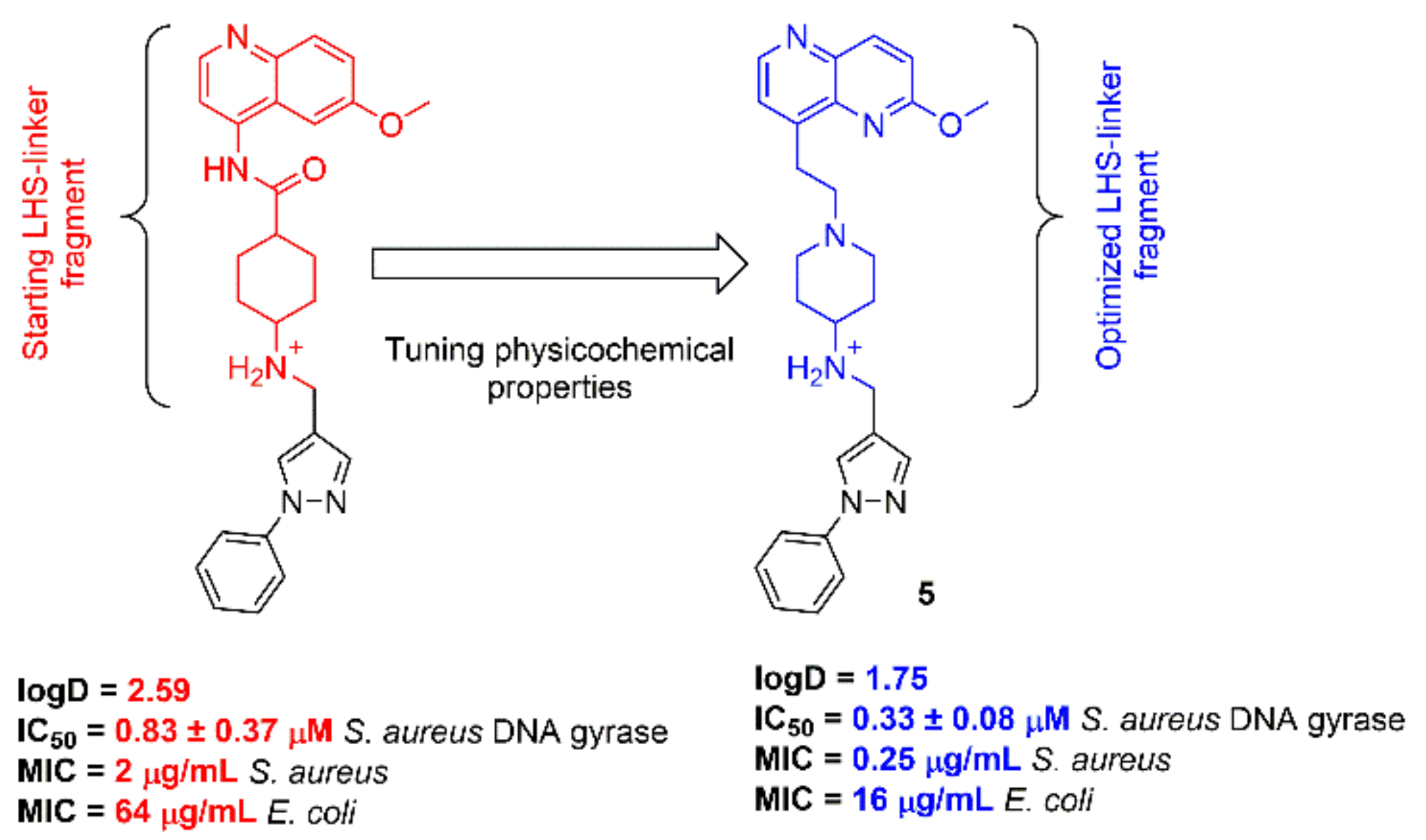
2.1. NBTIs Design Strategy
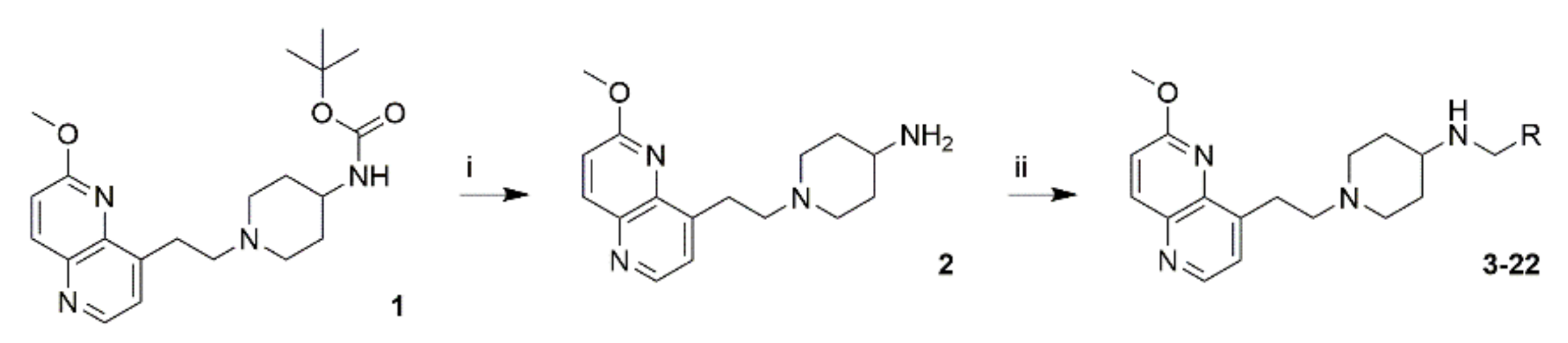
2.2. Synthesis
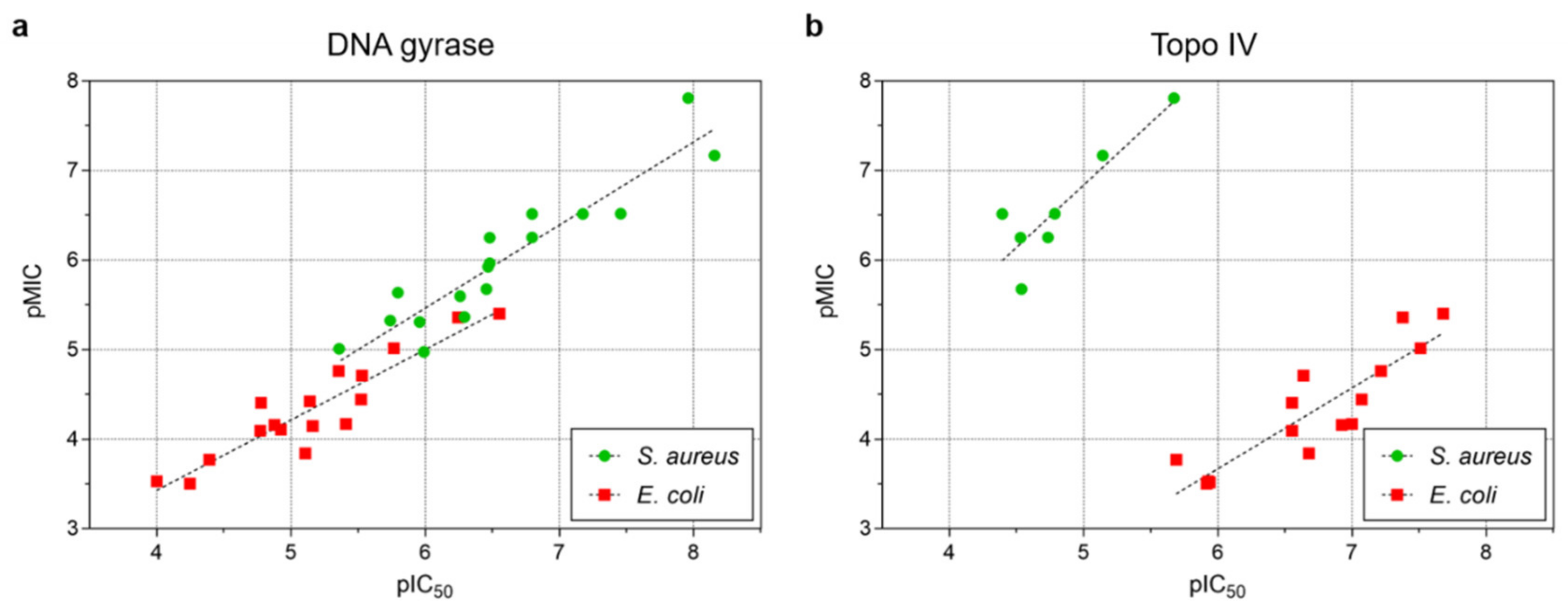
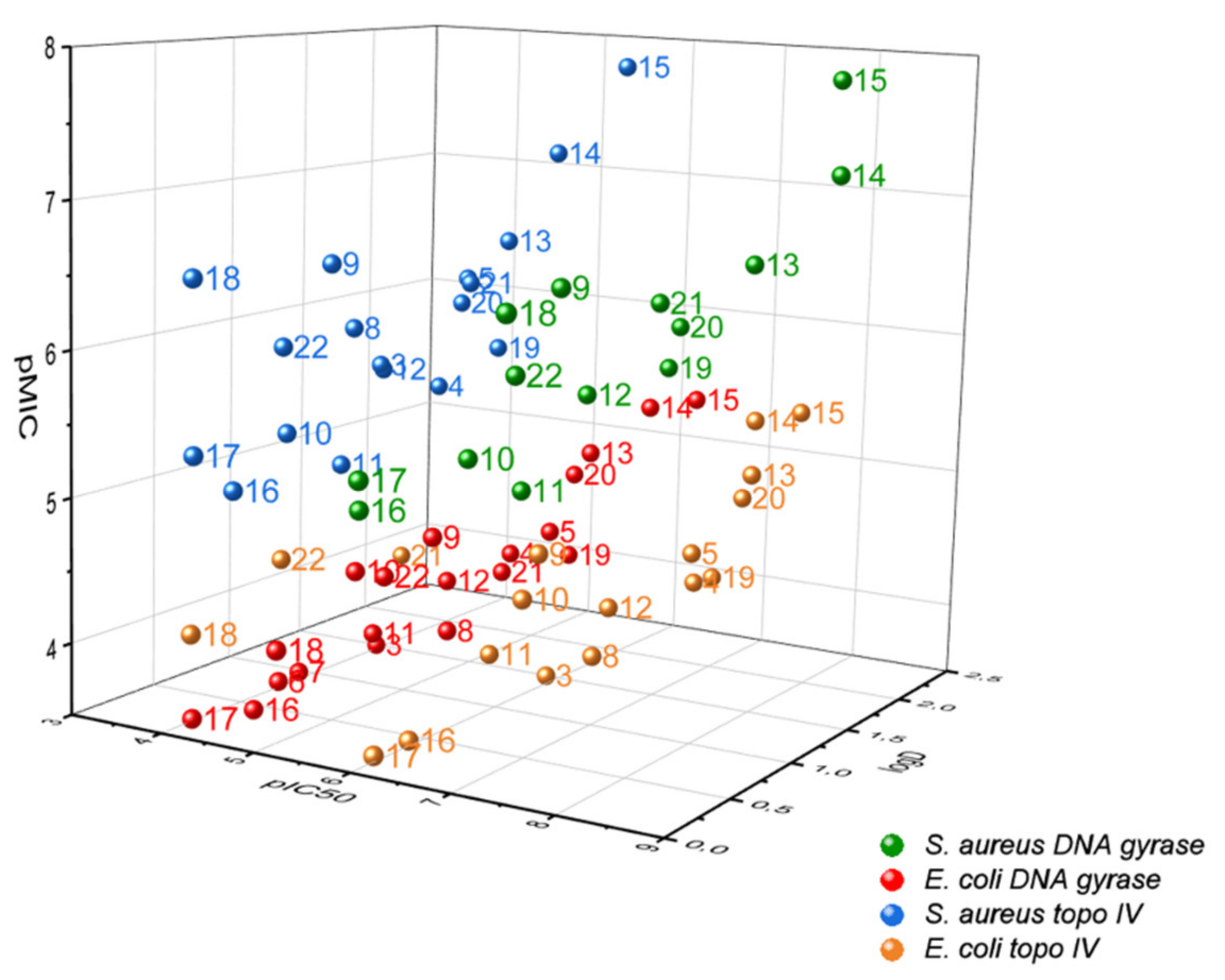
2.3. In Vitro Measurement of Inhibitor Potency and Antibacterial Activity
2.4. Cytotoxicity Studies
3. Conclusions
4. Materials and Methods
4.1. Biopharmaceutical Classification
4.1.1. Solubility
4.1.2. Permeability
4.2. Homology Modeling
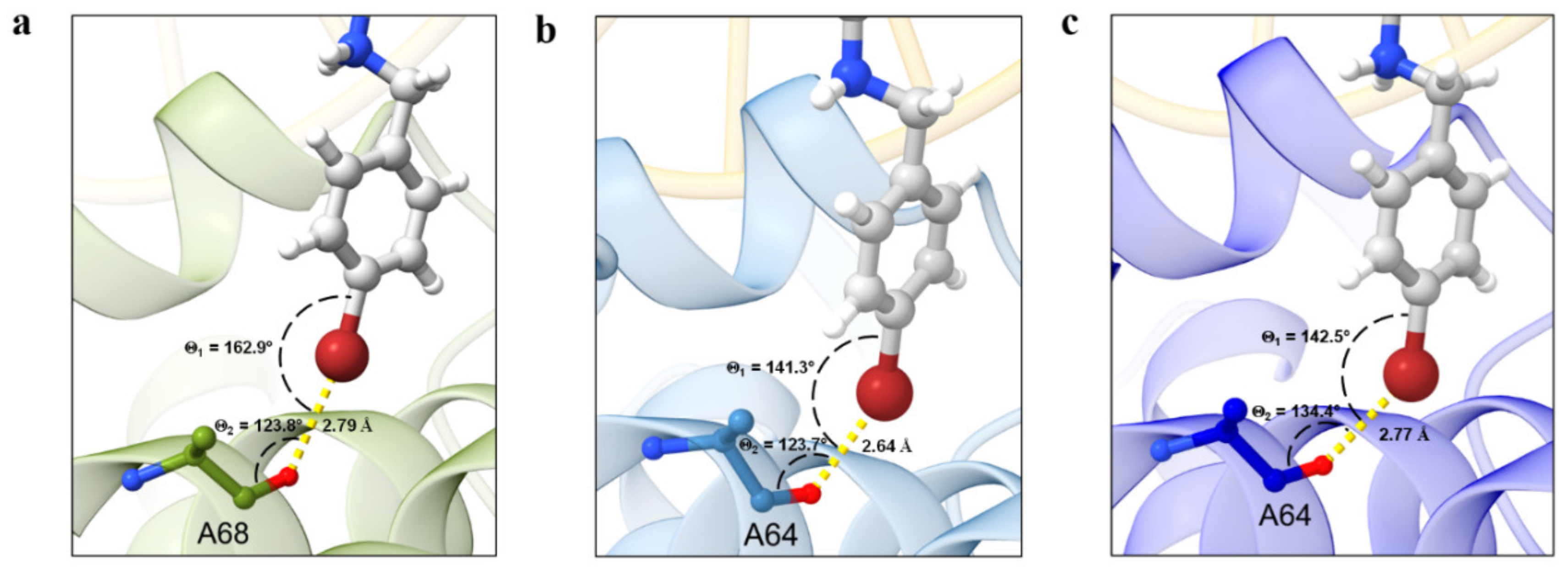
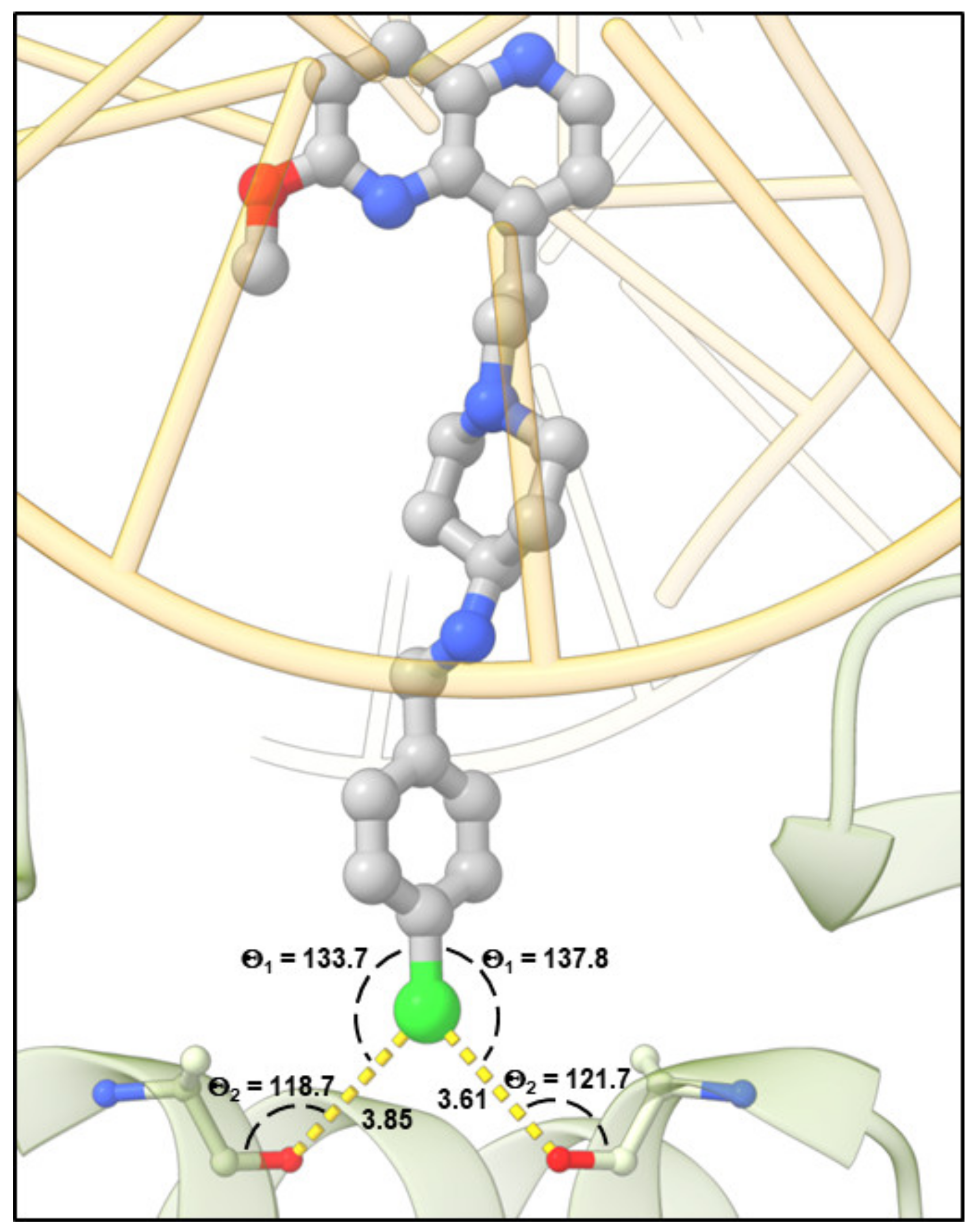
4.3. Molecular Docking Calculations
4.4. Analytical Techniques
4.5. Synthesis
4.5.1. 1-(2-(6-Methoxy-1,5-naphthyridin-4-yl)ethyl)piperidin-4-amine (2)
4.5.2. General Procedure for Reductive Amination
4.5.3. 1-(2-(6-Methoxy-1,5-naphthyridin-4-yl)ethyl)-N-((1-methyl-1H-benzo[d]imidazol-2-yl)methyl)piperidin-4-amine (3)
4.5.4. N-((1-(4-Fluorophenyl)-1H-pyrazol-4-yl)methyl)-1-(2-(6-methoxy-1,5-naphthyridin-4-yl)ethyl)piperidin-4-amine (4)
4.5.5. 1-(2-(6-Methoxy-1,5-naphthyridin-4-yl)ethyl)-N-((1-phenyl-1H-pyrazol-4-yl)methyl)piperidin-4-amine (5)
4.5.6. N-((2,8-Dimethylimidazo[1,2-a]pyridin-3-yl)methyl)-1-(2-(6-methoxy-1,5-naphthyridin-4-yl)ethyl)piperidin-4-amine (6)
4.5.7. 8-Cyano-N-(1-(2-(6-methoxy-1,5-naphthyridin-4-yl)ethyl)piperidin-4-yl)imidazo[1,2-a]pyridine-2-carboxamide (7)
4.5.8. 2-Phenyl-2H-1,2,3-triazole-4-carbaldehyde (23)
4.5.9. 1-(2-(6-Methoxy-1,5-naphthyridin-4-yl)ethyl)-N-((2-phenyl-2H-1,2,3-triazol-4-yl)methyl)piperidin-4-amine (8)
4.5.10. N-((1-Isopropyl-1H-pyrazol-4-yl)methyl)-1-(2-(6-methoxy-1,5-naphthyridin-4-yl)ethyl)piperidin-4-amine (9)
4.5.11. N-((1-Allyl-1H-pyrazol-4-yl)methyl)-1-(2-(6-methoxy-1,5-naphthyridin-4-yl)ethyl)piperidin-4-amine (10)
4.5.12. N-(4-(Difluoromethoxy)-3-methoxybenzyl)-1-(2-(6-methoxy-1,5-naphthyridin-4-yl)ethyl)piperidin-4-amine (19)
4.5.13. 2-(Difluoromethoxy)-5-((1-(2-(6-methoxy-1,5-naphthyridin-4-yl)ethyl)piperidin-4-ylamino)methyl)phenol (20)
4.5.14. 1-(2-(6-Methoxy-1,5-naphthyridin-4-yl)ethyl)-N-((6-(trifluoromethyl)pyridin-3-yl)methyl)piperidin-4-amine (21)
4.5.15. 1-(2-(6-Methoxy-1,5-naphthyridin-4-yl)ethyl)-N-((6-methoxypyridin-3-yl)methyl)piperidin-4-amine (22)
4.6. In Vitro DNA Gyrase Inhibitory Activity
4.7. In Vitro Topo IV Inhibitory Activity
4.8. Human Topo II Selectivity Evaluation
4.9. Antimicrobial Susceptibility Testing
4.10. Metabolic Activity Assessment
4.11. Cardiovascular hERG Inhibition
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rice, L.B. Federal funding for the study of antimicrobial resistance in nosocomial pathogens: No ESKAPE. J. Infect. Dis. 2008, 197, 1079–1081. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Available online: https://www.cdc.gov/drugresistance/pdf/ar-threats-2013-508.pdf (accessed on 29 March 2021).
- World Health Organization. Available online: https://www.who.int/news-room/detail/27-02-2017-who-publishes-list-of-bacteria-for-which-new-antibiotics-are-urgently-needed (accessed on 29 March 2021).
- Fair, R.J.; Tor, Y. Antibiotics and bacterial resistance in the 21st century. Perspect. Medicin. Chem. 2014, 6, 25–64. [Google Scholar] [CrossRef] [Green Version]
- Centers for Disease Control and Prevention. Available online: https://www.cdc.gov/drugresistance/pdf/threats-report/2019-ar-threats-report-508.pdf (accessed on 29 March 2021).
- Levasseur, P.; Delachaume, C.; Lowther, J.; Hodgson, J. Minimum Inhibitory Concentrations (MIC) And Mutation Prevention Concentrations (MPC) Of NXL101, A Novel Topoisomerase IV Inhibitor, Against Staphylococcus aureus Including Multi-resistant Strains. In Proceedings of the 45th Annual Interscience Conference on Antimicrobial Agents and Chemotherapy, Washington, DC, USA, 16–19 December 2005; American Society for Microbiology: Washington, DC, USA, 2005; p. 184. [Google Scholar]
- Gomez, L.; Hack, M.D.; Wu, J.; Wiener, J.J.M.; Venkatesan, H.; Santillaán, A., Jr.; Pippel, D.J.; Mani, N.; Morrow, B.J.; Motley, S.T.; et al. Novel pyrazolederivates as potent inhibitors of type II topoisomerases. Part 1: Synthesis and preliminary SAR analysis. Bioorg. Med. Chem. Lett. 2007, 17, 2723–2727. [Google Scholar] [PubMed]
- Wiener, J.J.M.; Gomez, L.; Venkatesan, H.; Santillaán, A., Jr.; Allison, B.D.; Schwarz, K.L.; Shinde, S.; Tang, L.; Hack, M.D.; Morrow, B.J.; et al. Tetrahydroindazole inhibitors of bacterial type II topoisomerases. Part 2: SAR development and potency against multidrug resistant strains. Bioorg. Med. Chem. Lett. 2007, 17, 2718–2722. [Google Scholar]
- Bax, B.D.; Chan, P.F.; Eggleston, D.S.; Fosberry, A.; Gentry, D.R.; Gorrec, F.; Giordano, I.; Hann, M.M.; Hennessy, A.; Hibbs, M.; et al. Type IIA topoisomerase inhibition by a new class of antibacterial agents. Nature 2010, 466, 935–940. [Google Scholar] [CrossRef]
- Kolarič, A.; Anderluh, M.; Minovski, N. Two Decades of Successful SAR-Grounded Stories of the Novel Bacterial Topoisomerase Inhibitors (NBTIs). J. Med. Chem. 2020, 63, 5664–5674. [Google Scholar] [CrossRef]
- Black, M.T.; Stachyra, T.; Platel, D.; Girard, A.-M.; Claudon, M.; Bruneau, J.-M.; Miossec, C. Mechanism of action of the antibiotic NXL101, a novel non-fluoroquinolone inhibitor of bacterial type II topoisomerases. Antimicrob. Agents Chemother. 2008, 52, 3339–3349. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.B.; Kaelin, D.E.; Wu, J.; Miesel, L.; Tan, C.M.; Meinke, P.T.; Olsen, D.; Lagrutta, A.; Bradley, P.; Lu, J.; et al. Oxabicyclooctane-linked novel bacterial topoisomerase inhibitors as broad spectrum antibacterial agents. ACS Med. Chem. Lett. 2014, 5, 609–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolarič, A.; Germe, T.; Hrast, M.; Stevenson, C.E.M.; Lawson, D.M.; Burton, N.P.; Vörös, J.; Maxwell, A.; Minovski, N.; Anderluh, M. Potent DNA gyrase inhibitors bind asymmetrically to their target using symmetrical bifurcated halogen bonds. Nat. Commun. 2021, 12, 1–13. [Google Scholar] [CrossRef]
- Bax, B.D.; Murshudov, G.; Maxwell, A.; Germe, T. DNA topoisomerase inhibitors: Trapping a DNA-cleaving machine in motion. J. Mol. Biol. 2019, 431, 3427–3449. [Google Scholar] [CrossRef]
- Kolaricč, A.; Minovski, N. Novel bacterial topoisomerase inhibitors: Challenges and perspectives in reducing hERG toxicity. Future Med. Chem. 2018, 10, 2241–2244. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Okumu, A.A.; Nolan, S.; English, A.; Vibhute, S.; Lu, Y.; Hervert-Thomas, K.; Seffernick, J.T.; Azap, L.; Cole, S.L.; et al. 1,3-Dioxane-linked bacterial topoisomerase inhibitors with enhanced antibacterial activity and reduced hERG inhibition. ACS Infect. Dis. 2019, 5, 1115–1128. [Google Scholar] [CrossRef]
- Okumu, A.; Lu, Y.; Dellos-Nolan, S.; Papa, J.L.; Koci, B.; Cockroft, N.T.; Gallucci, J.; Wozniak, D.J.; Yalowich, J.C.; Mitton-Fry, M.J. Novel bacterial topoisomerase inhibitors derived from isomannide. Eur. J. Med. Chem. 2020, 199, 112324. [Google Scholar] [CrossRef] [PubMed]
- Kolaricč, A.; Novak, D.; Weiss, M.; Hrast, M.; Zdovc, I.; Anderluh, M.; Minovski, N. Cyclohexyl amide-based novel bacterial topoisomerase inhibitors with prospective GyrA-binding fragments. Future Med. Chem. 2019, 11, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Kolaric, A.; Minovski, N. Structure-based design of novel combinatorially generated NBTIs as potential DNA gyrase inhibitors against various Staphylococcus aureus mutant strains. Mol. BioSyst. 2017, 13, 1406–1420. [Google Scholar] [CrossRef]
- Surivet, J.P.; Zumbrunn, C.; Bruyeère, T.; Bur, D.; Kohl, C.; Locher, H.H.; Seiler, P.; Ertel, E.A.; Hess, P.; Enderlin-Paput, M.; et al. Synthesis and characterization of tetrahydropyranbased bacterial topoisomerase inhibitors with antibacterial activity against Gram-negative bacteria. J. Med. Chem. 2017, 60, 3776–3794. [Google Scholar] [CrossRef]
- Rosenberger, J.; Butler, J.; Muenster, U.; Dressman, J. Application of a refined developability classification system. J. Pharm. Sci. 2019, 108, 1090–1100. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. Available online: https://www.fda.gov/media/70963/download (accessed on 29 March 2021).
- Surivet, J.P.; Zumbrunn, C.; Rueedi, G.; Bur, D.; Bruyeère, T.; Locher, H.; Ritz, D.; Seiler, P.; Kohl, C.; Ertel, E.A.; et al. Novel tetrahydropyran-based bacterial topoisomerase inhibitors with potent anti-gram positive activity and improved safety profile. J. Med. Chem. 2015, 58, 927–942. [Google Scholar] [CrossRef]
- Nayar, A.S.; Dougherty, T.J.; Reck, F.; Thresher, J.; Gao, N.; Shapiro, A.B.; Ehmann, D.E. Target-based resistance in Pseudomonas aeruginosa and Escherichia coli to NBTI 5463, a novel bacterial type II topoisomerase inhibitor. Antimicrob. Agents Chemother. 2015, 59, 331–337. [Google Scholar] [CrossRef] [Green Version]
- Sirimulla, S.; Bailey, J.B.; Vegesna, R.; Narayan, M. Halogen interactions in protein–ligand complexes: Implications of halogen bonding for rational drug design. J. Chem. Inf. Model. 2013, 53, 2781–2791. [Google Scholar] [CrossRef]
- Hopkins, A.L.; Keserü, G.M.; Leeson, P.D.; Rees, D.C.; Reynolds, C.H. The role of ligand efficiency metrics in drug discovery. Nat. Rew. 2014, 13, 105–121. [Google Scholar] [CrossRef]
- Silhavy, T.J.; Kahne, D.; Walker, S. The bacterial cell envelope. Cold Spring Harb. Perspect Biol. 2010, 2, a000414. [Google Scholar] [CrossRef]
- Nikaido, H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 2003, 67, 593–656. [Google Scholar] [CrossRef] [Green Version]
- Richter, M.F.; Drown, B.S.; Riley, A.P.; Garcia, A.; Shirai, T.; Svec, R.L.; Hergenrother, P.J. Predictive compound accumulation rules yield a broad-spectrum antibiotic. Nature 2017, 545, 299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manchester, J.I.; Dussault, D.D.; Rose, J.A.; Boriack-Sjodin, P.A.; Uria-Nickelsen, M.; Ioannidis, G.; Bist, S.; Fleming, P.; Hull, K.G. Discovery of a novel azaindole class of antibacterial agents targeting the ATPase domains of DNA gyrase and topoisomerase IV. Bioorg. Med. Chem. Lett. 2012, 22, 5150–5156. [Google Scholar] [CrossRef]
- Bissachi, G.S.; Manchester, J.I. A new-class antibacterial—almost. Lessons in drug discovery and development: A critical analysis of more than 50 years of effort toward ATPase inhibitors of DNA gyrase and topoisomerase IV. ACS Infect. Dis. 2015, 1, 4–41. [Google Scholar]
- Delcour, A.H. Outer membrane permeability and antibiotic resistance. Biochim. Biophys. Acta 2009, 1794, 808–816. [Google Scholar] [CrossRef] [Green Version]
- Jang, S. Multidrug efflux pumps in Staphylococcus aureus and their clinical implications. J. Microbiol. 2016, 54, 1–8. [Google Scholar] [CrossRef]
- Fullam, E.; Young, R.J. Physicochemical properties and Mycobacterium tuberculosis transporters: Keys to efficacious antitubercular drugs? RSC Med. Chem. 2021, 12, 43–56. [Google Scholar] [CrossRef]
- Nikaido, H. Multidrug efflux pumps of gram-negative bacteria. J. Bacteriol. 1996, 178, 5853–5859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Murawski, A.; Patel, K.; Crespi, C.; Balimane, P. A novel design of artificial membrane for improving the PAMPA model. Pharm. Res. 2008, 25, 1511–1520. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylo, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein-ligand docking using GOLD. Proteins Struct. Funct. Bioinf. 2003, 52, 609–623. [Google Scholar] [CrossRef]
- Gonzaga, D.; Senger, M.R.; da Silva, C.; Ferreira, V.F.; Silva, F.P., Jr. 1-Phenyl-1H- and 2-phenyl-2H-1,2,3-triazol derivatives: Design, synthesis and inhibitory effect on alpha-glycosidases. Eur. J. Med. Chem. 2014, 74, 461–476. [Google Scholar] [CrossRef]
- Patel, J.B.; Cockerill, F.R.; Badfort, P.A.; Elioppoulos, G.M.; Hindler, J.A.; Jenkins, S.G.; Lewis, J.S.; Limbago, B.; Miller, L.A.; Nicolau, D.P.; et al. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically, Approved Standard, 10th ed.; CLSI Document M07-A10; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2015; Available online: https://clsi.org/media/1632/m07a10_sample.pdf (accessed on 13 July 2021).
- The European Committee on Antimicrobial Susceptibility Testing. Breakpoint Tables for Interpretation of MICs and Zone Diameters. Version 5.0. 2015. Available online: www.eucast.org (accessed on 13 July 2021).
- Promega Corporation. Technical Bulletin: CellTiter 96® AQueous One Solution Cell Proliferation Assay; Promega: Madison, WI, USA, 2001; Available online: https://www.promega.com/-/media/files/resources/protocols/technical-bulletins/0/celltiter-96-aqueous-one-solution-cell-proliferation-assay-system-protocol.pdf (accessed on 13 July 2021).








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cmpd | Structure | DNA Gyrase IC50 (μM) 1 | Topoisomerase IV IC50 (μM) 1 | Human Topo II (% RA) 2 | ||
|---|---|---|---|---|---|---|
| S. aureus | E. coli | S. aureus | E. coli | |||
| 3 |  | 1.60 ± 0.35 | >100 | >100 | 1.18 ± 0.09 | 94.71 ± 4.82 |
| 4 |  | 0.51 ± 0.25 | 13.27 ± 2.84 | >100 | 0.12 ± 0.01 | 88.41 ± 0.15 |
| 5 |  | 0.33 ± 0.08 | 3.00 ± 0.55 | 29.59 ± 0.54 | 0.085 ± 0.002 | 82.10 ± 2.52 |
| 6 |  | >100 | >100 | >100 | >100 | 99.36 ± 9.93 |
| 7 |  | >100 | >100 | >100 | >100 | 90.35 ± 7.56 |
| 8 |  | 0.24 ± 0.04 | 7.83 ± 0.55 | >100 | 0.21 ± 0.00 | 94.17 ± 2.90 |
| 9 |  | 0.16 ± 0.01 | 2.95 ± 0.56 | 40.38 ± 0.09 | 0.23 ± 0.00 | 101.25 ± 6.82 |
| 10 |  | 1.10 ± 0.17 | 16.70 ± 0.93 | >100 | 0.28 ± 0.01 | 95.69 ± 6.42 |
| 11 |  | 1.02 ± 0.02 | 40.60 ± 1.97 | >100 | 2.04 ± 0.20 | 96.97 ± 9.58 |
| 12 |  | 0.55 ± 0.06 | 16.95 ± 1.69 | >100 | 0.28 ± 0.03 | 100.81 ± 11.22 |
| 13 |  | 0.035 ± 0.01 | 1.71 ± 0.05 | 16.37 ± 0.56 | 0.031 ± 0.00 | 101.98 ± 0.84 |
| 14 |  | 0.007 ± 0.00 3 | 0.57 ± 0.06 | 7.22 ± 0.55 | 0.042 ± 0.003 | 98.25 ± 2.72 |
| 15 |  | 0.011 ± 0.003 3 | 0.28 ± 0.02 | 2.13 ± 0.11 | 0.021 ± 0.001 | 88.26 ± 3.33 |
| 16 |  | 4.39 ± 0.55 | 56.56 ± 3.32 | >100 | 1.21 ± 0.05 | 97.73 ± 1.08 |
| 17 |  | 1.82 ± 0.11 | >100 | >100 | 1.15 ± 0.03 | 97.80 ± 2.21 |
| 18 |  | 0.067 ± 0.01 | 11.89 ± 0.83 | >100 | >100 | 94.19 ± 1.33 |
| 19 |  | 0.35 ± 0.03 | 3.90 ± 0.06 | 29.04 ± 1.89 | 0.10 ± 0.01 | 96.36 ± 2.30 |
| 20 |  | 0.33 ± 0.02 | 4.41 ± 0.16 | >100 | 0.06 ± 0.00 | 94.80 ± 0 |
| 21 |  | 0.16 ± 0.02 | 6.91 ± 1.62 | 18.45 ± 1.32 | >100 | 95.49 ± 2.39 |
| 22 |  | 0.34 ± 0.05 | 7.24 ± 0.34 | >100 | >100 | 97.05 ± 6.94 |
| Gepotidacin |  | 0.37 ± 0.02 | 0.24 ± 0.04 | 8.30 ± 0.37 | 0.05 ± 0.00 | ND |
| Compd | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | 21 | 22 | Gepotidacin |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MIC [μg/mL] | |||||||||||||||||||||
| S. aureus (ATCC 29213) | 1 | 2 | 0.25 | >128 | >128 | 0.5 | 0.125 | 2 | 4 | 1 | 0.125 | 0.031 | 0.0078 | 4 | 2 | 0.125 | 1 | 0.5 | 0.25 | 0.5 | 0.125 |
| MRSA | 2 | 2 | 0.5 | >128 | >128 | 0.5 | 0.25 | 2 | 8 | 2 | 0.25 | 0.062 | 0.015 | 4 | 4 | 0.25 | 2 | 1 | 0.5 | 1 | ND |
| MRSA 1 (QA-11.7) | 8 | 2 | 0.5 | >128 | >128 | 0.5 | 1 | 4 | 8 | 4 | 0.5 | 0.0625 | 0.0078 | 4 | 4 | 0.5 | 2 | 0.5 | 1 | 1 | ND |
| MRSA 2 (QA-11.6) | 2 | 4 | 1 | >128 | >128 | 0.5 | 1 | 4 | 8 | 4 | 0.25 | 0.0625 | 0.03125 | 4 | 4 | 0.25 | 2 | 1 | 0.5 | 2 | ND |
| MRSA 3 (QA-11.4) | 4 | 8 | 1 | >128 | >128 | 1 | 1 | 4 | 8 | 4 | 0.5 | 0.125 | 0.0625 | 8 | 8 | 0.5 | 2 | 2 | 1 | 2 | ND |
| MRSA 4 (QA-11.2) | 4 | 8 | 1 | >128 | >128 | 0.5 | 1 | 4 | 8 | 4 | 0.25 | 0.125 | 0.03125 | 8 | 8 | 0.5 | 2 | 1 | 0.5 | 2 | ND |
| E. faecalis DRK 057 (ATCC 29212) | 4 | 8 | 1 | >128 | >128 | 1 | 1 | 8 | 16 | 8 | 1 | 1 | 1 | 8 | 128 | 1 | 16 | 4 | 2 | 2 | ND |
| S. pneumoniae (QA-10/02) | 2 | 4 | 0.5 | >128 | 128 | 0.25 | 0.5 | 1 | 2 | 2 | 0.5 | 0.125 | 0.015 | 2 | 16 | 0.25 | 2 | 1 | 1 | 1 | ND |
| S. agalactiae RDK 047 (QA-990/02) | <1 | 2 | 1 | 128 | >128 | 0.5 | 0.5 | 4 | 16 | 8 | 1 | 0.5 | 0.125 | 8 | >128 | 0.25 | <1 | 2 | 4 | 1 | ND |
| E. coli (ATCC 25922) | >128 | 32 | 16 | >128 | >128 | 64 | 8 | 16 | 64 | 32 | 4 | 2 | 2 | 128 | >128 | 32 | 32 | 8 | 32 | 16 | 1 |
| E. coli 5 (QA-11.8) | 64 | 16 | 2 | >128 | >128 | 32 | 8 | 16 | 64 | 16 | 2 | 1 | 0.25 | 128 | >128 | 8 | 4 | 4 | 8 | 8 | ND |
| E. coli 6 (QA-11.7) | 64 | 16 | 2 | >128 | >128 | 4 | 2 | 4 | 32 | 8 | 1 | 1 | 1 | 32 | 128 | 4 | 2 | 2 | 2 | 2 | ND |
| E. coli 7 (QA-11.3) | 128 | 8 | 2 | 128 | >128 | 32 | 4 | 8 | 32 | 8 | 2 | 1 | 1 | 32 | 128 | 8 | 4 | 1 | 8 | 4 | ND |
| E. coli D228 | >128 | 16 | 1 | >128 | >128 | 32 | 1 | 8 | 16 | 16 | 2 | 0.125 | 0.125 | 16 | 128 | 8 | 16 | 2 | 4 | 4 | 0.125 |
| E. coli N43 9 (CGSC# 5583) | 16 | 2 | 1 | 128 | >128 | 1 | 0.25 | 1 | 8 | 4 | 0.5 | 0.125 | 0.078 | 16 | 16 | 1 | 1 | 0.5 | 1 | 1 | 0.016 |
| A. baumannii 8C6 (GES-14) | >128 | 32 | 4 | >128 | >128 | 32 | 2 | 32 | 64 | 32 | 4 | 1 | 0.25 | 128 | 128 | 8 | 8 | 4 | 16 | 4 | ND |
| P. aeruginosa RDK 184 (DSM 939) | >128 | >128 | >128 | >128 | >128 | >128 | >128 | >128 | >128 | >128 | >128 | 128 | 32 | >128 | >128 | >128 | >128 | >128 | >128 | >128 | ND |
| S. alachua RDK 030c (QA-1482/04) | >128 | 128 | 64 | >128 | >128 | >128 | 64 | 128 | >128 | 128 | 32 | 16 | 8 | >128 | >128 | 128 | 128 | 64 | 128 | 64 | ND |
| C. jejuni RDK 135 (ATCC 33560) | 1 | 128 | 16 | >128 | >128 | 4 | 0.5 | 8 | 32 | 1 | 2 | 0.5 | 0.015 | 32 | 128 | 4 | 2 | 0.015 | 128 | 2 | ND |
| M. smegmatis | 4 | ≤1 | 0.25 | 64 | 16 | 0.5 | 0.25 | ≤1 | 8 | 2 | 0.25 | 0.125 | 0.015 | 32 | 16 | 4 | 4 | 0.5 | 0.25 | 16 | ND |
| M. avium | >128 | 128 | 64 | >128 | >128 | 32 | 128 | >128 | >128 | 32 | 4 | 2 | 1 | >128 | 128 | 128 | 128 | 128 | 4 | >128 | ND |
| LE 1 | ||||
|---|---|---|---|---|
| Cmpd | DNA Gyrase | Topo IV | ||
| S. aureus | E. coli | S. aureus | E. coli | |
| 3 | 0.25 | NA | NA | 0.25 |
| 4 | 0.25 | 0.20 | NA | 0.28 |
| 5 | 0.27 | 0.23 | 0.19 | 0.29 |
| 6 | NA | NA | NA | NA |
| 7 | NA | NA | NA | NA |
| 8 | 0.27 | 0.21 | NA | 0.28 |
| 9 | 0.31 | 0.25 | 0.20 | 0.30 |
| 10 | 0.27 | 0.22 | NA | 0.30 |
| 11 | 0.29 | 0.21 | NA | 0.28 |
| 12 | 0.30 | 0.23 | NA | 0.31 |
| 13 | 0.35 | 0.27 | 0.23 | 0.35 |
| 14 | 0.39 | 0.29 | 0.24 | 0.35 |
| 15 | 0.38 | 0.31 | 0.27 | 0.36 |
| 16 | 0.24 | 0.19 | NA | 0.27 |
| 17 | 0.25 | NA | NA | 0.26 |
| 18 | 0.32 | 0.22 | NA | NA |
| 19 | 0.26 | 0.22 | 0.18 | 0.28 |
| 20 | 0.27 | 0.22 | NA | 0.30 |
| 21 | 0.29 | 0.22 | 0.20 | NA |
| 22 | 0.30 | 0.23 | NA | NA |
| Cmpd | HUVEC | HepG2 | hERG |
|---|---|---|---|
| IC50 [μM] | IC50 [μM] | IC50 [μM] | |
| 3 | >50 | Nd | 1.04 |
| 4 | 9.64 ± 0.03 | 4.64 ± 0.10 | 0.33 |
| 5 | 10.70 ± 0.01 | 6.33 ± 0.19 | 0.62 |
| 6 | >50 | ND | ND |
| 7 | ND | ND | ND |
| 8 | 39.41 ± 5.47 | 17.08 ± 0.98 | 0.37 |
| 9 | >50 | >50 | 9.82 |
| 10 | >50 | >50 | 6.62 |
| 11 | 54.08 ± 3.45 | 17.67 ± 2.16 | 1.05 |
| 12 | >50 | 12.67± 0.21 | 0.59 |
| 13 | 26.96 ± 0.38 | 10.16 ± 0.10 | 0.27 |
| 14 | 25.42 ± 0.01 | 9.37 ± 0.07 | 0.29 |
| 15 | 18.98 ± 0.85 | 8.47 ± 0.09 | 0.18 |
| 16 | >50 | ND | 3.24 |
| 17 | >50 | ND | 13.45 |
| 18 | 23.85 ± 0.09 | 10.03 ± 0.06 | 2.11 |
| 19 | 19.83 ± 2.58 | 6.31 ± 0.09 | 0.46 |
| 20 | 29.80 ± 1.13 | 10.86 ± 0.12 | 0.18 |
| 21 | >50 | >50 | 1.97 |
| 22 | >50 | 32.03 ± 0.30 | 3.05 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kolarič, A.; Kokot, M.; Hrast, M.; Weiss, M.; Zdovc, I.; Trontelj, J.; Žakelj, S.; Anderluh, M.; Minovski, N. A Fine-Tuned Lipophilicity/Hydrophilicity Ratio Governs Antibacterial Potency and Selectivity of Bifurcated Halogen Bond-Forming NBTIs. Antibiotics 2021, 10, 862. https://doi.org/10.3390/antibiotics10070862
Kolarič A, Kokot M, Hrast M, Weiss M, Zdovc I, Trontelj J, Žakelj S, Anderluh M, Minovski N. A Fine-Tuned Lipophilicity/Hydrophilicity Ratio Governs Antibacterial Potency and Selectivity of Bifurcated Halogen Bond-Forming NBTIs. Antibiotics. 2021; 10(7):862. https://doi.org/10.3390/antibiotics10070862
Chicago/Turabian StyleKolarič, Anja, Maja Kokot, Martina Hrast, Matjaž Weiss, Irena Zdovc, Jurij Trontelj, Simon Žakelj, Marko Anderluh, and Nikola Minovski. 2021. "A Fine-Tuned Lipophilicity/Hydrophilicity Ratio Governs Antibacterial Potency and Selectivity of Bifurcated Halogen Bond-Forming NBTIs" Antibiotics 10, no. 7: 862. https://doi.org/10.3390/antibiotics10070862
APA StyleKolarič, A., Kokot, M., Hrast, M., Weiss, M., Zdovc, I., Trontelj, J., Žakelj, S., Anderluh, M., & Minovski, N. (2021). A Fine-Tuned Lipophilicity/Hydrophilicity Ratio Governs Antibacterial Potency and Selectivity of Bifurcated Halogen Bond-Forming NBTIs. Antibiotics, 10(7), 862. https://doi.org/10.3390/antibiotics10070862