
An Update Review of Approaches to Multiple Action-Based Antibacterials

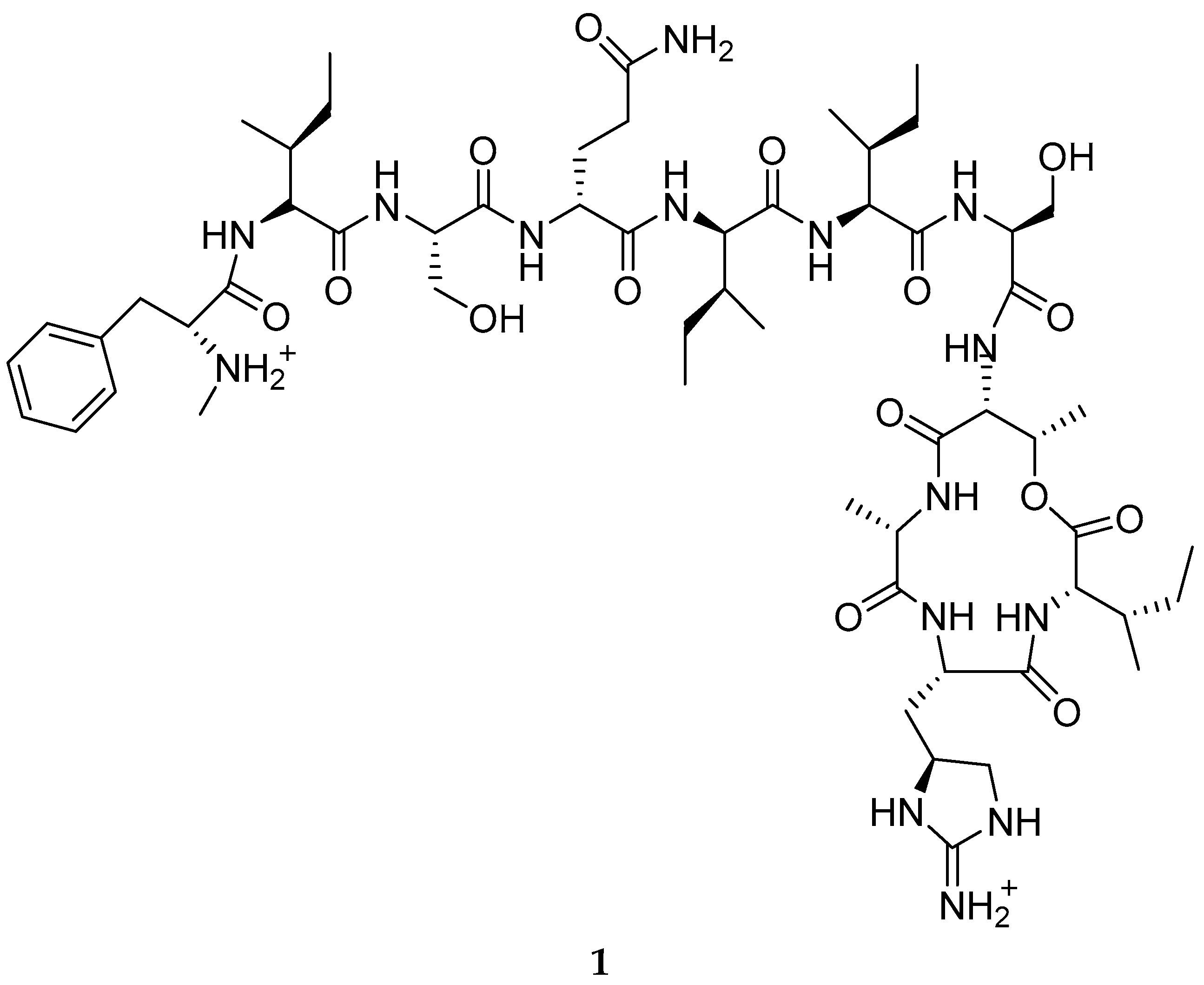{kind=link}
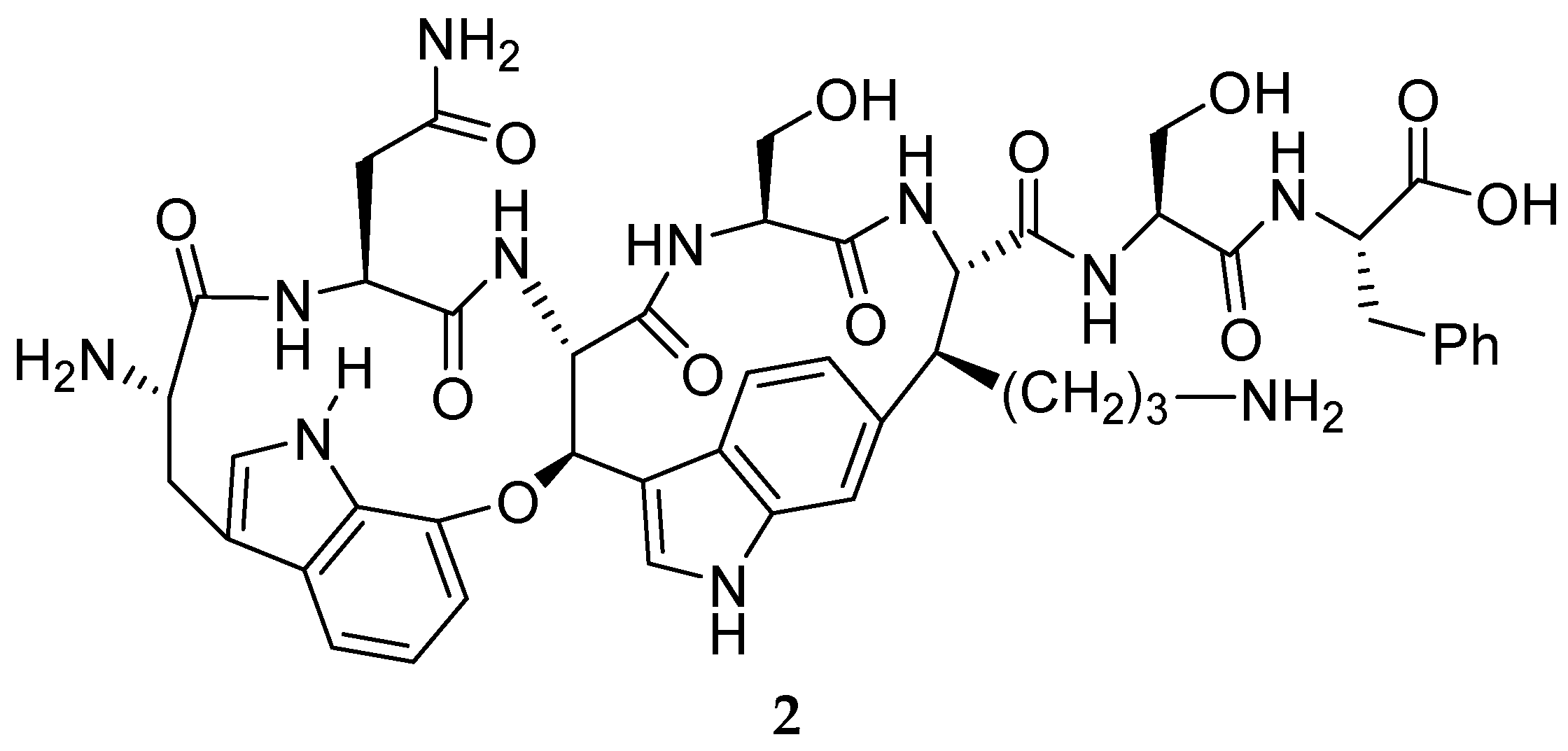{kind=link}
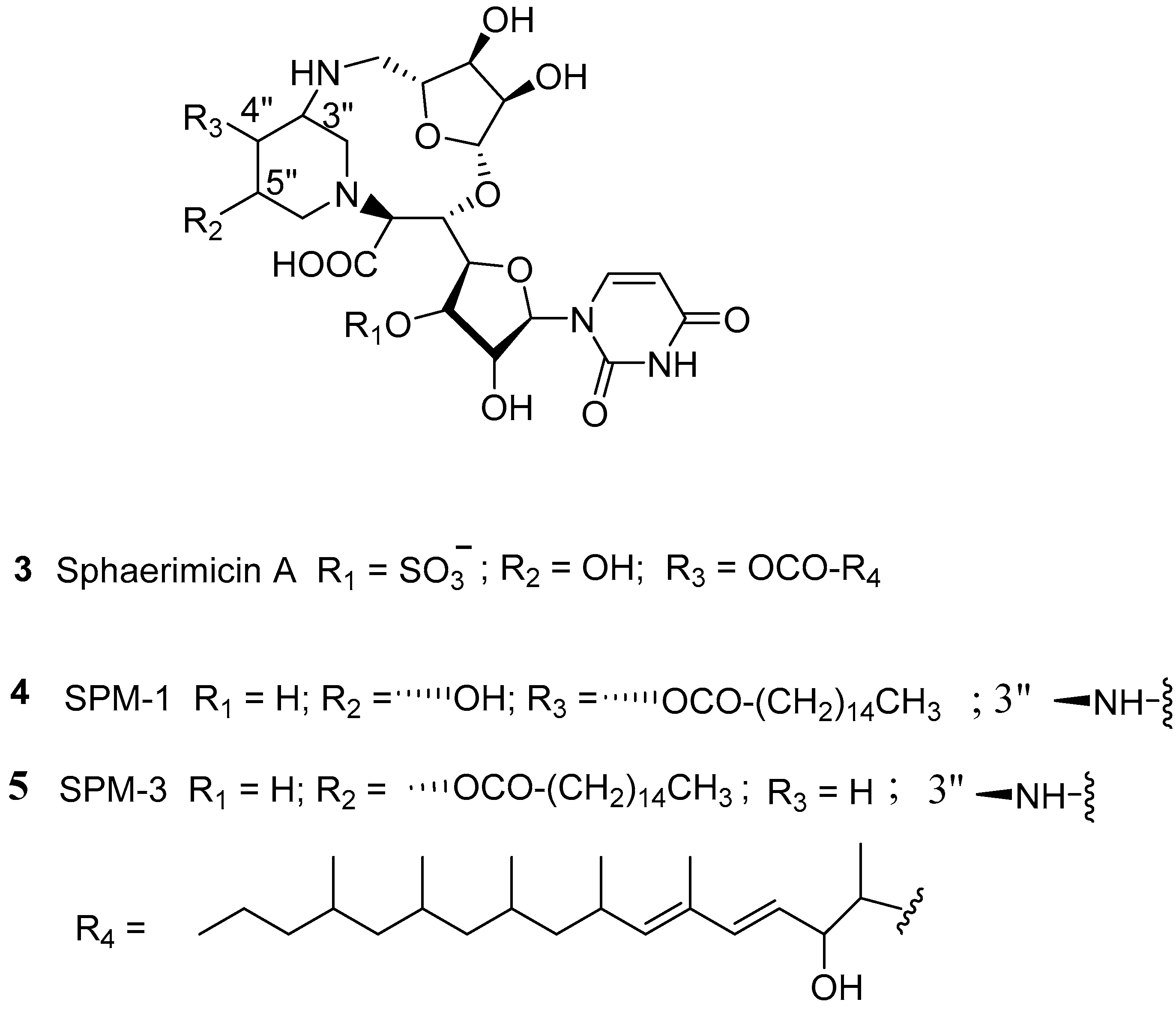{kind=link}
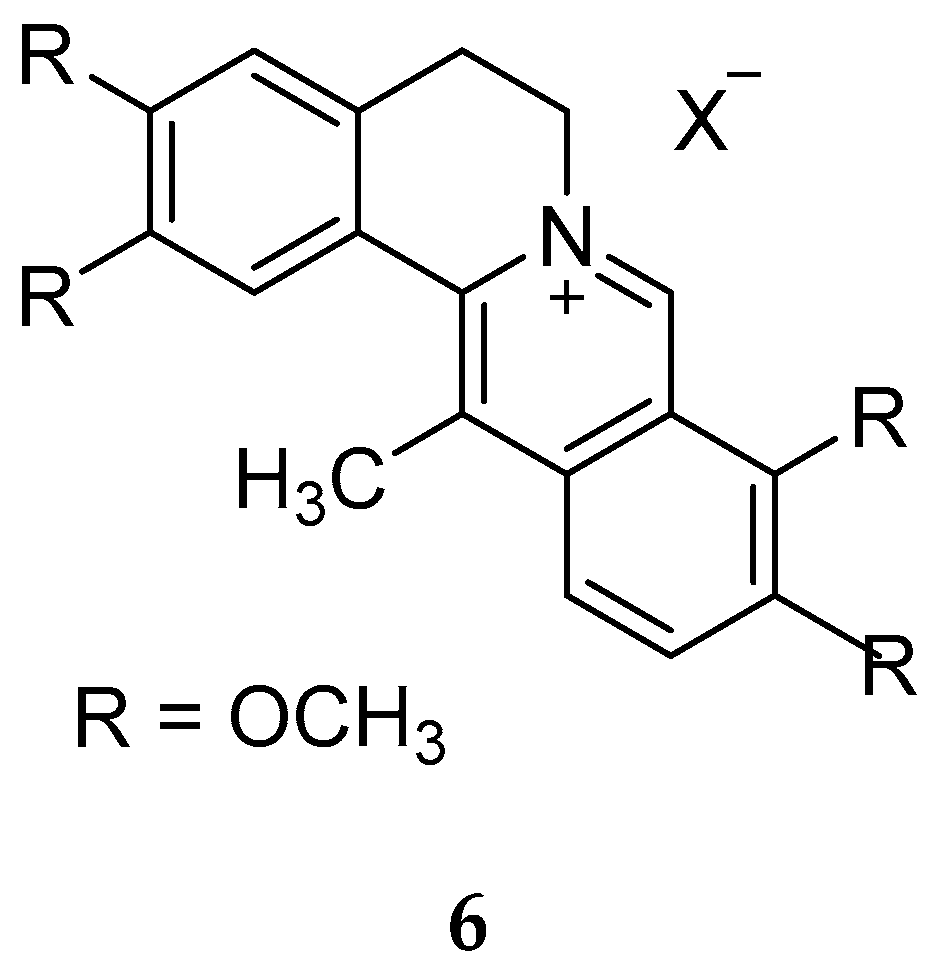{kind=link}
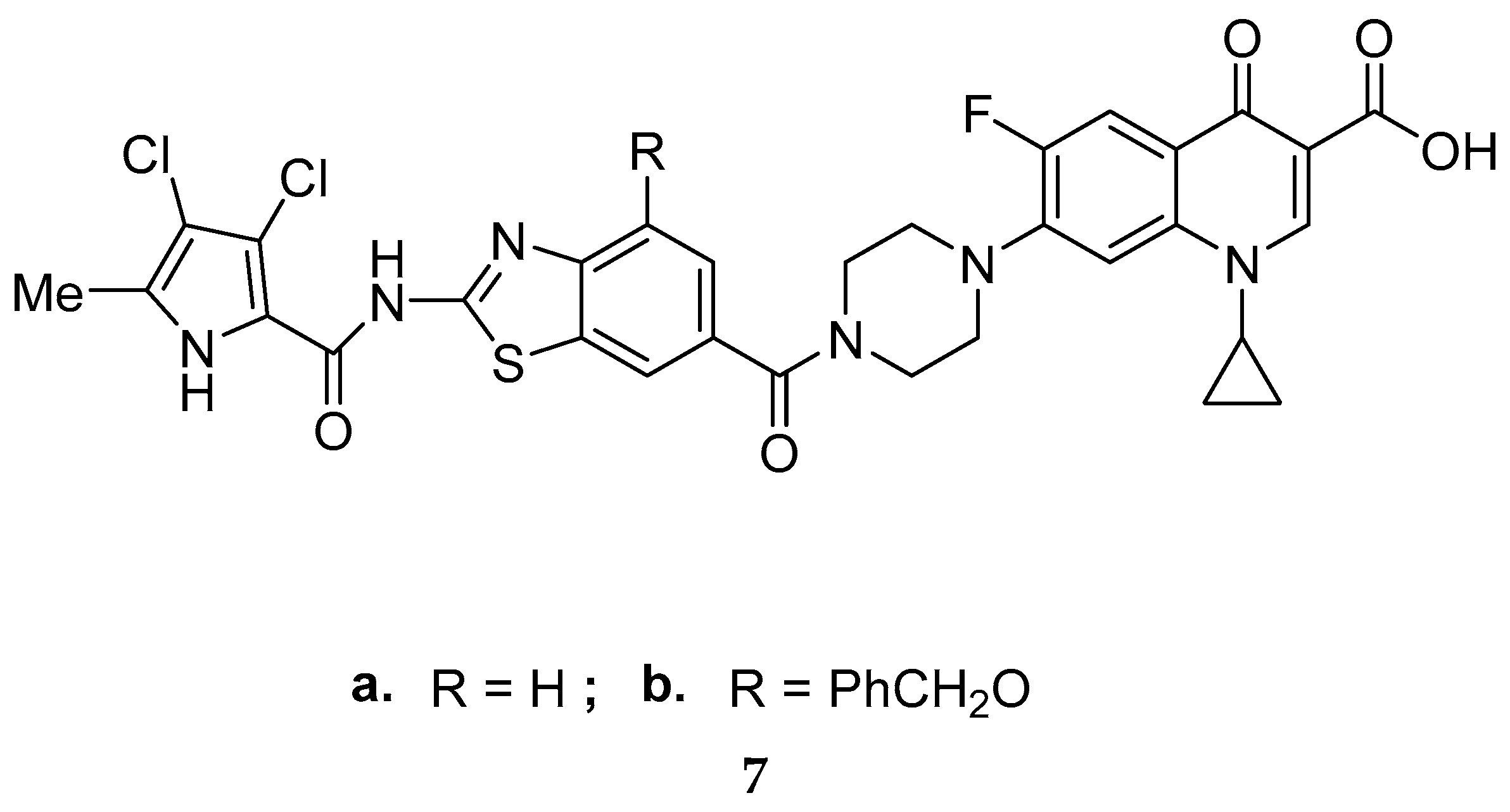{kind=link}
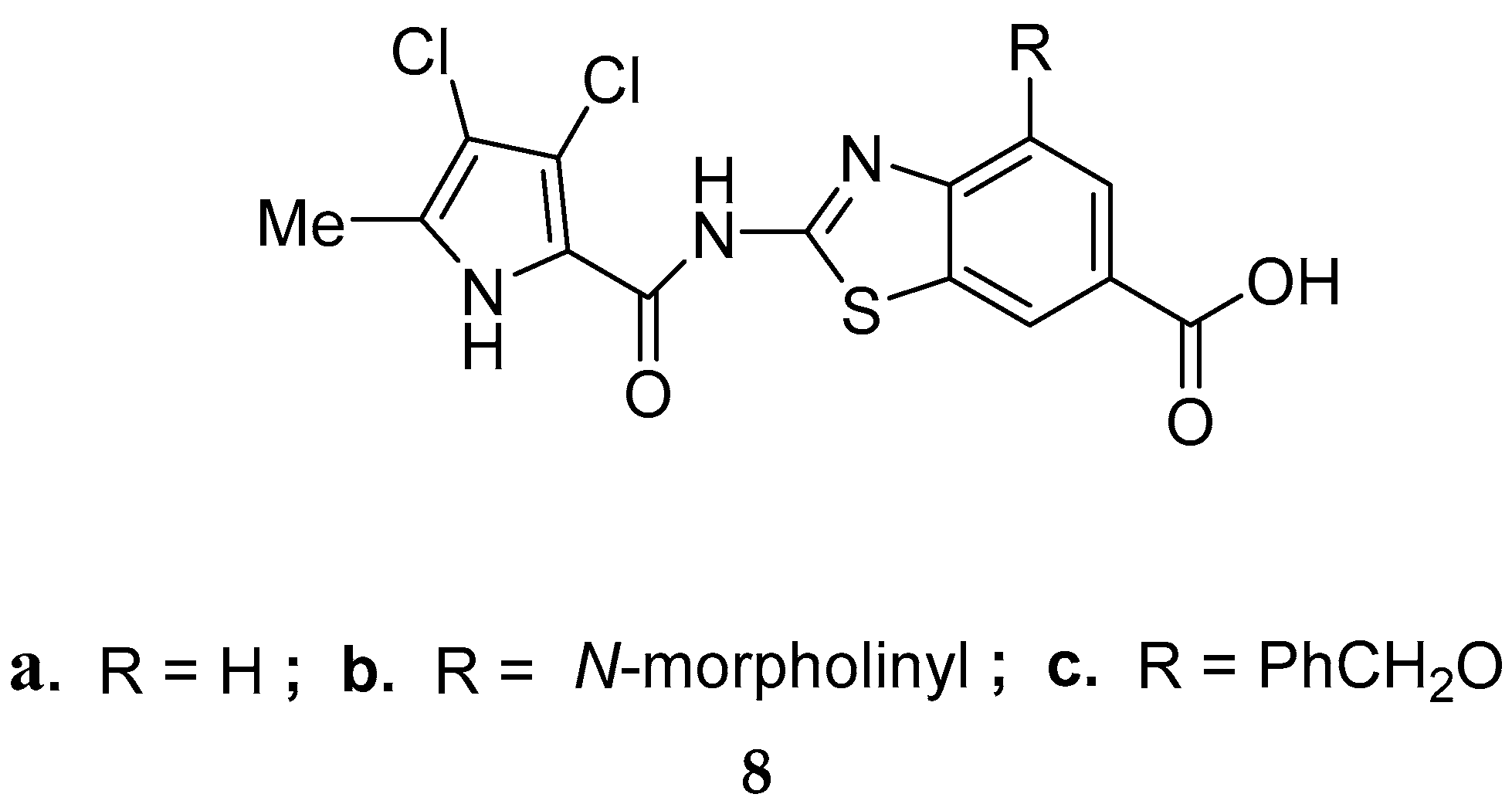{kind=link}
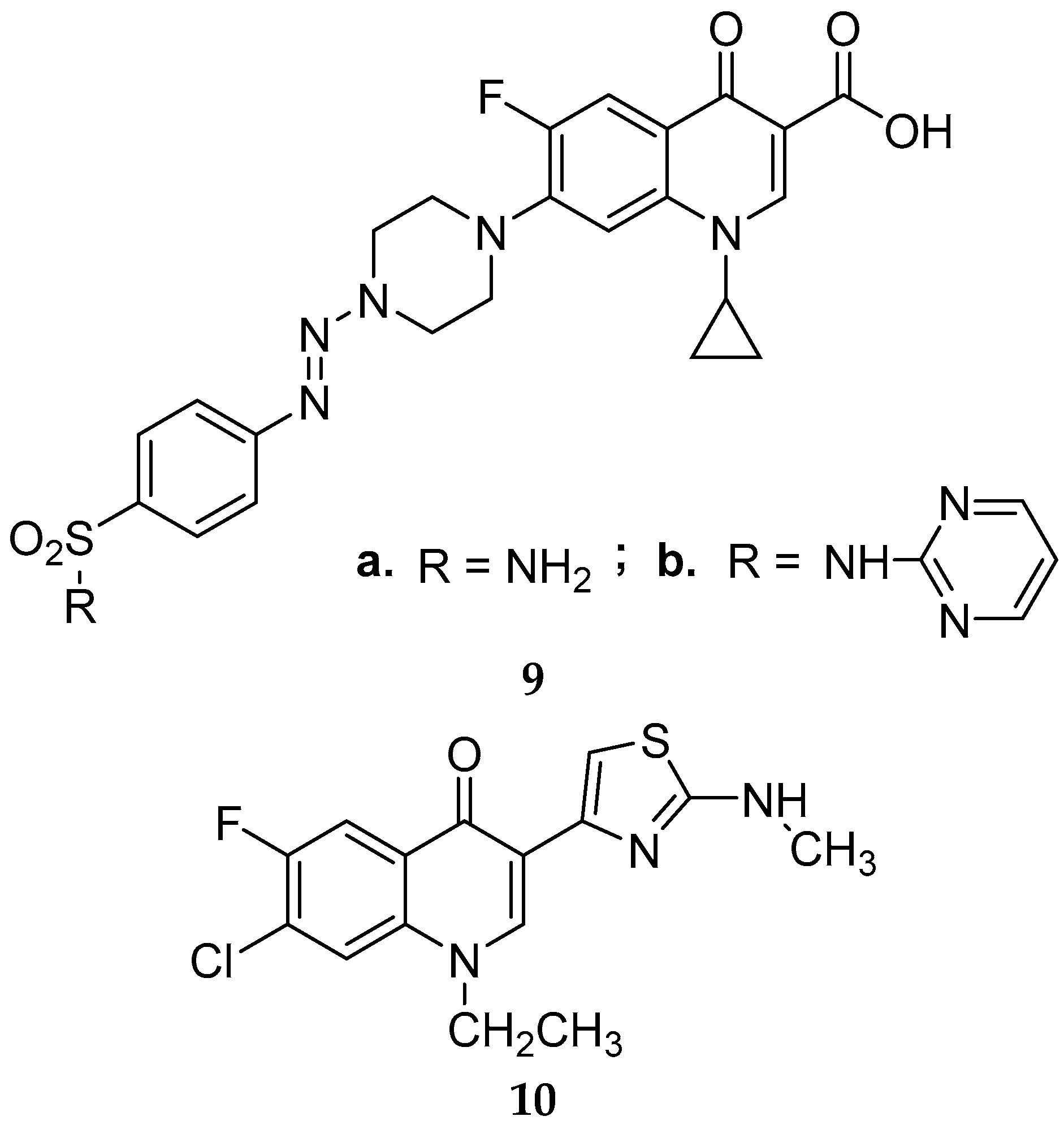{kind=link}
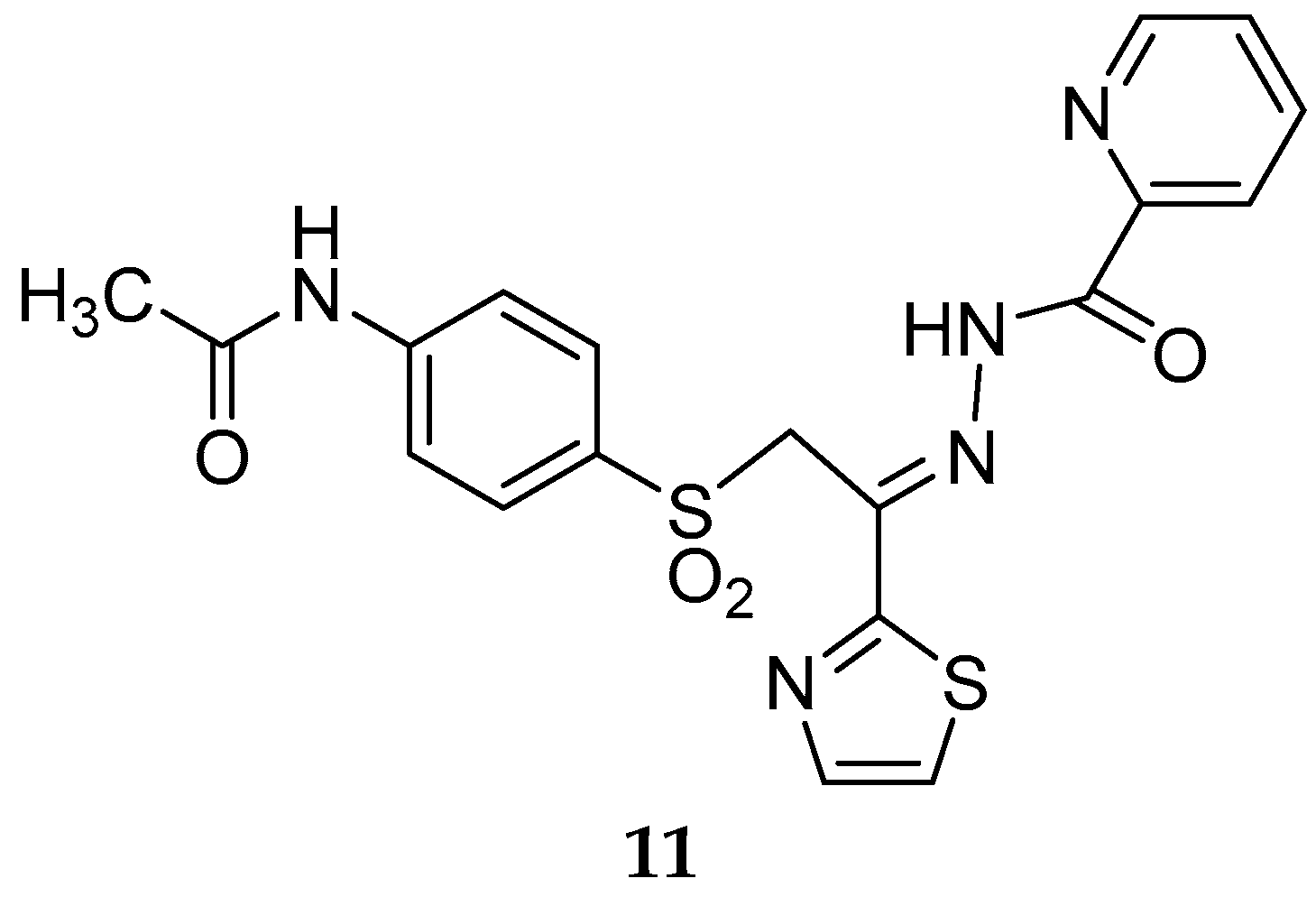{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Multi-Targeting Combinations
2.1. Recent Trends with Antibacterial Combinations
2.2. Nanoparticulates in Combinations
3. Single-Molecule Non-Cleavable Hybrids
3.1. Natural Products
3.2. Synthetic Multi-Targeting Non-Cleavable Compounds
4. Prodrug-Based Approaches
4.1. Enzyme-Mediated Triggering
4.2. Bio-Orthogonal Activation Approaches
4.3. Release of Sulfur Dioxide
4.4. Future Antibacterial Prodrug Developments
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Xie, J. Grand Challenge of Antibiotics Resistance: A Global, Multidisciplinary Effort is Needed. Front. Antibiot. 2022, 1, 984076. [Google Scholar] [CrossRef]
- World Health Organization. Global Antimicrobial Resistance and Use Surveillance System (GLASS) Report 2022; World Health Organization: Geneva, Switzerland, 2022. [Google Scholar]
- CNN.com. Available online: https://edition.cnn.com/2023/01/19/health/first-us-multidrug-resistant-gonorrhea/index.html (accessed on 23 March 2023).
- Wang, Y.; Zhigang, Y.; Ding, P.; Lu, J.; Mao, L.; Ngiam, L.; Yuan, Z.; Engelstädter, J.; Schembri, M.; Guo, J. Antidepressants Can Induce Mutation and Enhance Persistence toward Multiple Antibiotics. Proc. Natl. Acad. Sci. USA 2023, 120, e2208344120. [Google Scholar] [CrossRef] [PubMed]
- Drew, L. How Antidepressants Help Bacteria Resist Antibiotics. Nature 2023. [Google Scholar] [CrossRef] [PubMed]
- Miethke, M.; Pieroni, M.; Weber, T.; Brönstrup, M.; Hammann, P.; Halby, L.; Arimondo, P.B.; Glaser, P.; Aigle, B.; Bode, H.B.; et al. Towards the Sustainable Discovery and Development of New Antibiotics. Nat. Rev. Chem. 2021, 5, 726–749. [Google Scholar] [CrossRef] [PubMed]
- Schcolnik-Cabrera, A. Current Approaches to Overcome Antimicrobial Resistance. Curr. Med. Chem. 2022, 30, 3–4. [Google Scholar] [CrossRef]
- Ahmed, S.; Ahmed, M.Z.; Rafique, S.; Almasoudi, S.E.; Shah, M.; Jalil, N.A.C.; Ojha, S.C. Recent Approaches for Downplaying Antibiotic Resistance: Molecular Mechanisms. Biomed Res. Int. 2023, 2023, 5250040. [Google Scholar] [CrossRef]
- Eisinger, R.W.; Williams, M.P.; Choe, S.H.; Krofah, E. A Call to Action—Stopping Antimicrobial Resistance. JAC Antimicrob. Resist. 2023, 5, dlac142. [Google Scholar] [CrossRef]
- Canturri, A.M.; Smani, Y. Anthelmintic Drugs for Repurposing against Gram-Negative Bacilli Infections. Curr. Med. Chem. 2022, 30, 59–71. [Google Scholar] [CrossRef]
- Gray, D.A.; Wenzel, M. Multitarget Approaches against Multiresistant Superbugs. ACS Infect. Dis. 2020, 6, 1346–1365. [Google Scholar] [CrossRef]
- Privalsky, T.M.; Soohoo, A.M.; Wang, J.; Walsh, C.T.; Wright, G.D.; Gordon, E.M.; Gray, N.S.; Khosla, C. Prospects for Antibacterial Discovery and Development. J. Am. Chem. Soc. 2021, 143, 21127–21142. [Google Scholar] [CrossRef]
- Bremner, J. Multiple Action-Based Design Approaches to Antibacterials; Springer: Singapore, 2021. [Google Scholar]
- Si, Z.; Pethe, K.; Chan-Park, M.B. Chemical Basis of Combination Therapy to Combat Antibiotic Resistance. J. Amer. Chem. Soc. Au 2023, 3, 276–292. [Google Scholar] [CrossRef] [PubMed]
- Compagne, N.; Vieira Da Cruz, A.; Müller, R.T.; Hartkoorn, R.C.; Flipo, M.; Pos, K.M. Update on the Discovery of Efflux Pump Inhibitors against Critical Priority Gram-Negative Bacteria. Antibiotics 2023, 12, 180. [Google Scholar] [CrossRef] [PubMed]
- Lázár, V.; Snitser, O.; Barkan, D.; Kishony, R. Antibiotic Combinations Reduce Staphylococcus aureus Clearance. Nature 2022, 610, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Luo, J.; Deng, F.; Huang, Y.; Zhou, H. Antibiotic Combination Therapy: A Strategy to Overcome Bacterial Resistance to Aminoglycoside Antibiotics. Front. Pharmacol. 2022, 13, 839808. [Google Scholar] [CrossRef]
- Hussein, M.; Han, M.L.; Zhu, Y.; Zhou, Q.; Lin, Y.W.; Hancock, R.E.W.; Hoyer, D.; Creek, D.J.; Li, J.; Velkov, T. Metabolomics Study of the Synergistic Killing of Polymyxin B in Combination with Amikacin against Polymyxin-Susceptible and -Resistant Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2019, 64, e01587-19. [Google Scholar] [CrossRef]
- Rosenberg, C.R.; Fang, X.; Allison, K.R. Potentiating Aminoglycoside Antibiotics to Reduce Their Toxic Side Effects. PLoS ONE 2020, 15, e0237948. [Google Scholar] [CrossRef]
- Mei, J.A.; Johnson, W.; Kinn, B.; Laskey, E.; Nolin, L.; Bhamare, P.; Stalker, C.; Dunman, P.M.; Wozniak, R.A.F. Antimicrobial Activity of a Triple Antibiotic Combination Toward Ocular Pseudomonas aeruginosa Clinical Isolates. Trans. Vis. Sci. Technol. 2022, 11, 26. [Google Scholar] [CrossRef]
- Al-Madboly, L.A. A Novel Triple Combination to Combat Serious Infections with Carbapenem-Resistant Acinetobacter baumannii in a Mouse Pneumonia Model. Microbiol. Spectr. 2022, 10, e0271021. [Google Scholar] [CrossRef]
- Umar, H.; Wahab, H.A.; Gazzali, A.M.; Tahir, H.; Ahmad, W. Cubosomes: Design, Development, and Tumor-Targeted Drug Delivery Applications. Polymers 2022, 14, 3118. [Google Scholar] [CrossRef]
- Lai, X.; Han, M.-L.; Ding, Y.; Chow, S.H.; Le Brun, A.P.; Wu, C.-M.; Bergen, P.J.; Jiang, J.-h.; Hsu, H.-Y.; Muir, B.W.; et al. A Polytherapy Based Approach to Combat Antimicrobial Resistance Using Cubosomes. Nat. Commun. 2022, 13, 343. [Google Scholar] [CrossRef]
- Wang, H.; Wang, M.; Xu, X.; Gao, P.; Xu, Z.; Zhang, Q.; Li, H.; Yan, A.; Kao, R.Y.-T.; Sun, H. Multi-target Mode of Action of Silver against Staphylococcus aureus Endows It with Capability to Combat Antibiotic Resistance. Nat. Commun. 2021, 12, 3331. [Google Scholar] [CrossRef] [PubMed]
- Dash, S.; Kumar, M.; Pareek, N. Enhanced Antibacterial Potential of Berberine via Synergism with Chitosan Nanoparticles. Materials Today Proc. 2020, 31, 640–645. [Google Scholar] [CrossRef]
- Belakhov, V.V. Polyfunctional Drugs: Search, Development, Use in Medical Practice, and Environmental Aspects of Preparation and Application (A Review). Russ. J. Gen. Chem. 2022, 92, 3030–3055. [Google Scholar] [CrossRef]
- Kuppusamy, R.; Browne, K.; Suresh, D.; Do Rosario, R.M.; Chakraborty, S.; Yang, S.; Willcox, M.; Black, D.; Chen, R.; Kumar, N. Transition Towards Antibiotic Hybrid Vehicles: The Next Generation Antibacterials. Curr. Med. Chem. 2022, 30, 104–125. [Google Scholar] [CrossRef]
- Batchelder, J.I.; Hare, P.J.; Mok, W.W.K. Resistance-resistant Antibacterial Treatment Strategies. Front. Antibiot. 2023, 2, 1093156. [Google Scholar] [CrossRef]
- Wetzel, C.; Lonneman, M.; Wu, C. Polypharmacological Drug Actions of Recently FDA Approved Antibiotics. Eur. J. Med. Chem. 2021, 209, 112931. [Google Scholar] [CrossRef]
- Ling, L.L.; Schneider, T.; Peoples, A.J.; Spoering, A.L.; Engels, I.; Conlon, B.P.; Mueller, A.; Schäberle, T.F.; Hughes, D.E.; Epstein, S.; et al. A New Antibiotic Kills Pathogens without Detectable Resistance. Nature 2015, 517, 455–459. [Google Scholar] [CrossRef]
- Shukla, R.; Lavore, F.; Maity, S.; Derks, M.G.N.; Jones, C.R.; Vermeulen, B.J.A.; Melcrová, A.; Morris, M.A.; Becker, L.M.; Wang, X.; et al. Teixobactin Kills Bacteria by a Two-pronged Attack on the Cell Envelope. Nature 2022, 608, 390–396. [Google Scholar] [CrossRef]
- Imai, Y.; Meyer, K.J.; Iinishi, A.; Favre-Godal, Q.; Green, R.; Manuse, S.; Caboni, M.; Mori, M.; Niles, S.; Ghiglieri, M.; et al. A New Antibiotic Selectively Kills Gram-negative Pathogens. Nature 2019, 576, 459–464. [Google Scholar] [CrossRef]
- Seyfert, C.E.; Porten, C.; Yuan, B.; Deckarm, S.; Panter, F.; Bader, C.D.; Coetzee, J.; Deschner, F.; Tehrani, K.; Higgins, P.G.; et al. Darobactins Exhibiting Superior Antibiotic Activity by Cryo-EM Structure Guided Biosynthetic Engineering. Angew. Chem. Int. Ed. Engl. 2023, 62, e202214094. [Google Scholar] [CrossRef]
- Nakaya, T.; Yabe, M.; Mashalidis, E.H.; Sato, T.; Yamamoto, K.; Hikiji, Y.; Katsuyama, A.; Shinohara, M.; Minato, Y.; Takahashi, S.; et al. Synthesis of Macrocyclic Nucleoside Antibacterials and Their Interactions with MraY. Nat. Commun. 2022, 13, 7575. [Google Scholar] [CrossRef] [PubMed]
- Hegemann, J.D.; Birkelbach, J.; Walesch, S.; Müller, R. Current Developments in Antibiotic Discovery: Global Microbial Diversity as a Source for Evolutionary Optimized Anti-bacterials: Global Microbial Diversity as a Source for Evolutionary Optimized Anti-bacterials. EMBO Rep. 2023, 24, e56184. [Google Scholar] [CrossRef] [PubMed]
- Walesch, S.; Birkelbach, J.; Jézéquel, G.; Haeckl, F.P.J.; Hegemann, J.D.; Hesterkamp, T.; Hirsch, A.K.H.; Hammann, P.; Müller, R. Fighting Antibiotic Resistance-strategies and (Pre)clinical Developments to Find New Antibacterials. EMBO Rep. 2023, 24, e56033. [Google Scholar] [CrossRef] [PubMed]
- Surur, A.S.; Sun, D. Macrocycle-Antibiotic Hybrids: A Path to Clinical Candidates. Front. Chem. 2021, 9, 659845. [Google Scholar] [CrossRef]
- Pavlova, J.A.; Tereshchenkov, A.G.; Nazarov, P.A.; Lukianov, D.A.; Skvortsov, D.A.; Polshakov, V.I.; Vasilieva, B.F.; Efremenkova, O.V.; Kaiumov, M.Y.; Paleskava, A.; et al. Conjugates of Chloramphenicol Amine and Berberine as Antimicrobial Agents. Antibiotics 2022, 12, 15. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.; Xu, Y.; Zhang, J.; Sui, Z.; Corke, H. Antibacterial Activity and Multi-Targeting Mechanism of Dehydrocorydaline from Corydalis turtschaninovii Bess. Against Listeria monocytogenes. Front. Microbiol. 2021, 12, 799094. [Google Scholar] [CrossRef]
- Griffith, R.; Bremner, J.B. Computational Evaluation of N-Based Transannular Interactions in Some Model Fused Medium-Sized Heterocyclic Systems and Implications for Drug Design. Molecules 2023, 28, 1631. [Google Scholar] [CrossRef] [PubMed]
- Durcik, M.; Skok, Ž.; Ilaš, J.; Zidar, N.; Zega, A.; Szili, P.; Draskovits, G.; Révész, T.; Kikelj, D.; Nyerges, A.; et al. Hybrid Inhibitors of DNA Gyrase A and B: Design, Synthesis and Evaluation. Pharmaceutics 2020, 13, 6. [Google Scholar] [CrossRef]
- Durcik, M.; Cotman, A.E.; Toplak, Ž.; Možina, Š.; Skok, Ž.; Szili, P.E.; Czikkely, M.; Maharramov, E.; Vu, T.H.; Piras, M.V.; et al. New Dual Inhibitors of Bacterial Topoisomerases with Broad-Spectrum Antibacterial Activity and In Vivo Efficacy against Vancomycin-Intermediate Staphylococcus aureus. J. Med. Chem. 2023, 66, 3968–3994. [Google Scholar] [CrossRef]
- Nyerges, A.; Tomašič, T.; Durcik, M.; Revesz, T.; Szili, P.; Draskovits, G.; Bogar, F.; Skok, Ž.; Zidar, N.; Ilaš, J.; et al. Rational Design of Balanced Dual-targeting Antibiotics with Limited Resistance. PLoS Biol. 2020, 18, e3000819. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, N.; Fahim, S.; Hassan, M.; Farag, A.; Georgey, H. Design and Synthesis of Ciprofloxacin-Sulfonamide Hybrids to Manipulate Ciprofloxacin pharmacological Qualities: Potency and Side Effects. Eur. J. Med. Chem. 2021, 228, 114021. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Battini, N.; Ou, J.M.; Zhang, S.L.; Zhang, L.; Zhou, C.H. New Efforts toward Aminothiazolylquinolones with Multitargeting Antibacterial Potential. J. Agric. Food Chem. 2023, 71, 2322–2332. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.-M.; Hu, Y.-Y.; Fang, B.; Zhou, C.-H. Benzenesulfonyl Thiazoloimines as Unique Multitargeting Antibacterial Agents towards Enterococcus faecalis. Eur. J. Med. Chem. 2023, 248, 115088. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.C.; Zeng, C.M.; Avula, S.R.; Peng, X.M.; Geng, R.X.; Zhou, C.H. Novel Coumarin Aminophosphonates as Potential Multitargeting Antibacterial Agents against Staphylococcus aureus. Eur. J. Med. Chem. 2023, 245, 114891. [Google Scholar] [CrossRef]
- Suckling, C.J.; Hunter, I.S.; Scott, F.J. Multitargeted Anti-infective Drugs: Resilience to Resistance in the Antimicrobial Resistance Era. Future Drug Discov. 2022, 4, Fdd73. [Google Scholar] [CrossRef]
- Juárez-López, D.; Morales-Ruiz, E.; Herrera-Zúñiga, L.D.; González-Carrera, Z.; Cuevas-Reyes, E.; Corzo, G.; Schcolnik-Cabrera, A.; Villegas, E. The Resilience of Pseudomonas aeruginosa to Antibiotics and the Designing of Antimicrobial Peptides to Overcome Microbial Resistance. Curr. Med. Chem. 2022, 30, 72–103. [Google Scholar] [CrossRef] [PubMed]
- Teng, P.; Shao, H.; Huang, B.; Xie, J.; Cui, S.; Wang, K.; Cai, J. Small Molecular Mimetics of Antimicrobial Peptides as a Promising Therapy to Combat Bacterial Resistance. J. Med. Chem. 2023, 66, 2211–2234. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Yu, Q.; Zheng, Z.; Liu, J.; Cai, Q.; Liu, S.; Lin, S. Design and Synthesis of Phenyl Sulfide-Based Cationic Amphiphiles as Membrane-Targeting Antimicrobial Agents against Gram-Positive Pathogens. J. Med. Chem. 2022, 65, 14221–14236. [Google Scholar] [CrossRef]
- Recce Pharmaceuticals. Available online: https://recce.com.au/science/mechanism-of-action (accessed on 21 March 2023).
- Frei, A.; Verderosa, A.D.; Elliott, A.G.; Zuegg, J.; Blaskovich, M.A.T. Metals to Combat Antimicrobial Resistance. Nat. Rev. Chem. 2023, 7, 202–224. [Google Scholar] [CrossRef]
- Jiang, L.; Ma, Y.; Chen, Y.; Cai, M.; Wu, Z.; Xiong, Y.; Duan, X.; Liao, X.; Wang, J. Multi-target Antibacterial Mechanism of Ruthenium Polypyridine Complexes with Anthraquinone Groups against Staphylococcus aureus. RSC Med. Chem. 2023, 14, 700–709. [Google Scholar] [CrossRef]
- Jubeh, B.; Breijyeh, Z.; Karaman, R. Antibacterial Prodrugs to Overcome Bacterial Resistance. Molecules 2020, 25, 1543. [Google Scholar] [CrossRef] [PubMed]
- Evans, L.E.; Krishna, A.; Ma, Y.; Webb, T.E.; Marshall, D.C.; Tooke, C.L.; Spencer, J.; Clarke, T.B.; Armstrong, A.; Edwards, A.M. Exploitation of Antibiotic Resistance as a Novel Drug Target: Development of a β-Lactamase-Activated Antibacterial Prodrug. J. Med. Chem. 2019, 62, 4411–4425. [Google Scholar] [CrossRef] [PubMed]
- Rineh, A.; Soren, O.; McEwan, T.; Ravikumar, V.; Poh, W.H.; Azamifar, F.; Naimi-Jamal, M.R.; Cheung, C.Y.; Elliott, A.G.; Zuegg, J.; et al. Discovery of Cephalosporin-3′-diazeniumdiolates that Show Dual Antibacterial and Antibiofilm Effects against Pseudomonas aeruginosa Clinical Cystic Fibrosis Isolates and Efficacy in a Murine Respiratory Infection Model. ACS Infect.Dis. 2020, 6, 1460–1479. [Google Scholar] [CrossRef] [PubMed]
- Meiers, J.; Rox, K.; Titz, A. Lectin-Targeted Prodrugs Activated by Pseudomonas aeruginosa for Self-Destructive Antibiotic Release. J. Med. Chem. 2022, 65, 13988–14014. [Google Scholar] [CrossRef]
- Miller, J.J.; Shah, I.T.; Hatten, J.; Barekatain, Y.; Mueller, E.A.; Moustafa, A.M.; Edwards, R.L.; Dowd, C.S.; Hoops, G.C.; Johnson, R.J.; et al. Structure-guided Microbial Targeting of Antistaphylococcal Prodrugs. Elife 2021, 10, e66657. [Google Scholar] [CrossRef]
- Bryan, E.; Ferrer-González, E.; Sagong, H.Y.; Fujita, J.; Mark, L.; Kaul, M.; LaVoie, E.J.; Matsumura, H.; Pilch, D.S. Structural and Antibacterial Characterization of a New Benzamide FtsZ Inhibitor with Superior Bactericidal Activity and in vivo Efficacy against Multidrug-Resistant Staphylococcus aureus. ACS Chem. Biol. 2023, 18, 629–642. [Google Scholar] [CrossRef] [PubMed]
- Weng, Y.; Chen, H.; Chen, X.; Yang, H.; Chen, C.-H.; Tan, H. Adenosine Triphosphate-activated Prodrug System for On-demand Bacterial Inactivation and Wound Disinfection. Nat. Commun. 2022, 13, 4712. [Google Scholar] [CrossRef] [PubMed]
- Li, C.H.; Huang, R.; Makabenta, J.M.; Schmidt-Malan, S.; Patel, R.; Rotello, V.M. In situ Generation of Antibiotics using Bioorthogonal “Nanofactories”. Microbiol. Insights 2021, 14, 1178636121997121. [Google Scholar] [CrossRef]
- Southwell, J.W.; Herman, R.; Raines, D.J.; Clarke, J.E.; Böswald, I.; Dreher, T.; Gutenthaler, S.M.; Schubert, N.; Seefeldt, J.; Metzler-Nolte, N.; et al. Siderophore-Linked Ruthenium Catalysts for Targeted Allyl Ester Prodrug Activation within Bacterial Cells. Chemistry 2023, 29, e202202536. [Google Scholar] [CrossRef]
- Sobotta, L.; Skupin-Mrugalska, P.; Piskorz, J.; Mielcarek, J. Non-porphyrinoid Photosensitizers Mediated Photodynamic Inactivation against Bacteria. Dye. Pigm. 2019, 163, 337–355. [Google Scholar] [CrossRef]
- Rineh, A.; Bremner, J.B.; Hamblin, M.R.; Ball, A.R.; Tegos, G.P.; Kelso, M.J. Attaching NorA Efflux Pump Inhibitors to Methylene Blue Enhances Antimicrobial Photodynamic Inactivation of Escherichia coli and Acinetobacter baumannii in vitro and in vivo. Bioorg. Med. Chem. Lett. 2018, 28, 2736–2740. [Google Scholar] [CrossRef] [PubMed]
- Rineh, A.; Dolla, N.K.; Ball, A.R.; Magana, M.; Bremner, J.B.; Hamblin, M.R.; Tegos, G.P.; Kelso, M.J. Attaching the NorA Efflux Pump Inhibitor INF55 to Methylene Blue Enhances Antimicrobial Photodynamic Inactivation of Methicillin-Resistant Staphylococcus aureus in vitro and in vivo. ACS Infect. Dis. 2017, 3, 756–766. [Google Scholar] [CrossRef] [PubMed]
- Black, C.M.; Chu, A.J.; Thomas, G.H.; Routledge, A.; Duhme-Klair, A.K. Synthesis and Antimicrobial Activity of an SO2-releasing Siderophore Conjugate. J. Inorg. Biochem. 2022, 234, 111875. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.Y.; Zhu, X.Y.; Wu, F.G. Antibacterial Gas Therapy: Strategies, Advances, and Prospects. Bioact. Mater. 2023, 23, 129–155. [Google Scholar] [CrossRef]
- Venkatesh, Y.; Kiran, K.S.; Shah, S.S.; Chaudhuri, A.; Dey, S.; Singh, N.P. One-and Two-photon Responsive Sulfur Dioxide (SO2) Donors: A Combinatorial Drug Delivery for Improved Antibiotic Therapy. Org. Biomol. Chem. 2019, 17, 2640–2645. [Google Scholar] [CrossRef]
- Hou, D.; Wang, R.; Wang, Z.; Yang, G.; Xu, Z.; Zeng, Q.; Chen, Y. A light-activatable Antibiotic with High Activation Efficiency and Uncompromised Bactericidal Potency in the Activated State. J. Leather Sci. Eng. 2021, 3, 8. [Google Scholar] [CrossRef]
- Venkatesan, J.; Murugan, D.; Rangasamy, L. A Perspective on Newly Emerging Proteolysis-Targeting Strategies in Antimicrobial Drug Discovery. Antibiotics 2022, 11, 1717. [Google Scholar] [CrossRef]
- Burslem, G.M. BacPROTACs to Basics: Targeted Protein Degradation in Bacteria. Cell 2022, 185, 2203. [Google Scholar] [CrossRef]
- Morreale, F.E.; Kleine, S.; Leodolter, J.; Junker, S.; Hoi, D.M.; Ovchinnikov, S.; Okun, A.; Kley, J.; Kurzbauer, R.; Junk, L.; et al. BacPROTACs Mediate Targeted Protein Degradation in Bacteria. Cell 2022, 185, 2338–2353.e18. [Google Scholar] [CrossRef]
- Santos, A.L.; Liu, D.; Reed, A.K.; Wyderka, A.M.; van Venrooy, A.; Li, J.T.; Li, V.D.; Misiura, M.; Samoylova, O.; Beckham, J.L.; et al. Light-activated Molecular Machines are Fast-acting Broad-spectrum Antibacterials that Target the Membrane. Sci. Adv. 2022, 8, eabm2055. [Google Scholar] [CrossRef]












Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bremner, J.B. An Update Review of Approaches to Multiple Action-Based Antibacterials. Antibiotics 2023, 12, 865. https://doi.org/10.3390/antibiotics12050865
Bremner JB. An Update Review of Approaches to Multiple Action-Based Antibacterials. Antibiotics. 2023; 12(5):865. https://doi.org/10.3390/antibiotics12050865
Chicago/Turabian StyleBremner, John B. 2023. "An Update Review of Approaches to Multiple Action-Based Antibacterials" Antibiotics 12, no. 5: 865. https://doi.org/10.3390/antibiotics12050865
APA StyleBremner, J. B. (2023). An Update Review of Approaches to Multiple Action-Based Antibacterials. Antibiotics, 12(5), 865. https://doi.org/10.3390/antibiotics12050865