Identification of Small Molecule Inhibitors of Staphylococcus aureus RnpA

 , , and
, , and
Abstract
:1. Introduction
2. Results
2.1. Mupirocin Synergy Screening to Identify Inhibitors of RnpA-Dependent ptRNA Processing
2.2. Effects of RnpA ptRNA Processing Inhibitors on Messenger RNA Degradation and Human Cytotoxicity
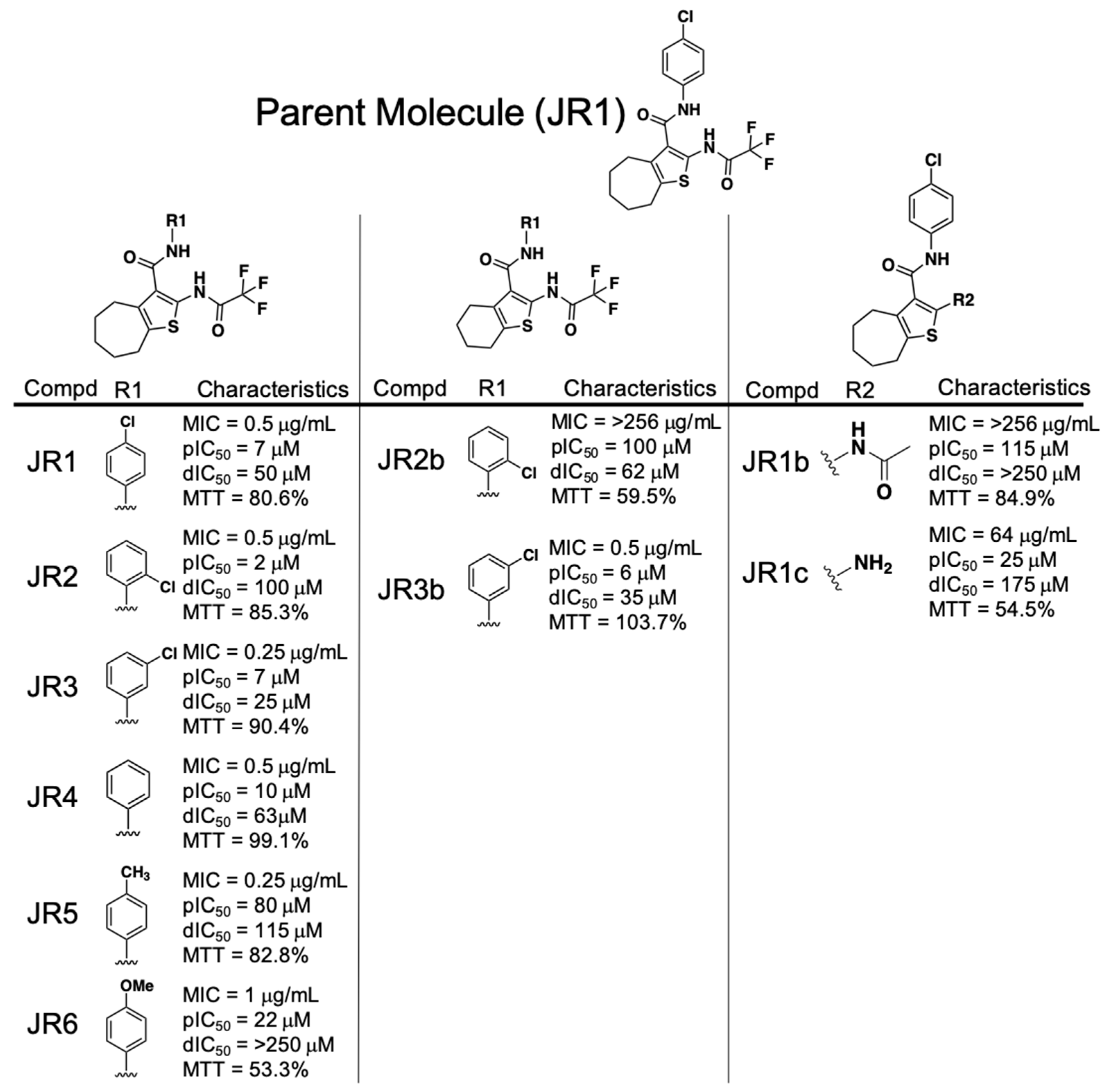
2.3. Performance of the Phenylcarbamoyl Cyclic Thiophene Class of RnpA Inhibitors
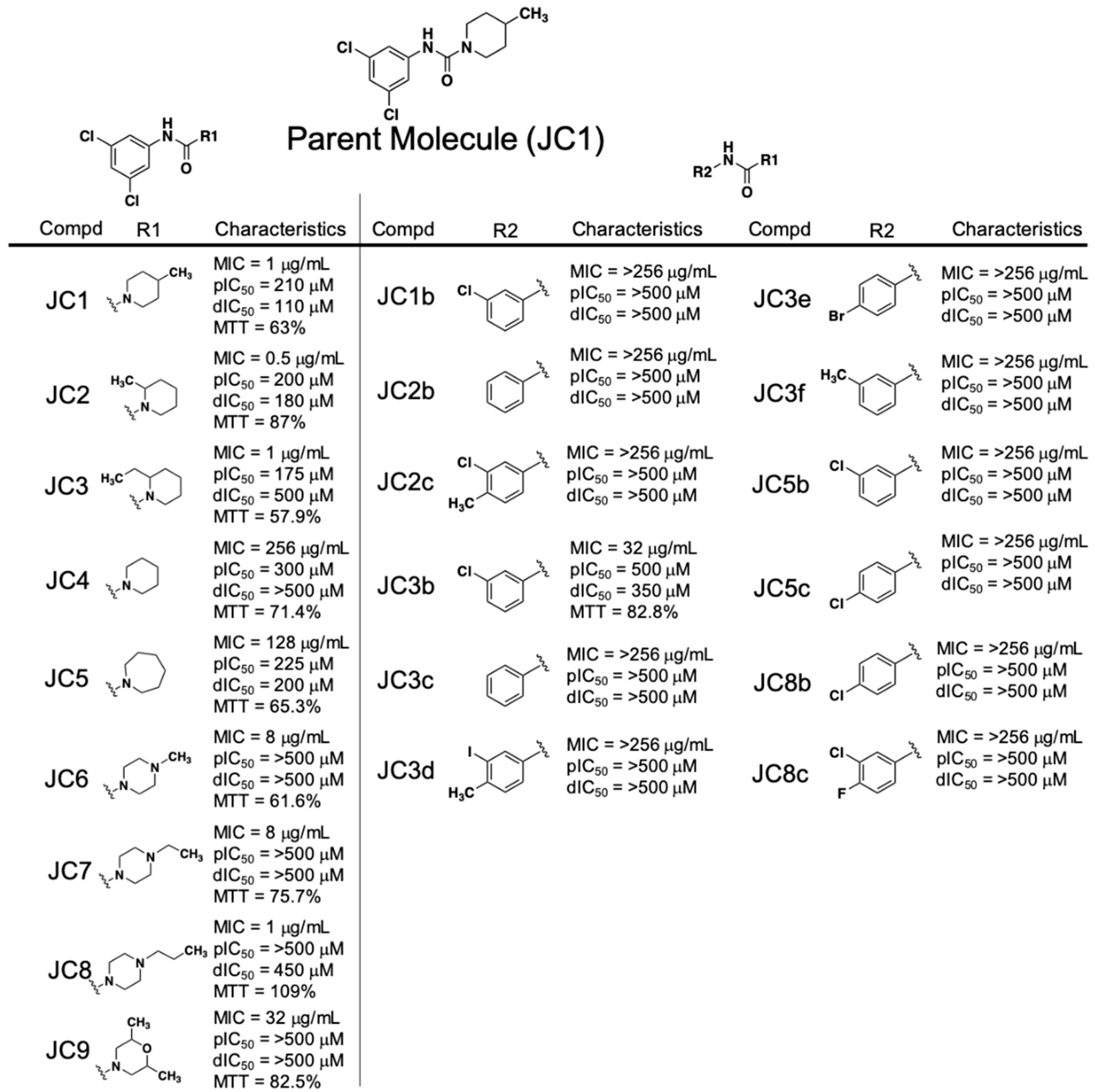
2.4. Performance of the Piperidinecarboxamide Class of RnpA Inhibitors
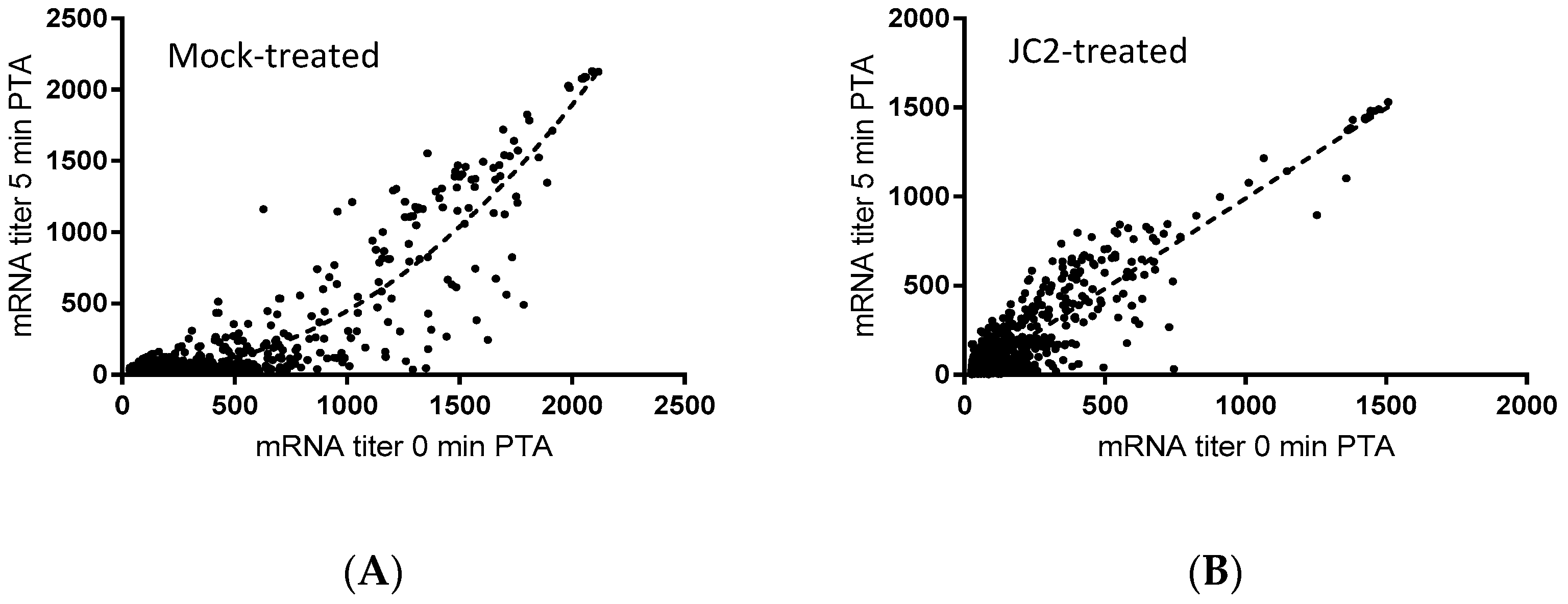
2.5. Cellular Activities of the Piperidinecarboxamide Class of RnpA Inhibitors
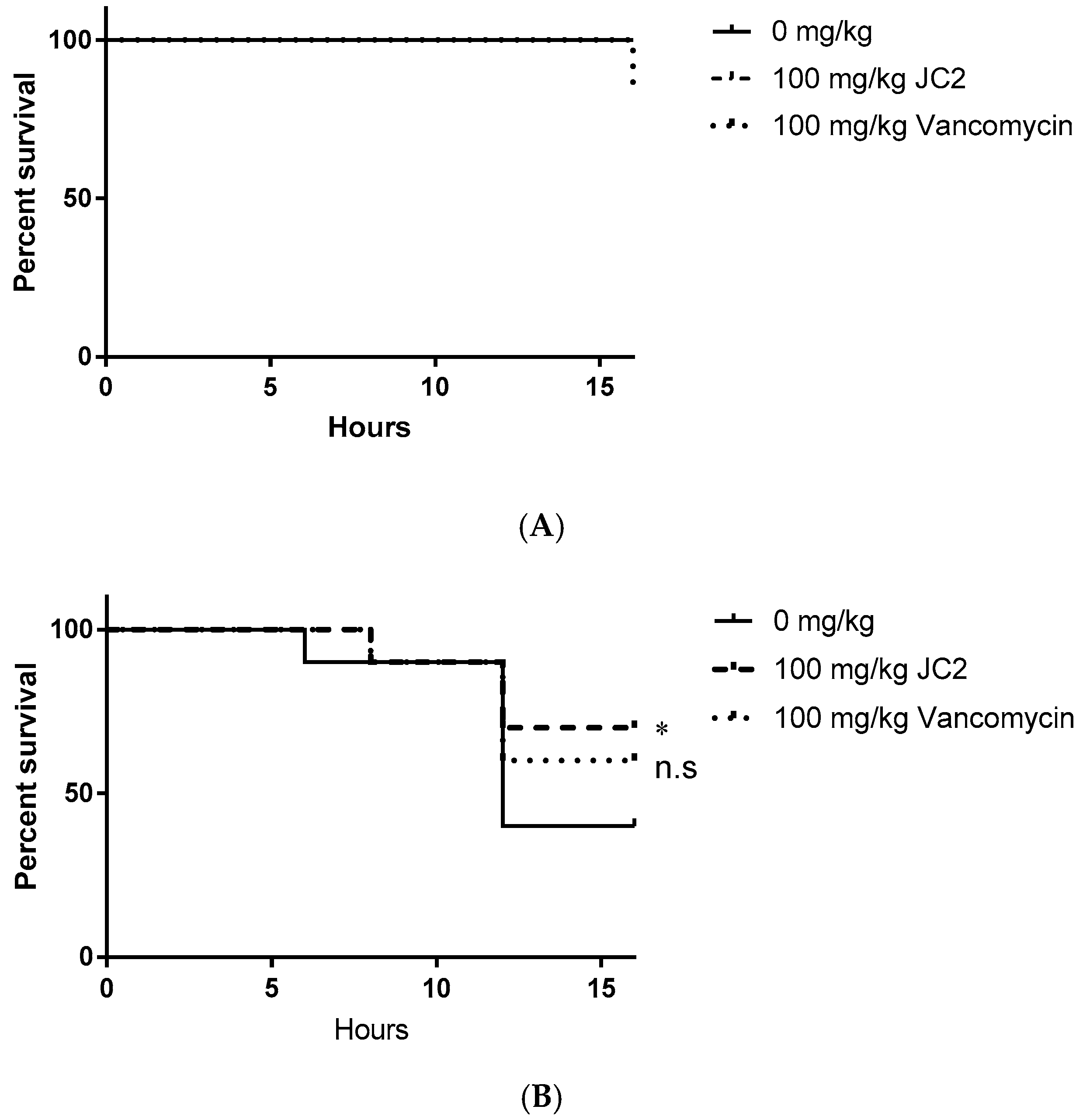
2.6. The RnpA Inhibitor, JC2, Displays Antimicrobial Efficacy in an in Vivo Model of Lethal S. aureus Infection
3. Discussion
3.1. Identification of Compounds that Potentiate Mupirocin
3.2. SAR Trends of Phenylcarbamoyl Cyclic Thiophene and Piperidinecarboxamide Class Scaffolds
3.3. Cellular Activities of Piperidinecarboxamide Class Inhibitors
3.4. Potential for Therapeutic Development of the Putative RnpA Inhibitors Identified
4. Materials and Methods
4.1. Bacterial and Human Cell Lines Growth Conditions and Chemicals
4.2. Antimicrobial Susceptibility Testing
4.3. Human Cell Cytotoxicity Testing
4.4. Cellular mRNA Turnover Assays
4.5. Cellular tRNATyr Population Measures
4.6. Bacterial RNA Isolation and Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
4.7. Affymetrix GeneChip® Analysis
4.8. In Vitro Transcription of RNA Species
4.9. RnpA Protein Purification
4.10. In Vitro mRNA Degradation Assays
4.11. In Vitro ptRNATyr Processing Assays
4.12. Cellular RnpA Depletion Hypersusceptibility to Compounds
4.13. Galleria Mellonella Model of Single Tolerated Dose and S. aureus Infection
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Schlievert, P.M.; Tripp, T.J.; Peterson, M.L. Reemergence of staphylococcal toxic shock syndrome in Minneapolis-St. Paul, Minnesota, during the 2000–2003 surveillance period. J. Clin. Microbiol. 2004, 42, 2875–2876. [Google Scholar] [CrossRef] [PubMed]
- Biedenbach, D.J.; Moet, G.J.; Jones, R.N. Occurrence and antimicrobial resistance pattern comparisons among bloodstream infection isolates from the SENTRY Antimicrobial Surveillance Program (1997–2002). Diagn. Microbiol. Infect. Dis. 2004, 50, 59–69. [Google Scholar] [CrossRef]
- Shurland, S.; Zhan, M.; Bradham, D.D.; Roghmann, M.C. Comparison of mortality risk associated with bacteremia due to methicillin-resistant and methicillin-susceptible Staphylococcus aureus. Infect. Control Hosp. Epidemiol. 2007, 28, 273–279. [Google Scholar] [CrossRef]
- Lowy, F.D. Staphylococcus aureus infections. N Engl. J. Med. 1998. 339, 520–532.
- WHO. Antimicrobial resistance: Global report on surveillance 2014. Available online: https://www.who.int/drugresistance/documents/surveillancereport/en/ (accessed on 2 January 2019).
- Holmes, N.E.; Johnson, P.D.; Howden, B.P. Relationship between vancomycin-resistant Staphylococcus aureus, vancomycin-intermediate S. aureus, high vancomycin MIC, and outcome in serious S. aureus infections. J. Clin. Microbiol. 2012, 50, 2548–2552. [Google Scholar] [CrossRef]
- Foster, T.J. Antibiotic resistance in Staphylococcus aureus. Current status and future prospects. FEMS Microbiol. Rev. 2017, 41, 430–449. [Google Scholar] [PubMed]
- Boucher, H.W.; Talbot, G.H.; Bradley, J.S.; Edwards, J.E.; Gilbert, D.; Rice, L.B.; Scheld, M.; Spellberg, B.; Bartlett, J. Bad bugs, no drugs: No ESKAPE! An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 2009, 48, 1–12. [Google Scholar] [CrossRef]
- Condon, C.; Putzer, H. The phylogenetic distribution of bacterial ribonucleases. Nucleic Acids Res. 2002, 30, 5339–5346. [Google Scholar] [CrossRef] [Green Version]
- Eidem, T.M.; Roux, C.M.; Dunman, P.M. RNA decay: A novel therapeutic target in bacteria. Wiley Interdiscip. Rev. RNA 2012, 3, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, R.R.; Allen, A.G.; Owen, P.J.; Shalom, G.; Stone, K.; Harrison, M.; Burgis, T.A.; Lockyer, M.; Garcia-Lara, J.; Foster, S.J.; et al. Comprehensive identification of essential Staphylococcus aureus genes using Transposon-Mediated Differential Hybridisation (TMDH). BMC Genomics 2009, 10, 291. [Google Scholar] [CrossRef] [PubMed]
- Reich, C.; Olsen, G.J.; Pace, B.; Pace, N.R. Role of the protein moiety of ribonuclease P, a ribonucleoprotein enzyme. Science 1988, 239, 178–181. [Google Scholar] [CrossRef] [PubMed]
- Kurz, J.C.; Fierke, C.A. The affinity of magnesium binding sites in the Bacillus subtilis RNase P x pre-tRNA complex is enhanced by the protein subunit. Biochemistry 2002, 41, 9545–9558. [Google Scholar] [CrossRef]
- Olson, P.D.; Kuechenmeister, L.J.; Anderson, K.L.; Daily, S.; Beenken, K.E.; Roux, C.M.; Reniere, M.L.; Lewis, T.L.; Weiss, W.J.; Pulse, M.; et al. Small molecule inhibitors of Staphylococcus aureus RnpA alter cellular mRNA turnover, exhibit antimicrobial activity, and attenuate pathogenesis. PLoS Pathog. 2011, 7, e1001287. [Google Scholar] [CrossRef]
- Eidem, T.M.; Lounsbury, N.; Emery, J.F.; Bulger, J.; Smith, A.; Abou-Gharbia, M.; Childers, W.; Dunman, P.M. Small-molecule inhibitors of Staphylococcus aureus RnpA-mediated RNA turnover and tRNA processing. Antimicrob. Agents Chemother. 2015, 59, 2016–2028. [Google Scholar] [CrossRef]
- Roux, C.M.; DeMuth, J.P.; Dunman, P.M. Characterization of components of the Staphylococcus aureus mRNA degradosome holoenzyme-like complex. J. Bacteriol. 2011, 193, 5520–5526. [Google Scholar] [CrossRef]
- Blanchard, C.; Brooks, L.; Beckley, A.; Colquhoun, J.; Dewhurst, S.; Dunman, P.M. Neomycin Sulfate Improves the Antimicrobial Activity of Mupirocin-Based Antibacterial Ointments. Antimicrob. Agents Chemother. 2016, 60, 862–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohni, E. Chemotherapeutic activity of the combination of trimethoprim and sulphamethoxazole in infections of mice. Postgrad. Med. J. 1969, 45, S18–S21. [Google Scholar]
- Lounsbury, N.; Eidem, T.; Colquhoun, J.; Mateo, G.; Abou-Gharbia, M.; Dunman, P.M.; Childers, W.E. Novel inhibitors of Staphylococcus aureus RnpA that synergize with mupirocin. Bioorg. Med. Chem. Lett. 2018, 28, 1127–1131. [Google Scholar] [CrossRef]
- Eubank, T.D.; Biswas, R.; Jovanovic, M.; Litovchick, A.; Lapidot, A.; Gopalan, V. Inhibition of bacterial RNase P by aminoglycoside-arginine conjugates. FEBS Lett. 2002, 511, 107–112. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Chen, Y.; Fierke, C.A. A real-time fluorescence polarization activity assay to screen for inhibitors of bacterial ribonuclease P. Nucleic Acids Res. 2014, 42, e159. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, N.E.; Brannvall, M.; Virtanen, A.; Kirsebom, L.A. Inhibition of RNase P RNA cleavage by aminoglycosides. Proc. Natl. Acad. Sci. USA 1999, 96, 6155–6160. [Google Scholar] [CrossRef] [Green Version]
- Satoh, H.; Gillette, J.R.; Davies, H.W.; Schulick, R.D.; Pohl, L.R. Immunochemical evidence of trifluoroacetylated cytochrome P-450 in the liver of halothane-treated rats. Mol. Pharmacol. 1985, 28, 468–474. [Google Scholar] [PubMed]
- Njoku, D.; Laster, M.J.; Gong, D.H.; Eger, E.I., 2nd; Reed, G.F.; Martin, J.L. Biotransformation of halothane, enflurane, isoflurane, and desflurane to trifluoroacetylated liver proteins: Association between protein acylation and hepatic injury. Anesth. Analg. 1997, 84, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Payne, D.J.; Gwynn, M.N.; Holmes, D.J.; Pompliano, D.L. Drugs for bad bugs: Confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov. 2007, 6, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Vestergaard, M.; Leng, B.; Haaber, J.; Bojer, M.S.; Vegge, C.S.; Ingmer, H. Genome-Wide Identification of Antimicrobial Intrinsic Resistance Determinants in Staphylococcus aureus. Front. Microbiol. 2016, 7, 2018. [Google Scholar] [CrossRef]
- Coughlin, D.J.; Pleiss, J.A.; Walker, S.C.; Whitworth, G.B.; Engelke, D.R. Genome-wide search for yeast RNase P substrates reveals role in maturation of intron-encoded box C/D small nucleolar RNAs. Proc. Natl. Acad. Sci. USA 2008, 105, 12218–12223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, D.; Marquez, S.M.; Pace, N.R. RNase P: Interface of the RNA and protein worlds. Trends Biochem. Sci. 2006, 31, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Guerrier-Takada, C.; Altman, S. A physical assay for and kinetic analysis of the interactions between M1 RNA and tRNA precursor substrates. Biochemistry 1993, 32, 7152–7161. [Google Scholar] [CrossRef] [PubMed]
- Peck-Miller, K.A.; Altman, S. Kinetics of the processing of the precursor to 4.5 S RNA, a naturally occurring substrate for RNase P from Escherichia coli. J. Mol. Biol. 1991, 221, 1–5. [Google Scholar] [CrossRef]
- CLSI. Performance Standards for Antimicrobial Susceptibility Testing; Twenty-Third Informational Supplement. Available online: https://clsi.org/standards/products/microbiology/documents/m100/ (accessed on 26 October 2018).
- Odds, F.C. Synergy, antagonism, and what the chequerboard puts between them. J. Antimicrob. Chemother. 2003, 52, 1. [Google Scholar] [CrossRef]
- Anderson, K.L.; Roberts, C.; Disz, T.; Vonstein, V.; Hwang, K.; Overbeek, R.; Olson, P.D.; Projan, S.J.; Dunman, P.M. Characterization of the Staphylococcus aureus heat shock, cold shock, stringent, and SOS responses and their effects on log-phase mRNA turnover. J. Bacteriol. 2006, 188, 6739–6756. [Google Scholar] [CrossRef]
- Bischoff, M.; Dunman, P.; Kormanec, J.; Macapagal, D.; Murphy, E.; Mounts, W.; Berger-Bachi, B.; Projan, S. Microarray-based analysis of the Staphylococcus aureus sigmaB regulon. J. Bacteriol. 2004, 186, 4085–4099. [Google Scholar] [CrossRef]
- Dunman, P.M.; Murphy, E.; Haney, S.; Palacios, D.; Tucker-Kellogg, G.; Wu, S.; Brown, E.L.; Zagursky, R.J.; Shlaes, D.; Projan, S.J. Transcription profiling-based identification of Staphylococcus aureus genes regulated by the agr and/or sarA loci. J. Bacteriol. 2001, 183, 7341–7353. [Google Scholar] [CrossRef] [PubMed]
- Pornillos, O.; Chen, Y.J.; Chen, A.P.; Chang, G. X-ray structure of the EmrE multidrug transporter in complex with a substrate. Science 2005, 310, 1950–1953. [Google Scholar] [CrossRef] [PubMed]
- Niranjanakumari, S.; Kurz, J.C.; Fierke, C.A. Expression, purification and characterization of the recombinant ribonuclease P protein component from Bacillus subtilis. Nucleic Acids Res. 1998, 26, 3090–3096. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound ID | Processing IC50 (μM) | Degradation IC50 (μM) | MIC (μg·mL−1) | % HepG2 Cell Survival 1 |
|---|---|---|---|---|
| ST029261 | 8 | 25 | 0.5 | 21.2 |
| ST016037 | 12 | 62.5 | 0.5 | 80.6 |
| ST028024 | 45 | >500 | 0.5 | 34.3 |
| ST029248 | 45 | 250 | 0.25 | 29.7 |
| ST034398 | 125 | >500 | 0.5 | 27.6 |
| ST027948 | 125 | >500 | 2 | 27.1 |
| ST011920 | 125 | >500 | 3.125 | 38.1 |
| 9044688 | 175 | 500 | 1 | 57.9 |
| ST030677 | 200 | >500 | 1 | 36.8 |
| 9049625 | 200 | 180 | 1 | 87 |
| 9044791 | 210 | 110 | 1 | 63 |
| ST009509 | 250 | >500 | 0.5 | 42.1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Colquhoun, J.M.; Ha, L.; Beckley, A.; Meyers, B.; Flaherty, D.P.; Dunman, P.M. Identification of Small Molecule Inhibitors of Staphylococcus aureus RnpA. Antibiotics 2019, 8, 48. https://doi.org/10.3390/antibiotics8020048
Colquhoun JM, Ha L, Beckley A, Meyers B, Flaherty DP, Dunman PM. Identification of Small Molecule Inhibitors of Staphylococcus aureus RnpA. Antibiotics. 2019; 8(2):48. https://doi.org/10.3390/antibiotics8020048
Chicago/Turabian StyleColquhoun, Jennifer M., Lisha Ha, Andrew Beckley, Brinkley Meyers, Daniel P. Flaherty, and Paul M. Dunman. 2019. "Identification of Small Molecule Inhibitors of Staphylococcus aureus RnpA" Antibiotics 8, no. 2: 48. https://doi.org/10.3390/antibiotics8020048
APA StyleColquhoun, J. M., Ha, L., Beckley, A., Meyers, B., Flaherty, D. P., & Dunman, P. M. (2019). Identification of Small Molecule Inhibitors of Staphylococcus aureus RnpA. Antibiotics, 8(2), 48. https://doi.org/10.3390/antibiotics8020048