

Passivation and pH-Induced Precipitation during Anodic Polarization of Steel in Aluminate Electrolytes as a Precondition for Plasma Electrolytic Oxidation

 and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
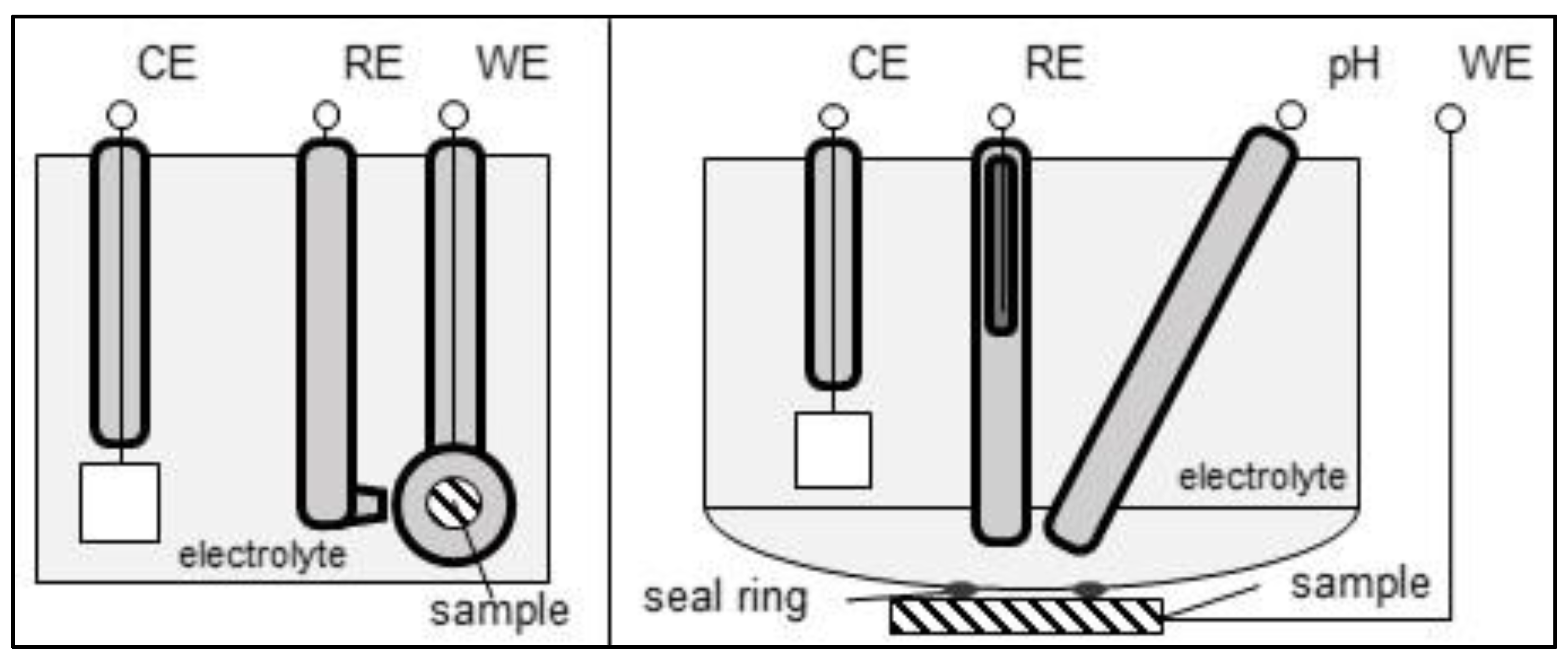
2.2. Electrochemical Polarization
2.3. Microstructural Analysis
3. Results
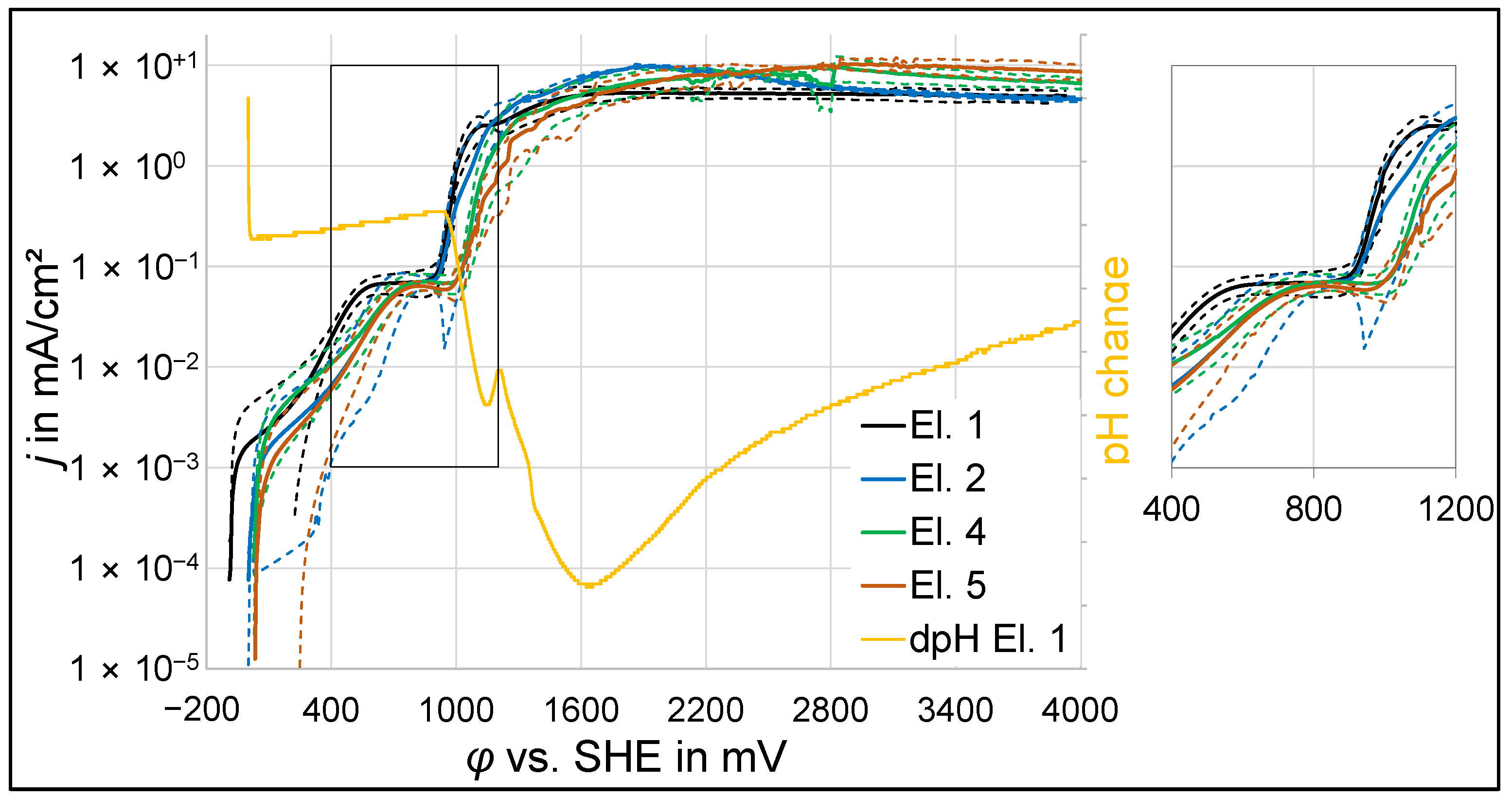
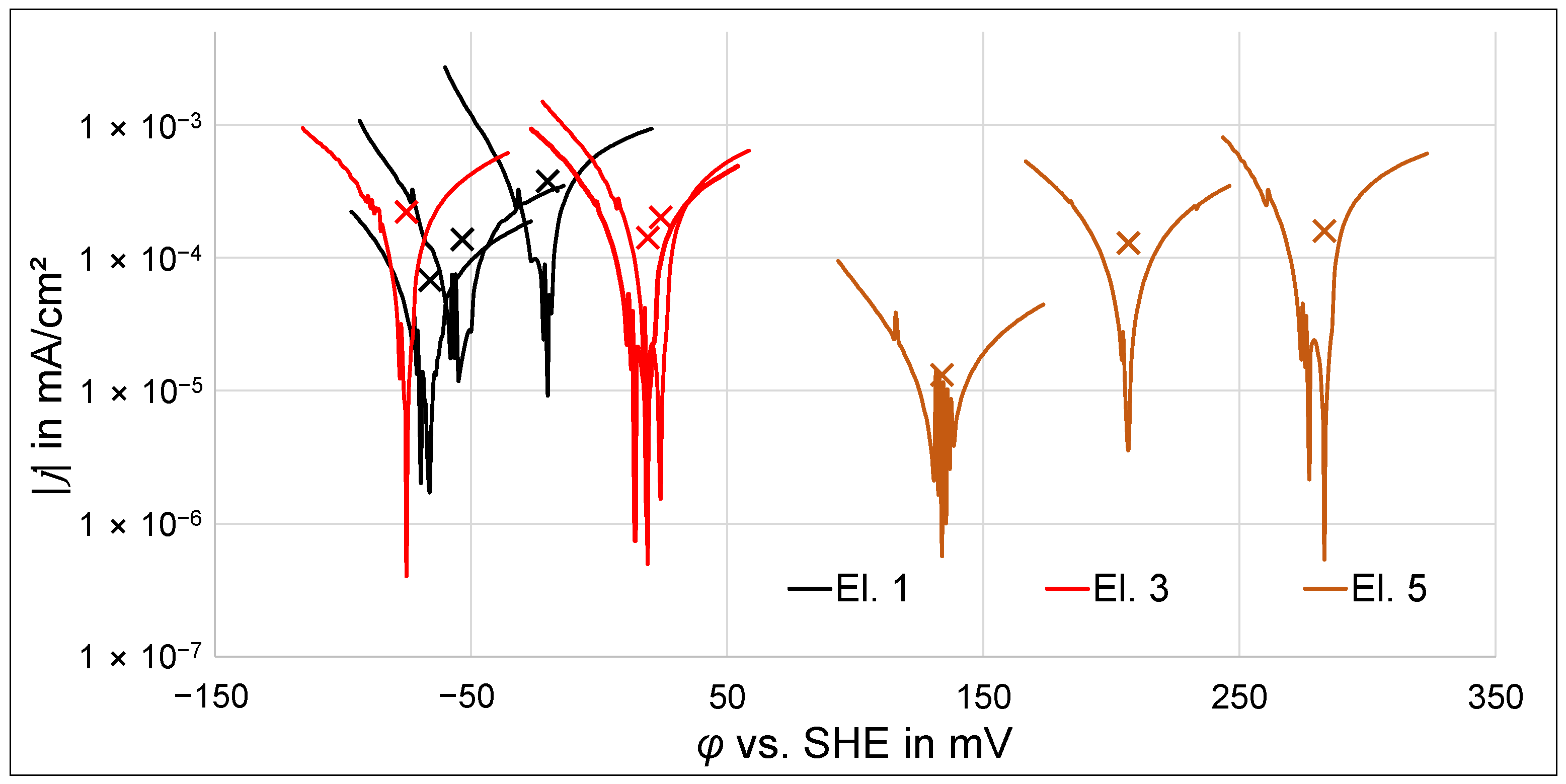
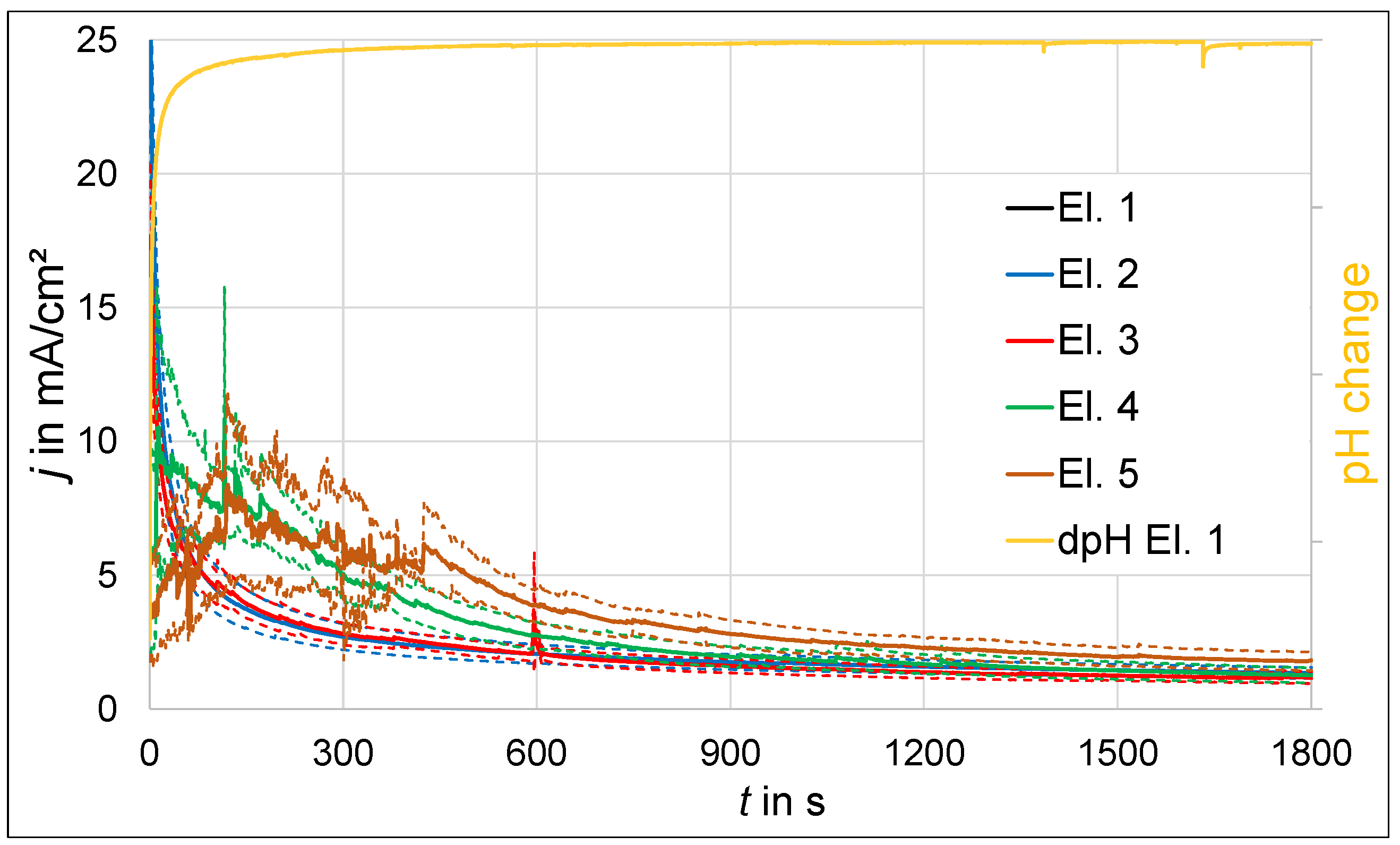
3.1. Polarization Experiments
3.1.1. Potentiodynamic Polarization
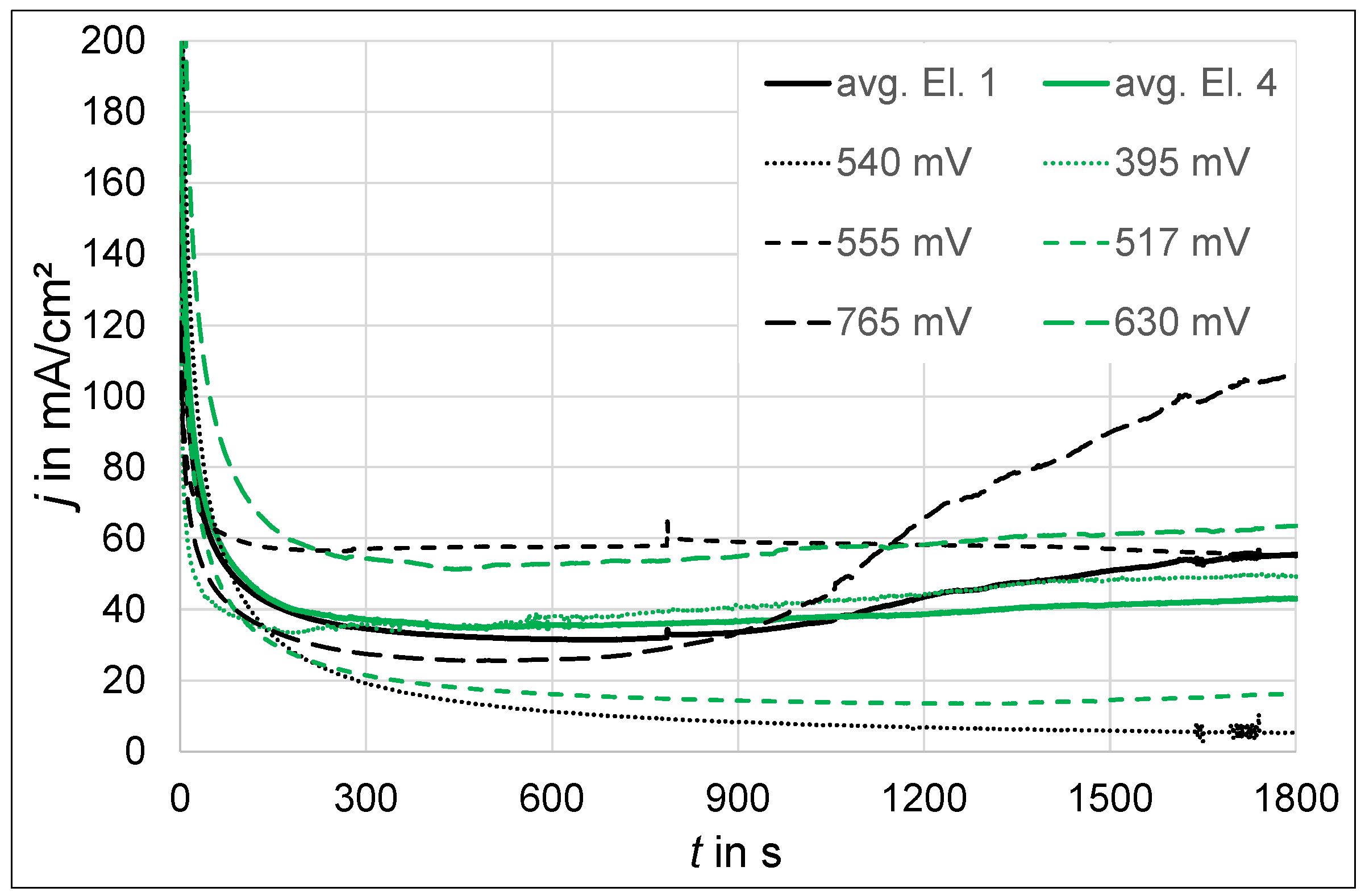
3.1.2. Polarization at Constant Potential
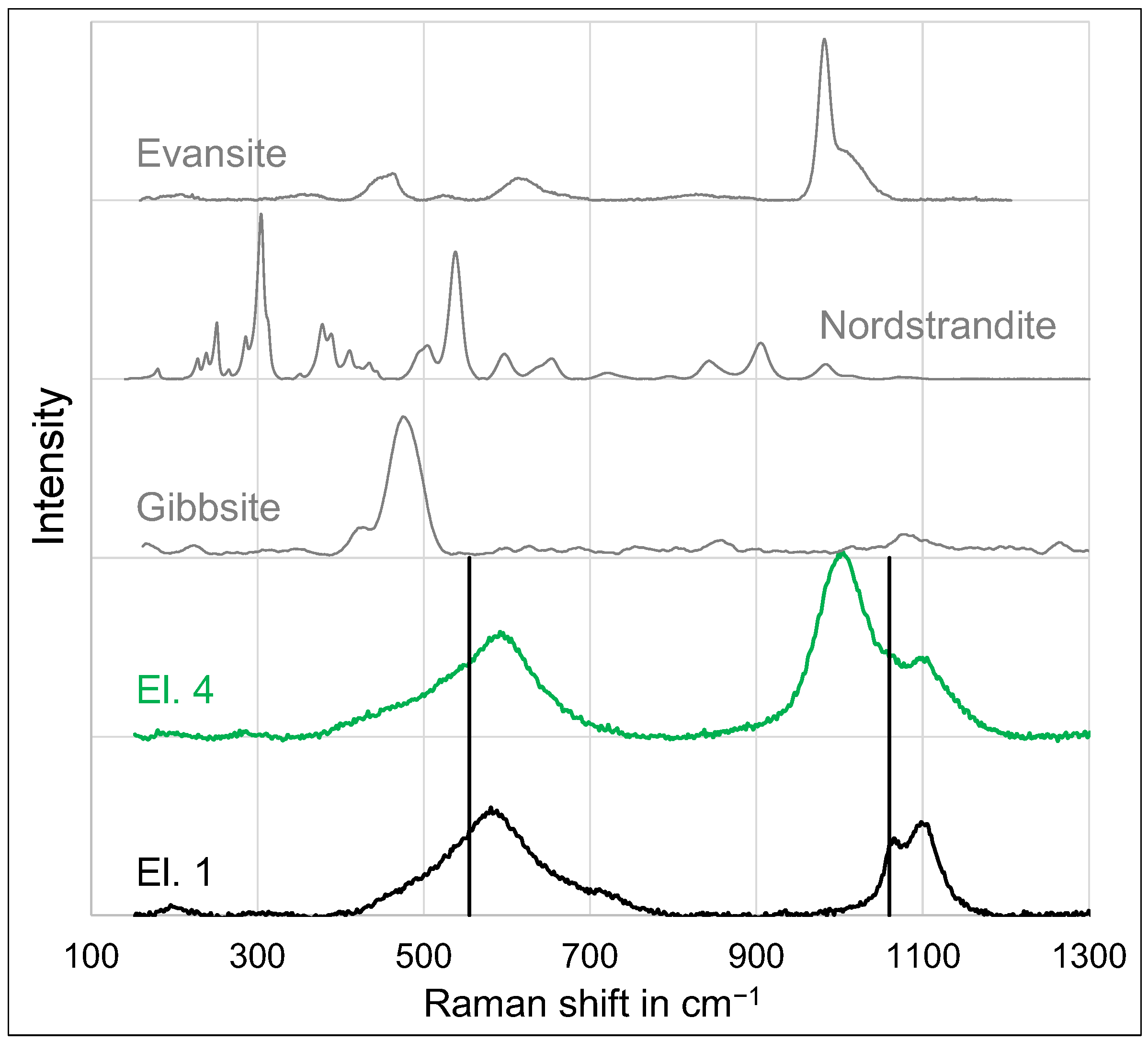
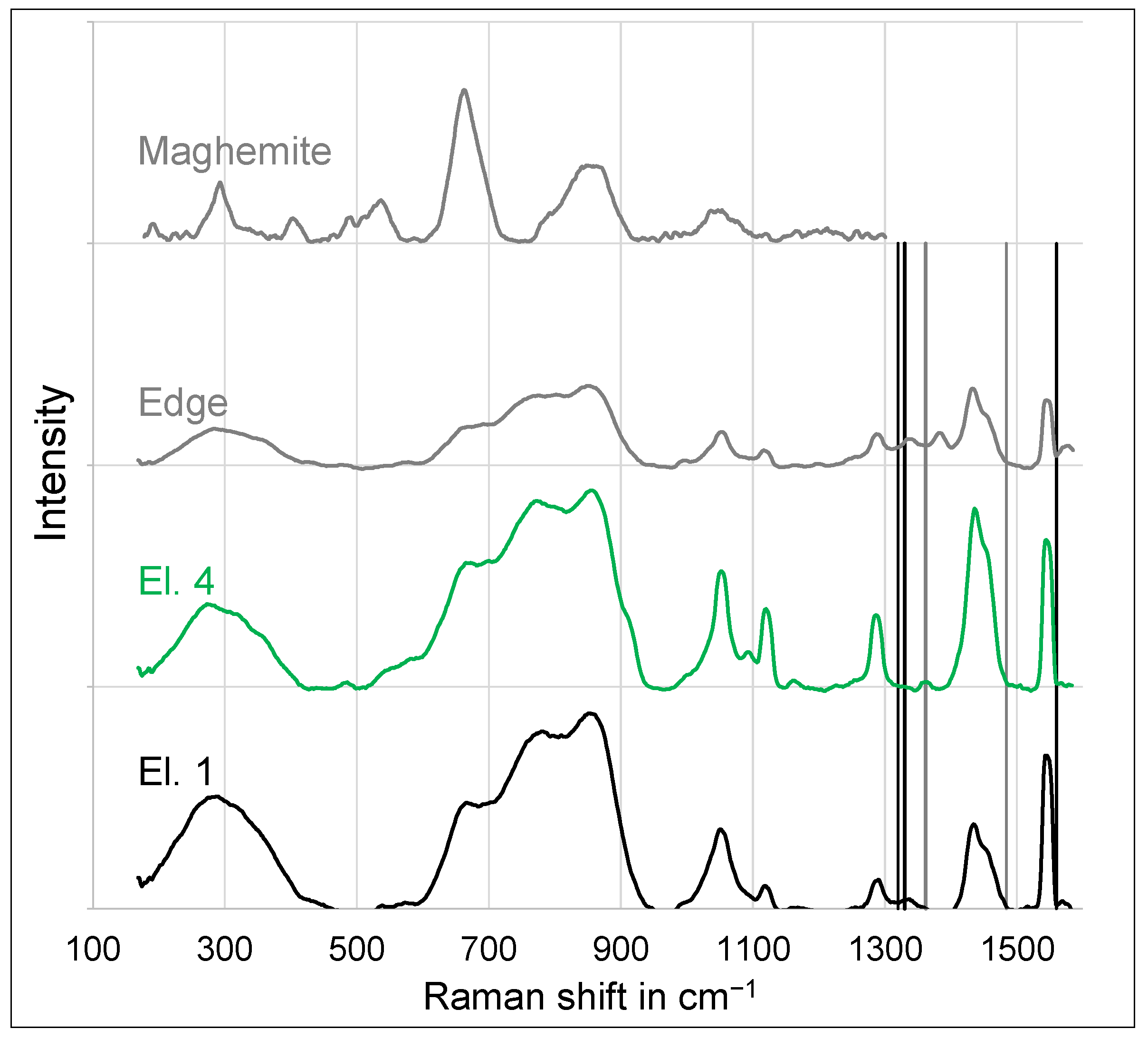
3.2. Microstructure
4. Discussion
5. Conclusions
- At an anodic potential of about 500 mV vs. SHE, slightly below the potential of oxygen evolution, electrochemical passivation takes place by the formation of an iron oxide, which probably consists of the maghemite phase.
- In the potential range between about 550 mV and 900 mV vs. SHE, passivation is still apparent. However, the passive layer is increasingly damaged with rising anodic potential due to oxygen evolution.
- At anodic potentials above about 1 V vs. SHE, oxygen evolution causes a sufficiently high pH drop at the anode surface, leading to the precipitation of a thick and porous oxide layer, which predominantly consists of amorphous alumina or nanocrystalline γ-alumina and, in the case of phosphate-containing electrolytes, the hydrous phosphate evansite.
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Krysmann, W. Beitrag Zur Anodischen Oxydation von Aluminium Unter Funkenentladung. Ph.D. Thesis, Technische Hochschule Karl-Marx-Stadt, Karl-Marx-Stadt, Germany, 1982. [Google Scholar]
- Simchen, F.; Sieber, M.; Mehner, T.; Lampke, T. Characterisation method of the passivation mechanisms during the pre-discharge stage of plasma electrolytic oxidation indicating the mode of action of fluorides in peo of magnesium. Coatings 2020, 10, 965. [Google Scholar] [CrossRef]
- Holze, R. Leitfaden der Elektrochemie, 1st ed.; Teubner: Leipzig, Germany, 1998; p. 190. [Google Scholar]
- Lohrengel, M.M. Thin anodic oxide layers on aluminium and other valve metals: High field regime. Mater. Sci. Eng. R Rep. 1993, 11, 243–294. [Google Scholar] [CrossRef]
- Yerokhin, A.L.; Snizhko, L.O.; Gurevina, N.L.; Leyland, A.; Pilkington, A.; Matthews, A. Discharge characterization in plasma electrolytic oxidation of aluminium. J. Phys. D Appl. Phys. 2003, 36, 2110–2120. [Google Scholar] [CrossRef]
- Guo, S.; Si, R.; Dai, Q.; You, Z.; Ma, Y.; Wang, J. A critical review of corrosion development and rust removal techniques on the structural/environmental performance of corroded steel bridges. J. Clean. Prod. 2019, 233, 126–146. [Google Scholar] [CrossRef]
- Joiret, S.; Keddam, M.; Nóvoa, X.R.; Pérez, M.C.; Rangel, C.; Takenouti, H. Use of EIS, ring-disk electrode, EQCM and Raman spectroscopy to study the film of oxides formed on iron in 1M NaOH. Cem. Concr. Compos. 2002, 24, 7–15. [Google Scholar] [CrossRef]
- Zou, J.Y.; Chin, D.T. Anodic Behaviour of Carbon Steel in Concentrated NaOH Solutions. Electrochim. Acta 1988, 33, 477–485. [Google Scholar] [CrossRef]
- Hugot-Le Goff, A.; Flis, J.; Boucherit, N.; Joiret, S.; Wilinski, J. Use of Raman Spectroscopy and Rotating Split Ring Disk Electrode for Identification of Surface Layers on Iron in 1M NaOH. J. Electrochem. Soc. 1990, 137, 2684–2690. [Google Scholar] [CrossRef]
- Freire, L.; Nóvoa, X.R.; Montemor, M.F.; Carmezim, M.J. Study of passive films formed on mild steel in alkaline media by the application of anodic potentials. Mater. Chem. Phys. 2009, 114, 962–972. [Google Scholar] [CrossRef]
- DorMohammadi, H.; Pang, Q.; Murkutec, P.; Árnadóttir, L.; Isgor, O.B. Investigation of iron passivity in highly alkaline media using reactive-force field molecular dynamics. Corros. Sci. 2019, 157, 31–40. [Google Scholar] [CrossRef]
- Nanda, T.; Singh, V.; Singh, V.; Chakraborty, A.; Sharma, S. Third generation of advanced high-strength steels: Processing routes and properties. Proc. Inst. Mech. Eng. Pt. L J. Mater. Des. Appl. 2019, 233, 209–238. [Google Scholar] [CrossRef]
- Sarkar, P.P.; Kumar, P.; Kumar Manna, M.; Chakraborti, P.C. Microstructural influence on the electrochemical corrosion behaviour of dual-phase steels in 3.5% NaCl solution. Mater. Lett. 2005, 59, 2488–2491. [Google Scholar] [CrossRef]
- Nadlene, R.; Esah, H.; Norliana, S.; Mohd Irwan, M.A. Study on the Effect of Volume Fraction of Dual Phase Steel to Corrosion Behaviour and Hardness. Int. J. Sci. Res. 2011, 5, 393–396. [Google Scholar]
- Hai, C.; Cheng, X.; Du, C.; Li, X. Role of Martensite Structural Characteristics on Corrosion Features in Ni-Advanced Dual-Phase Low-Alloy Steels. Acta Metall. Sin. (Engl. Lett.) 2021, 34, 802–812. [Google Scholar] [CrossRef]
- Chen, H.; Lv, Z.; Lu, L.; Huang, Y.; Li, X. Correlation of micro-galvanic corrosion behavior with corrosion rate in the initial corrosion process of dual phase steel. J. Mater. Res. Technol. 2021, 15, 3310–3320. [Google Scholar] [CrossRef]
- Abdo, H.S.; Seikh, A.H.; Mohammed, J.A.; Luqman, M.; Ragab, S.A.; Almotairy, S.M. Influence of Chloride Ions on Electrochemical Corrosion Behavior of Dual-Phase Steel over Conventional Rebar in Pore Solution. Appl. Sci. 2020, 10, 4568. [Google Scholar] [CrossRef]
- Amaral, S.T.; Müller, I.L. A RRDE study of the electrochemical behavior of iron in solutions containing silicate and sulphate at pH 10–13. Corros. Sci. 1999, 41, 759–771. [Google Scholar] [CrossRef]
- Simchen, F.; Masoud-Nia, N.; Mehner, T.; Lampke, T. Formation of corundum-rich alumina coatings on low-carbon steel by plasma electrolytic oxidation. IOP Conf. Ser. Mater. Sci. Eng. 2021, 1147, 012007. [Google Scholar] [CrossRef]
- Kurze, P. Herstellung, Charakterisierung und Anwendung von Al2O3-Schichten insbesondere auf Aluminium- und Eisenwerkstoffen. Ph.D. Thesis, Technische Hochschule Karl-Marx-Stadt, Karl-Marx-Stadt, Germany, 1981. [Google Scholar]
- Karpushenkov, S.A.; Shchukin, G.L.; Belanovich, A.L.; Savenko, V.P.; Kulak, A.I. Plasma electrolytic ceramic-like aluminum oxide coatings on iron. J. Appl. Electrochem. 2010, 40, 365–374. [Google Scholar] [CrossRef]
- Eremin, N.I.; Volokhov, Y.A.; Mironov, V.E. Structure and Behaviour of Aluminate Ions in Solution. Russ. Chem. Rev. 1974, 43, 92–104. [Google Scholar] [CrossRef]
- Ginsberg, H.; Hüttig, W.; Stiehl, H. Über die Bildung von kristallinem AI(OH)3 und die Umwandlung von Bayerit in Hydrargillit. Z. Anorg. Allg. Chem. 1962, 318, 238–256. [Google Scholar] [CrossRef]
- Dao Huu, N. Untersuchungen zur Anodischen Abscheidung von Schutzschichten aus Wäßrigen Aluminatlösungen auf Eisenwerkstoffen. Ph.D. Thesis, Technische Hochschule Karl-Marx-Stadt, Karl-Marx-Stadt, Germany, 1979. [Google Scholar]
- Li, Z.; Cheng, Y.; Kang, S.H.; Tu, W.; Cheng, Y. Re-understanding of the breakdown theory from the study of the plasma electrolytic oxidation of a carbon steel—A non-valve metal. Electrochim. Acta 2018, 284, 681–695. [Google Scholar] [CrossRef]
- Klapkiv, M.D.; Nykyforchyn, H.M.; Posuvailo, V.M. Spectral Analaysis of an Elctrolytic Plasma in the Process of Synthesis of Aluminum Oxide. Mater. Sci. 1995, 30, 333–343. [Google Scholar] [CrossRef]
- Klapkiv, M.D. Simulation of Sythesis of Oxide-Ceramic Coatings in Discharge Channels of a Metal-Electrolyte System. Mater. Sci. 1999, 35, 279–283. [Google Scholar] [CrossRef]
- Attarzadeh, N.; Molaei, M.; Babaei, K.; Fattah-Alhosseini, A. New Promising Ceramic Coatings for Corrosion and Wear Protection of Steels: A Review. Surf. Interfaces 2021, 23, 100997. [Google Scholar] [CrossRef]
- Yang, W.; Li, Q.; Liu, W.; Liang, J.; Peng, Z.; Liu, B. Characterization and properties of plasma electrolytic oxidation coating on low carbon steel fabricated from aluminate electrolyte. Vacuum 2017, 144, 207–216. [Google Scholar] [CrossRef]
- Stern, M. A Method For Determining Corrosion Rates From Linear Polarization Data. Corrosion 1958, 9, 60–64. [Google Scholar] [CrossRef]
- Database of Raman Spectroscopy, X-ray Diffraction and Chemistry Data for Minerals. Available online: https://rruff.info (accessed on 29 January 2023).
- Ruan, H.D.; Frost, R.L.; Kloprogge, J.T. Comparison of the Raman spectra of Bayerite, Boehmite, Diaspore and Gibbsite. J. Raman Spectrosc. 2001, 32, 745–750. [Google Scholar] [CrossRef] [Green Version]
- Sudare, T.; Zenzai, A.; Tamura, S.; Kiyama, M.; Hayashi, F.; Teshima, K. Hierarchical spheres of Mg–Al LDH for the removal of phosphate ions: Effect of alumina polymorph as precursor. CrystEngComm 2019, 47, 7211–7216. [Google Scholar] [CrossRef]
- Majzlan, J.; Lang, B.E.; Stevens, R.; Navrotsky, A.; Woodfield, B.F.; Boerio-Goates, J. Thermodynamics of Fe oxides: Part I. Entropy at standard temperature and pressure and heat capacity of goethite (α-FeOOH), lepidocrocite (γ-FeOOH), and maghemite (γ-Fe2O3). Am. Mineral. 2003, 88, 846–854. [Google Scholar] [CrossRef]
- Hanesch, M. Raman spectroscopy of iron oxides and (oxy)hydroxides at low laser power and possible applications in environmental magnetic studies. Geophys. J. Int. 2009, 177, 941–948. [Google Scholar] [CrossRef]
- Mazzetti, L.; Thistlethwaite, P.J. Raman spectra and thermal transformations of ferrihydrite and schwertmannite. J. Raman Spectrosc. 2002, 33, 104–111. [Google Scholar] [CrossRef]
- Modesto Lopez, L.B.; Pasteris, J.D.; Biswas, P. Sensitivity of Micro-Raman Spectrum to Crystallite Size of Electrospray-Deposited and Post-Annealed Films of Iron-Oxide Nanoparticle Suspensions. Appl. Spectrosc. 2009, 63, 627–635. [Google Scholar] [CrossRef]
- Etteyeb, N.; Dhouibi, L.; Sanchez, M.; Alonso, C.; Andrade, C.; Triki, E. Electrochemical study of corrosion inhibition of steel reinforcement in alkaline solutions containing phosphates based components. J. Mater. Sci. 2007, 42, 4721–4730. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Electrolyte No. | Concentration in mol/L | pH | |
|---|---|---|---|
| 1 | 0.2 | 0 | 12.0 |
| 2 | 0.2 | 0.05 | 11.7 |
| 3 | 0.2 | 0.05 | 12.0 * |
| 4 | 0.2 | 0.1 | 11.5 |
| 5 | 0.2 | 0.1 | 12.0 * |
| Electrolyte No. | in mV | in 10−5∙mA/cm2 |
|---|---|---|
| 1 | −47 ± 24 | 19 ± 16 |
| 2 | −70 ± 150 | 13 ± 7 |
| 3 | −10 ± 60 | 19 ± 5 |
| 4 | 90 ± 80 | 9 ± 7 |
| 5 | 210 ± 80 | 10 ± 8 |
| Electrolyte | Molar Fraction in % | |||
|---|---|---|---|---|
| No. | Al | O | P | Fe |
| 1 | 38.4 ± 0.8 | 61.4 ± 0.7 | <0.1 | 0.3 ± 0.1 |
| 3 | 25.0 ± 1.9 | 66 ± 3 | 7.9 ± 0.9 | 0.8 ± 0.2 |
| 5 | 20.5 ± 0.7 | 68.7 ± 2.1 | 9.3 ± 0.1 | 1.5 ± 1.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morgenstern, R.; Albero Rojas, C.; Simchen, F.; Meinhold, V.; Mehner, T.; Lampke, T. Passivation and pH-Induced Precipitation during Anodic Polarization of Steel in Aluminate Electrolytes as a Precondition for Plasma Electrolytic Oxidation. Coatings 2023, 13, 656. https://doi.org/10.3390/coatings13030656
Morgenstern R, Albero Rojas C, Simchen F, Meinhold V, Mehner T, Lampke T. Passivation and pH-Induced Precipitation during Anodic Polarization of Steel in Aluminate Electrolytes as a Precondition for Plasma Electrolytic Oxidation. Coatings. 2023; 13(3):656. https://doi.org/10.3390/coatings13030656
Chicago/Turabian StyleMorgenstern, Roy, Claudia Albero Rojas, Frank Simchen, Vanessa Meinhold, Thomas Mehner, and Thomas Lampke. 2023. "Passivation and pH-Induced Precipitation during Anodic Polarization of Steel in Aluminate Electrolytes as a Precondition for Plasma Electrolytic Oxidation" Coatings 13, no. 3: 656. https://doi.org/10.3390/coatings13030656
APA StyleMorgenstern, R., Albero Rojas, C., Simchen, F., Meinhold, V., Mehner, T., & Lampke, T. (2023). Passivation and pH-Induced Precipitation during Anodic Polarization of Steel in Aluminate Electrolytes as a Precondition for Plasma Electrolytic Oxidation. Coatings, 13(3), 656. https://doi.org/10.3390/coatings13030656