1. Introduction
As fish live in aquatic environments and are constantly exposed to microorganisms, their skin is a critical first line of defense against pathogen infection. The epidermis and the mucus layers of fish skin act as barriers that protect fish from pathogens in water. Moreover, they also provide niches for symbiotic microorganisms, which have positive effects on the host’s health [
1,
2,
3]. The symbiotic microorganisms on fish skin are considered to have protective effects against pathogens. Basic studies of fish skin microflora have demonstrated that it is altered by handling in fish farms and by changes in environmental conditions, including pH, salt concentration, and temperature [
4,
5,
6,
7,
8]. However, our understanding of the relationship between the fish skin bacterial flora and fish pathogen infection is lacking [
9]. Uncovering this relationship could facilitate infection control utilizing the fish skin bacterial flora.
A laboratory infection model is necessary to obtain reproducible and reliable experimental data from analyses of biological samples taken from individuals in different growth conditions. Infection experiments on fish should be conducted under controlled environments. However, it is quite difficult to control environmental parameters in the field affected by the climate and by seasonal weather. In addition, the use of fish pathogens in open environments such as fish farms is legally regulated because of pandemic potential. Experiments with pathogens must therefore be performed in special facilities to limit biohazardous risks. However, it is difficult to collect many individuals of common farmed fish with identical physiological conditions, such as growth stage, size, and genetic background, for experiments in laboratories.
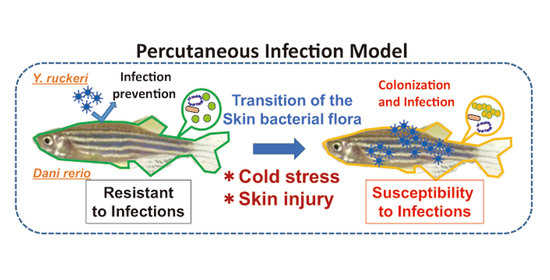
The purpose of this study is to establish a percutaneous infection model using zebrafish (
Danio rerio) and
Yersinia ruckeri.
D. rerio is a commonly used experimental model fish that is easy to grow in a small lab space, and is suitable for studies concerning infectious disease and the role of microbiota in infectious disease [
10,
11]. Moreover, genetic, physiological, and immunological information about zebrafish is abundant and useful for understanding host response to pathogens during infection.
Y. ruckeri is known to cause enteric redmouth disease (ERM) in fish, mainly among salmon and trout [
12]. The incidence of
Y. ruckeri infection among farmed fish is rising around the world. Although gills are the main infection route for
Y. ruckeri [
12], this bacterium was recently found on the skin of infected rainbow trout [
13]. We searched experimental manipulations that reproducibly caused
Y. ruckeri infection of zebrafish and performed 16S rDNA metagenomics of fish skin microflora to confirm the percutaneous infection accompanied by changes in the skin bacterial flora.
2. Materials and Methods
2.1. Maintenance and Handling of Zebrafish
Adult zebrafish used for the infection experiments (about 1 year old, 3–4 cm, 2:3 of male: female ratio, 300–400 fish) were acquired from the lab stock of Mie University. They were kept in a CROSS MINI (NWC-341; NISSO, Osaka, Japan) aquarium system for more than 2 weeks before being subjected to infection experiments. The fish (40 fish/6.8 L tank) were fed TetraMin Super 17653 (Spectrum Brands Japan, Kanagawa, Japan) every 12 h using a Tetra Auto Feeder AF-3 (Spectrum Brands Japan). A 12:12-h light-dark cycle was established using a Tetra LED Mini Light (Spectrum Brands Japan) for a tank and a digital timer PT70DW (REVEX, Saitama, Japan). Breeding water (6.8 of pH, 5° dH of carbonate hardness (KH), 0° dH of total hardness (GH), more than 7.0 mg/L of dissolve oxygen (DO), 18 L/tank) was maintained at 28 °C by using Safe Cover Heat Navi SH80 (GEX, Osaka, Japan).
When treating zebrafish with antibiotics, ampicillin (final concentration: 100 μg/mL), kanamycin (final concentration: 50 μg/mL), and amphotericin B (final concentration: 100 μg/mL) were added to 200 mL of sterile breeding water in a 500 mL flask, and five zebrafish were placed in it for 24 h at 20 °C. The breeding water was replaced with new water before pathogen challenge at 20 °C. To euthanize zebrafish, fish were placed in a tricaine (3-amino benzoic acid ethyl ester: TCI, Tokyo Japan) solution (600 mg/L) and allowed to swim until they stopped moving.
2.2. Bacterial Strains
Y. ruckeri NVH 3758 was isolated from rainbow trout with ERM in Norway, and kindly provided to us by Dr. Linke. For infection experiments, the strain was cultured in LB medium (Miller) at 28 °C for 24 h with shaking at 115 rpm. Aeromonas hydrophila NRIA14 and Vibrio ordalii NRIA90 were provided from the Japan Fisheries Research and Education Agency. Aeromonas caviae JCM1043 was purchased from the Japan Collection of Microorganisms, RIKEN BioResource Center.
Y. ruckeri transformation was performed by conjugal transfer from
Escherichia coli SM10 (λpir [
14] harboring pLBT::mini-Tn
10:
lac:
kan transposon vector [
15].
Y. ruckeri transformants were isolated as previously described [
16].
To isolate bacterial strains from the intrinsic skin microflora of zebrafish, euthanatized fish were laid on an aluminum foil disinfected with 70% ethanol, and the mucus on the fish epidermis was scraped with cotton swabs. The swabs were immersed in 1 mL Ultrapure deionized water. The collected mucus was resuspended using a vortex mixer. An aliquot (200 μL) of either the original suspension or their dilutions were spread on a nutrient agar medium, enriched Cytophaga agar medium, modified Zobell 2216E [
17] agar medium (0.8% NaCl), or tryptone soya broth agar medium, and incubated at either 28 °C or 20 °C for 2 d to grow bacterial colonies.
2.3. Detection of Bacteria
PCR detection of
Y. ruckeri was performed using 16S ribosomal DNA (rDNA) V1–V2 region specific primers or
Y. ruckeri glnA (glutamine synthetase) specific primers [
18]. Primer sequences are summarized in
Table S1. Amplification of target genes was verified by agarose gel electrophoresis and with an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA).
Y. ruckeri adhering to fish skin surface was observed through in vivo imaging of lacZ-expressing Y. ruckeri. For the imaging, zebrafish were euthanized, as described above, and fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) (pH 7.2) at 4 °C for 30 min. After washing the fish once with distilled water, the fish were placed in PBS containing 0.1% Triton-X 100 for 1 h. The fish were subsequently transferred to an X-gal staining solution (30 mM K3[Fe(CN)6], 30 mM K4[Fe(CN)6], 2 mM MgCl2, 1 mg/mL X-gal, 0.05% Tween-20 in PBS at pH 7.2) and maintained for 5 h at 30 °C. Fixed fish were observed under a fluorescence stereomicroscope (Axio Zoom V16, Carl Zeiss, Oberkochen, Germany).
2.4. Growth Inhibition Tests
To determine the growth inhibition activities of fish pathogens,
A. caviae JCM1043,
A. hydrophila NRIA14,
V. ordalii NRIA90,
Y. ruckeri NVH 3758, and isolated strains from the fish skin above were pre-cultured in a liquid medium overnight. The cross-streak method was performed, as described previously [
19]. In short, one of the isolated strains was streaked vertically on the solid medium that was used for its isolation, and the fish pathogens were streaked horizontally to cross the line of the isolated strain on the same agar medium. The plates were incubated thereafter at 28 °C or 20 °C for a few days.
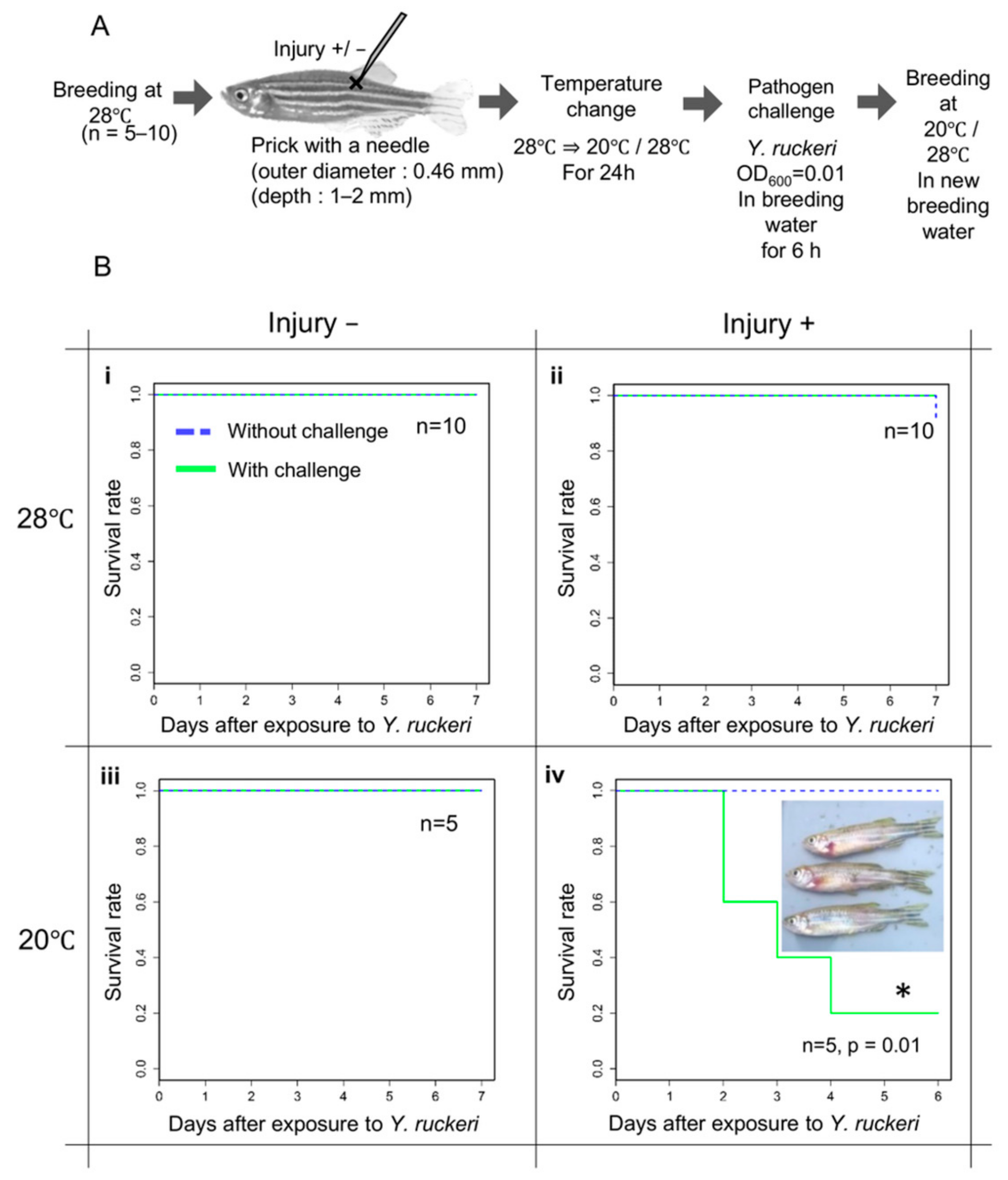
2.5. Infection Experiments
Sterile Instant Ocean Sea Salt (NAPQO, Concord, OH) solution was added to sterile water to prepare sterile breeding water, which contains 3 g/L seawater salt (10% the salt content of seawater). Three mL of a tricaine (3-amino benzoic acid ethyl ester: TCI, Tokyo Japan) solution (2 mg tricaine dissolved in 1 mL of 0.021 M Tris) was added to 100 mL of sterile breeding water to make an anesthetic solution. The fish were allowed to swim in the anesthetic solution for several minutes. Anesthetized fish were laid on an aluminum foil disinfected with 70% ethanol. Fish were injured by stinging at a dorsal ridge behind the dorsal fin with an injection needle (NN-2613S, 26 gauge, 13 mm; Terumo, Tokyo, Japan). The five injured fish were transferred into a 500 mL flask with 200 mL of the sterile breeding water, and maintained until they recovered from anesthesia. They were kept for 24 h in the flask with aeration at either 20 °C or 28 °C. As a control group, fish without injury and pathogen challenge (injury−,
Y. ruckeri−) were also simultaneously maintained in a flask under the above conditions. After 24 h, a bacterial suspension of
Y. ruckeri at an OD
600 (optical density at 600 nm) of 1.0 in Ultrapure deionized water was added to the flasks containing a fish group with pathogen challenge so that the OD
600 in breeding water was reduced to 0.01. After exposure to the pathogen for 6 h, the fish were transferred to fresh sterile breeding water in a new flask and maintained up to seven days at either 20 °C or 28 °C for the observation of an infected state. Feeding was discontinued for the duration of the infection experiment. Of five to ten fish, the numbers of dead and live fish were counted in each experimental set to calculate the survival rate and Kaplan-Meier curves were figured by executing R script on Rstudio (
https://rstudio.com/, accessed on 20 September 2019). The log rank tests were also executed on Rstudio. The infection experiments were repeated three times in each experimental set.
Infection by
Y. ruckeri was determined by either the observation of symptoms specific to ERM or by death of the fish. Fish were examined for disease symptoms every 12 h. Symptoms included slow swimming caused by debilitation as well as redness or petechiae at the lower jaw or the base of the fin [
12].
Y. ruckeri infection was diagnosed when these symptoms were observed. Fish with ERM symptoms and dead fish were captured immediately from the flasks. At the same time, the same number of fish in the control group were captured. All fish that survived for one week after pathogen challenge were also captured for further experiments. In addition, the breeding water was collected at the end of the infection experiment and filtered with suction through a membrane filter (mixed cellulose, 0.45 μm pore size, 47 mm diameter; ADVANTEC, Tokyo, Japan).
2.6. Next-Generation Sequencing of 16S rDNA Amplicon Libraries
A total of 45 zebrafish from ten experimental conditions (4–5 fish par each condition) were euthanized, as described above, rinsing off breeding water around the fish skin. Skin bacterial floras were collected by peeling off the skin of the fish, and skin samples containing the floras were transferred to sterile 1.5 mL tubes and stored at −30 °C. Bacteria in breeding water from seven experimental conditions were collected by filtration with 0.45 µm filter. A NucleoSpin Tissue Kit (Takara, Otsu, Japan) was used to extract and purify genomic DNA from skin samples containing surface microorganisms. According to the protocol for recovering bacterial genomic DNA from difficult-to-lyse bacteria, such as Gram-positive bacteria, the sample was pretreated with a pre-lysis solution (20 mg/mL lysozyme; 127-06724; Wako Pure Chemical Industries, 20 mM Tris-HCl, 2 mM EDTA, 1% Triton X-100) at 37 °C for 1 h. Bacterial 16S rDNA amplicon libraries were prepared from the extracted genomic DNA for DNA sequencing using iSeq 100 (Illumina, San Diego, CA, USA) according to the manual (16S Metagenomic Sequencing Library Preparation; 15044223 Rev. A; Illumina) with slight modifications. Ex Taq (Takara Bio, Kusatsu, Shiga, Japan) was used to amplify either the 16S rDNA V4 region from the extracted genomic DNA. Primer sequences for specific amplification of the V4 region were selected from sequences introduced in the Earth Microbiome Project (
Table S1). Overhang sequences for adaptor extension were added to the region-specific primer sequence. Primer sequences are summarized in
supplemental Table S1. To eliminate the contaminated DNA in reagents, 12.5 µg/mL of 5-methoxypsoralen was added to a PCR reagent mix, and the contaminated DNAs were cross-linked by exposing 365 nm UV for 10 min. Reagent samples without DNA were subjected to PCR for negative controls. The temperatures for denaturation, annealing, and extension reaction were set at 95 °C, 50.7 °C, and 72 °C, respectively. After 25 cycles of PCR, 11 of 52 samples were randomly selected, and the sizes of the amplification products (about 250–270 bp without primer sequences) were confirmed using an Agilent 2100 Bioanalyzer. The amplicons were purified using Agencourt AMPure XP beads (A63881; Beckman Coulter, Brea, CA, USA). In eight cycles of PCR using the Nextera XT Index Kit (Illumina) and 2× KAPA HiFi HotStart ReadyMix (Roche Diagnostics, Basel, Switzerland), the adapter and index sequences for Illumina sequencing analysis were added to the amplicons. The temperature conditions for this second PCR were as follow: 95 °C for denaturation, 55 °C for annealing, and 72 °C for extension reaction. The adapter-added amplicon libraries were again purified using AMPure XP beads. A part of the libraries was analyzed by the Bioanalyzer to confirm the library size. The library concentration was measured using the Qubit 4 and its dsDNA BR Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA). The library concentration was finally adjusted to 50 pM for analysis by the iSeq100 Sequencing System (1000000036024 v03 JPN; Illumina). Twenty µL of PhiX Control v3 (Illumina) was added to 100 µL of the library (about 16% of the library volume) to enhance sequence data quality.
2.7. In Silico Analysis Based on 16S rDNA Amplicon Sequencing
CLC Genomic Workbench (Qiagen Japan, Tokyo, Japan) with the Microbial genomic module was used for bacterial flora analysis and statistical analysis based on 16S rDNA sequences obtained by the iSeq100. The analyses were performed using sense reads (about 150 bp) of the pair-end fastq, because the merged sequences were not adequately obtained. Sequence data were imported into the software, and the length of the reads was trimmed using the ‘Trimming Sequence’ tool. As parameters, the length of the sequence after trimming was set to a minimum of 140 bp and a maximum of 160 bp, the base calling error probability limit was set to 0.05, and the maximum number of indeterminate bases was set to 1. Reads that matched the zebrafish reference genome (mitochondrial DNA etc.) were removed by the ‘Clean Host DNA’ tool. In order to classify reads according to experimental conditions, metadata files that included parameters for sample ID, water temperature, the presence or absence of injury, presence or absence of pathogen challenge, and live versus dead were created.
Reference-based operational taxonomic unit (OTU) clustering was performed utilizing the ‘OTU Clustering’ tool. The function of the elimination of chimera reads was executed during ‘OTU Clustering’, and the reads from 16S rDNA (1500–20,000 reads) were classified into OTUs. The Greengenes database (
http://greengenes.lbl.gov, accessed on 14 December 2018) was used as the reference database using a similarity threshold of 97%. The abundance tables including OTU data were exported as a Microsoft Excel spreadsheet (Data S1), and the total number of reads, the number of OTUs, and the number of OTUs duplicated between samples were calculated. In order to identify bacterial species from the read sequences, we searched for sequences similar to the read sequences in the NCBI 16s rDNA database using BLAST.
Analyses of β-diversity among experimental groups were performed as follows. An abundance table was created from a set of the sequence data to be compared, and the reads classified into OTUs were aligned using the ‘Align OTUs with MUSCLE’ tool. A phylogenetic tree was generated based on the alignment of reads using the ‘Create tree’ tool and the neighborhood linkage method. The K80 model [
20] was used to measure distances based on nucleic acid sequences’ alignment. The calculated weighted Unifrac distance between each sample [
21] was obtained from the phylogenetic tree generated using the ‘Beta diversity’ tool, and three-dimensional principal coordinate (3D PCoA) analysis was performed.
PERMANOVA (PER mutational Multivariate Analysis Of Variance) analysis for determining the intergroup variability and the statistical analysis for intergroup difference was performed. The weighted Unifrac distance was used for distance metrics. The permutation (number of permutations) was set to 99,999. The statistical value was calculated by the method of Anderson et al. [
22].
Heat maps were generated using the ‘Create Heat Map for Abundance Table’ tool based on the abundance of OTUs in the respective samples. An abundance table was generated from the sequence data of the samples to be compared as described above. The Euclidean distance was used for distance metrics between two samples, and the perfect connection method was selected as a clustering method.
Alpha-diversity indices were calculated using specific tools in CLC Genomic Workbench [
23,
24,
25]. To generate rarefaction curves, the range of the sub-sampling depth was set from 1 to 1500 reads, the number of different depths for sampling was set to 50, and the number of replicates for each depth was set to 100. The results of rarefaction curves were exported as Microsoft Excel spreadsheets (Data S2) and the results at the maximum sub-sampling depth (1500 reads) were presented as box plots.
4. Discussion
In this study, we found that pricking zebrafish with a needle did not result in Y. ruckeri infection following pathogen challenge. This bacterium had not been reported to be pathogenic toward zebrafish when we started this study. The infection of injured zebrafish by Y. ruckeri was caused by cold stress due to a shift in breeding water temperature from the optimum 28 °C to 20 °C. Most fish subjected to these experimental manipulations died and symptoms of ERM were observed in the dead fish, indicating that Y. ruckeri can cause the fatal infection of zebrafish.
We verified the adhesion of this bacterium to the skin of infected fish (
Figure 2 and
Figure S1), despite low numbers of
Y. ruckeri in the breeding water (
Figure 3).
Y. ruckeri is known to form biofilms on solid surfaces, and such biofilms are believed to be the source of recurrent infection in aquaculture facilities [
12]. The
in vivo observation of
Y. ruckeri (
Figure 2) suggests that a substantial population of
Y. ruckeri directly colonized the skin of injured zebrafish at 20 °C. Furthermore,
Y. ruckeri either occupied zebrafish skin or dominated the skin bacterial flora under infectious conditions (injury+, pathogen challenge+, 20 °C). Disruption of skin bacterial flora by antibiotic treatment also resulted in either the
Y. ruckeri occupying zebrafish skin or becoming predominant among the fish skin bacterial flora. This result suggests that the intrinsic skin bacterial flora protects zebrafish from pathogen adhesion and colonization.
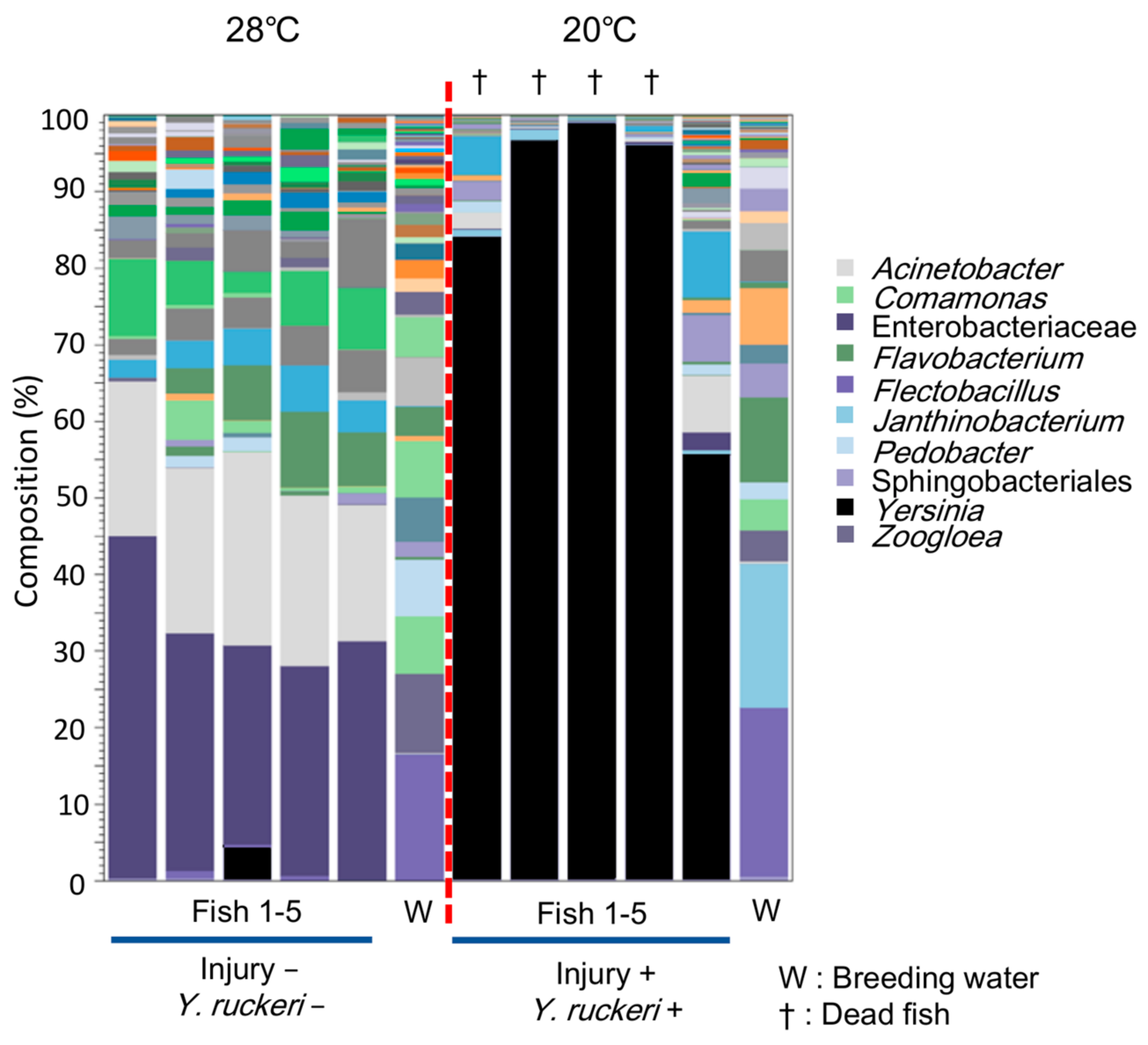
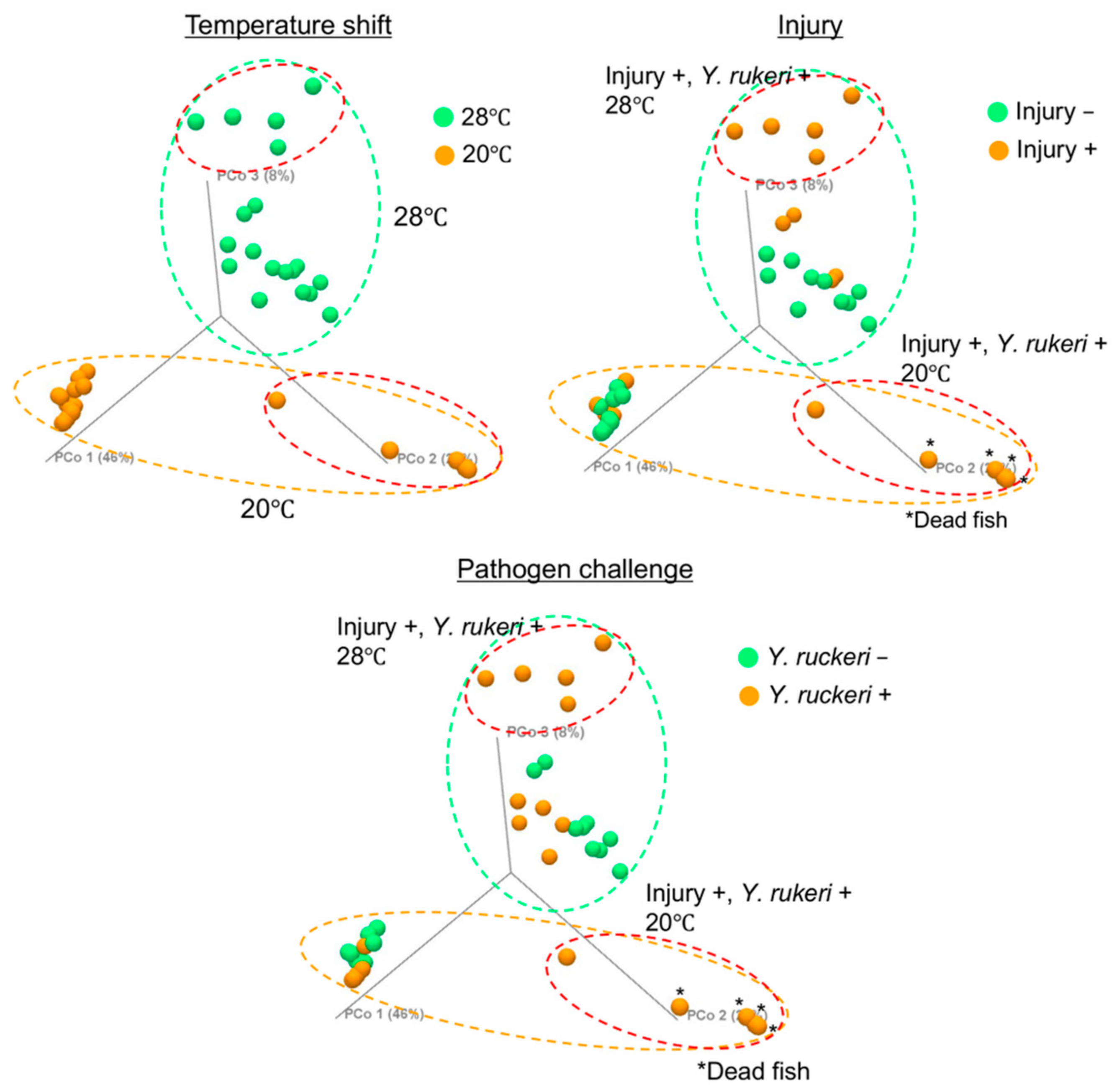
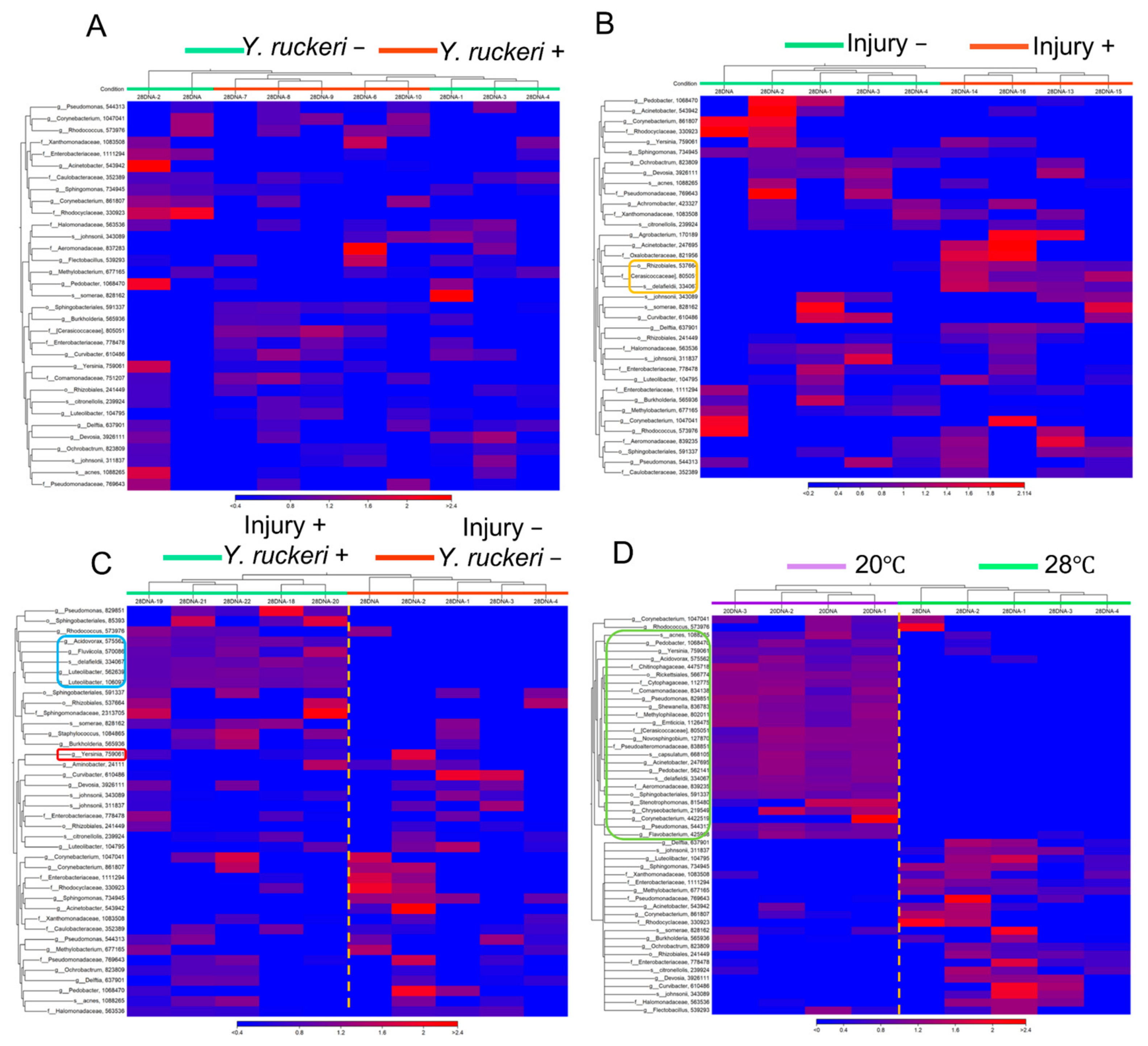
As we have demonstrated,
Y. ruckeri infection of zebrafish requires cold stress, which changes the composition of the skin bacterial flora drastically, including the major genera represented (
Figure 4 and
Figure 5 and
Figure S2, Table S2). This transition may allow the pathogen to dominate the fish skin bacterial flora. The change in bacterial floras on zebrafish was small or ambiguous by pathogen challenge or injury alone, which did not specifically increase
Y. ruckeri on fish skin (
Figure 5A,B). Both injury and pathogen challenge only had slight effects on the skin bacterial flora: population ratios of the genera
Acidovorax and
Luteolibacter, whose OTUs were not recognized without injury and pathogen challenge, increased, but the population ratio of the genus
Yersinia did not increase significantly (
Figure 5C). Neither
Acidovorax nor
Luteolibacter have been reported to antagonize fish pathogens’ growth as far as we know. However, these bacteria may play a role in repairing damage to the skin bacterial flora by pathogen challenge as a phenomenon of microflora resilience by colonizing a niche on the skin.
Environmental perturbations often challenge bacterial flora homeostasis. After such perturbations, a resilient bacterial flora will return to its original equilibrium or else dysbacteriosis would result [
26]. The resilience of microflora may be suppressed by a shift in temperature to 20 °C. The trends of the α-diversity indices at 20 °C (
Figure 6) indicate that cold stress destabilized the skin microflora of zebrafish and enabled the pathogen to affect the fish more easily. Once
Y. ruckeri colonizes a niche delivered by a change in the microflora, this bacterium may increase in proportion exclusively on the fish skin, resulting in its occupation or domination of the microflora as well as a decrease in α-diversity (
Figure 6). The reason for
Y. ruckeri’s occupation or domination of the skin microflora may be due to its ability to inhibit the growth of some bacteria present on zebrafish skin (
Figure S3), especially at 20 °C (
Table 1).
The reason for the drastic change in the skin bacterial flora caused by the change in temperature remains unclear. This is probably mainly because the optimum growth temperature varies among bacterial species. Alteration of the host’s immune system by cold stress may also be a factor. Fish are cold-blooded animals, and water temperature is known to affect immune responses, including antibody production [
27,
28,
29]. Zebrafish immune activity could be negatively impacted by lowering the water temperature. For example, skin mucus secreted from the epithelial cells contains biological defense factors such as lectin [
1,
2,
30,
31,
32,
33]. Such immune responses, including the secretion of defense factors, may be less active at 20 °C, leading to changes in the composition of the skin bacteria flora.
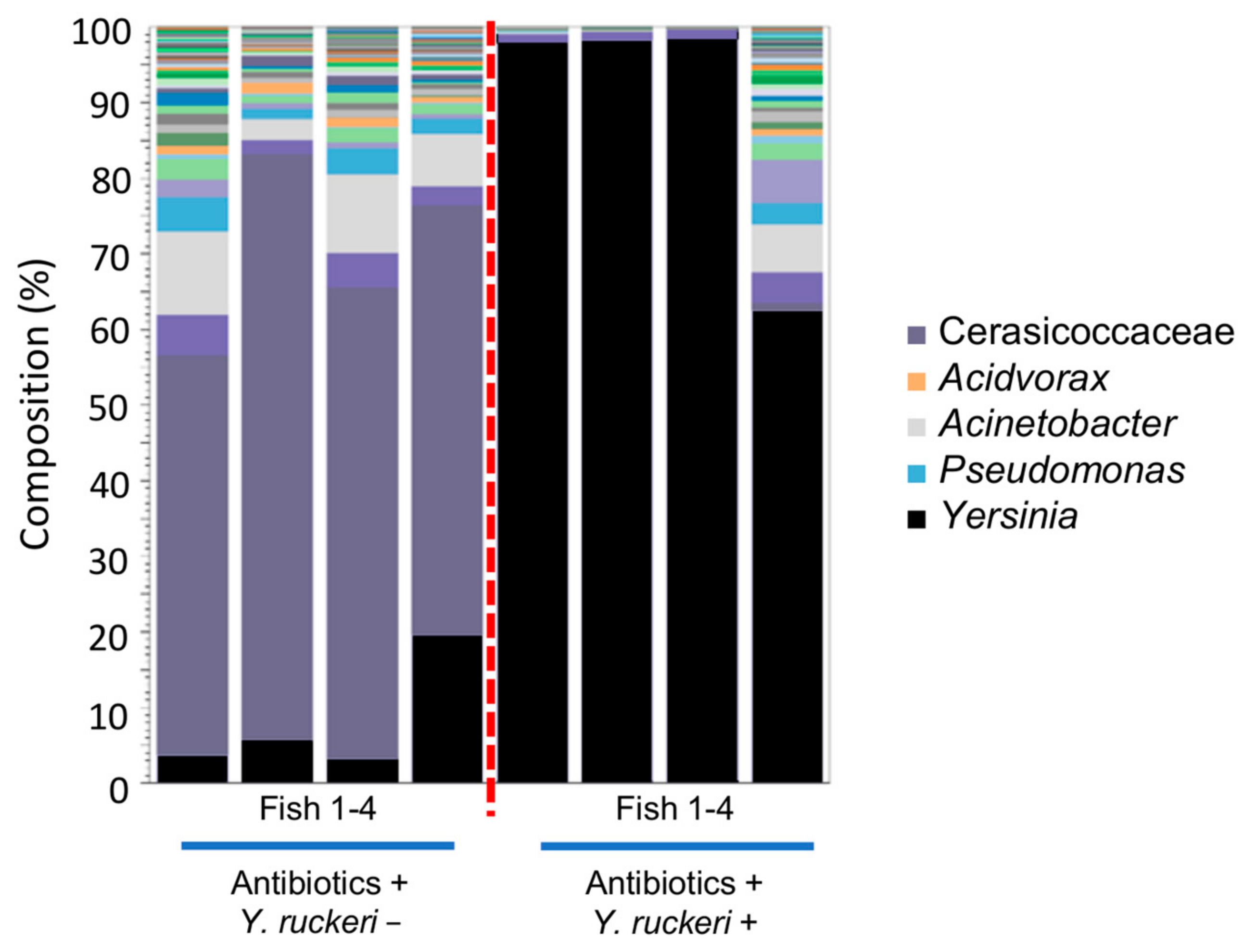
The result of the antibiotic treatment experiment shown in
Figure 7 indicates that disruption of the intrinsic skin bacterial flora promotes
Y. ruckeri adhesion to the skin and made it the predominant species in bacterial flora reconstructed on the fish skin following disruption. Although the fish skin was occupied by
Y. ruckeri following disruption of the skin bacterial flora with antibiotics (
Figure 7), the fish survived (
Figure S5). This is likely due to the fact that there was no injury to provide an invasion route for this pathogen through the skin. Invasion into the fish body is necessary for
Y. ruckeri to cause ERM in zebrafish. However, the abundance of
Yersinia on the skin did not increase unless the injury was inflicted even at 20 °C (
Figure S6), suggesting that the injury not only provided an invasion route for
Y. ruckeri but also imposed stress on the zebrafish, which together with the temperature shift, allowed this pathogen to dominate the skin bacterial flora.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}