
Lipid Metabolism and Ferroptosis



{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
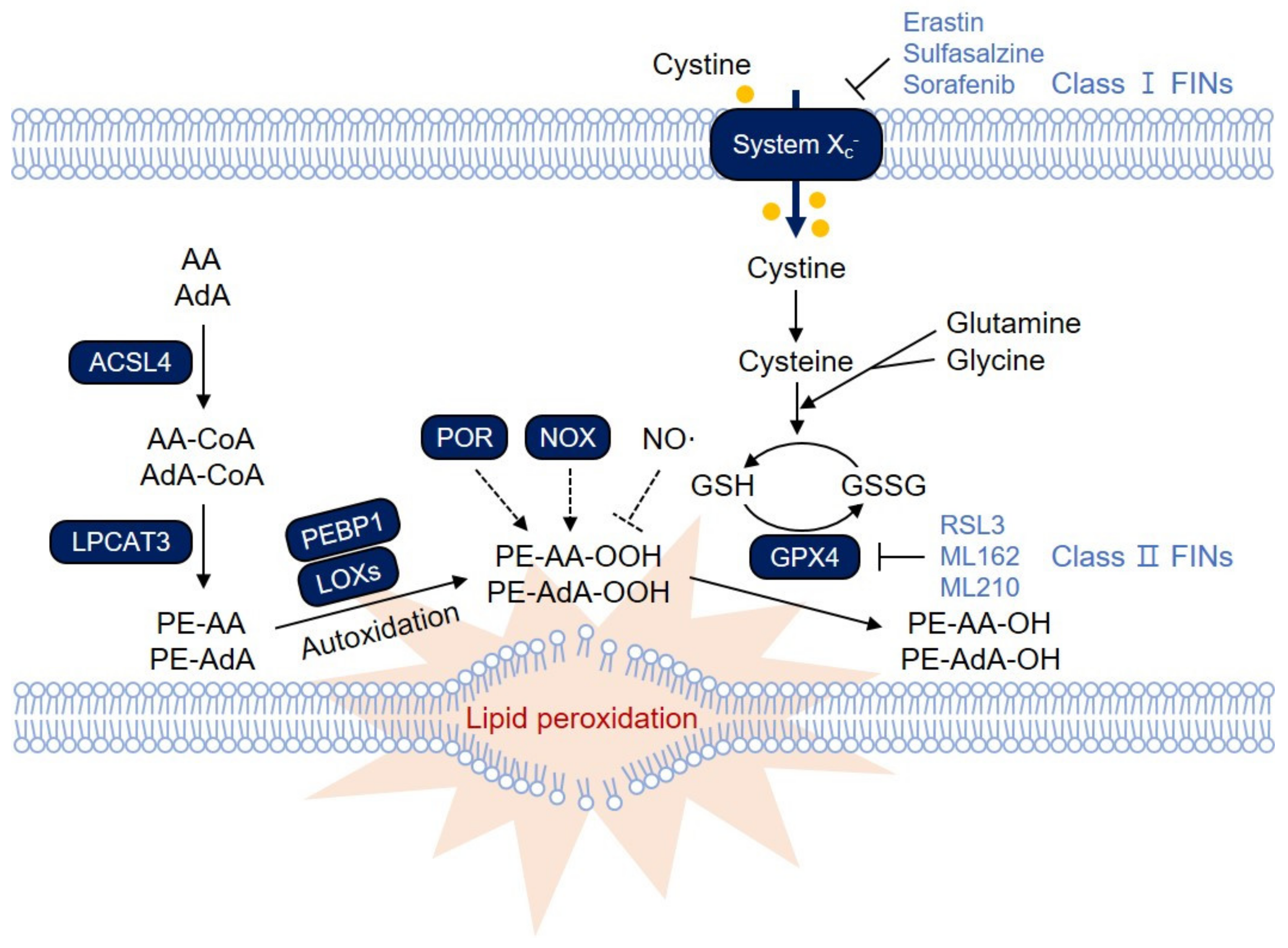
2. Lipid Peroxidation and Ferroptosis
2.1. PE-AA and PE-AdA, the Most Susceptible Substrates for Lipid Peroxidation
2.2. ACSL4 and LPCAT3 for Membrane Phospholipids
2.3. LOXs in Lipid Peroxidation
2.4. Phosphatidylethanolamine-Binding Protein 1 (PEBP1), a Key Regulator of Lipid Peroxidation
2.5. Other Oxygenases in Ferroptosis
2.6. Ether Phospholipids in Ferroptosis
3. Pathways That Regulate Lipid Metabolism and Ferroptosis
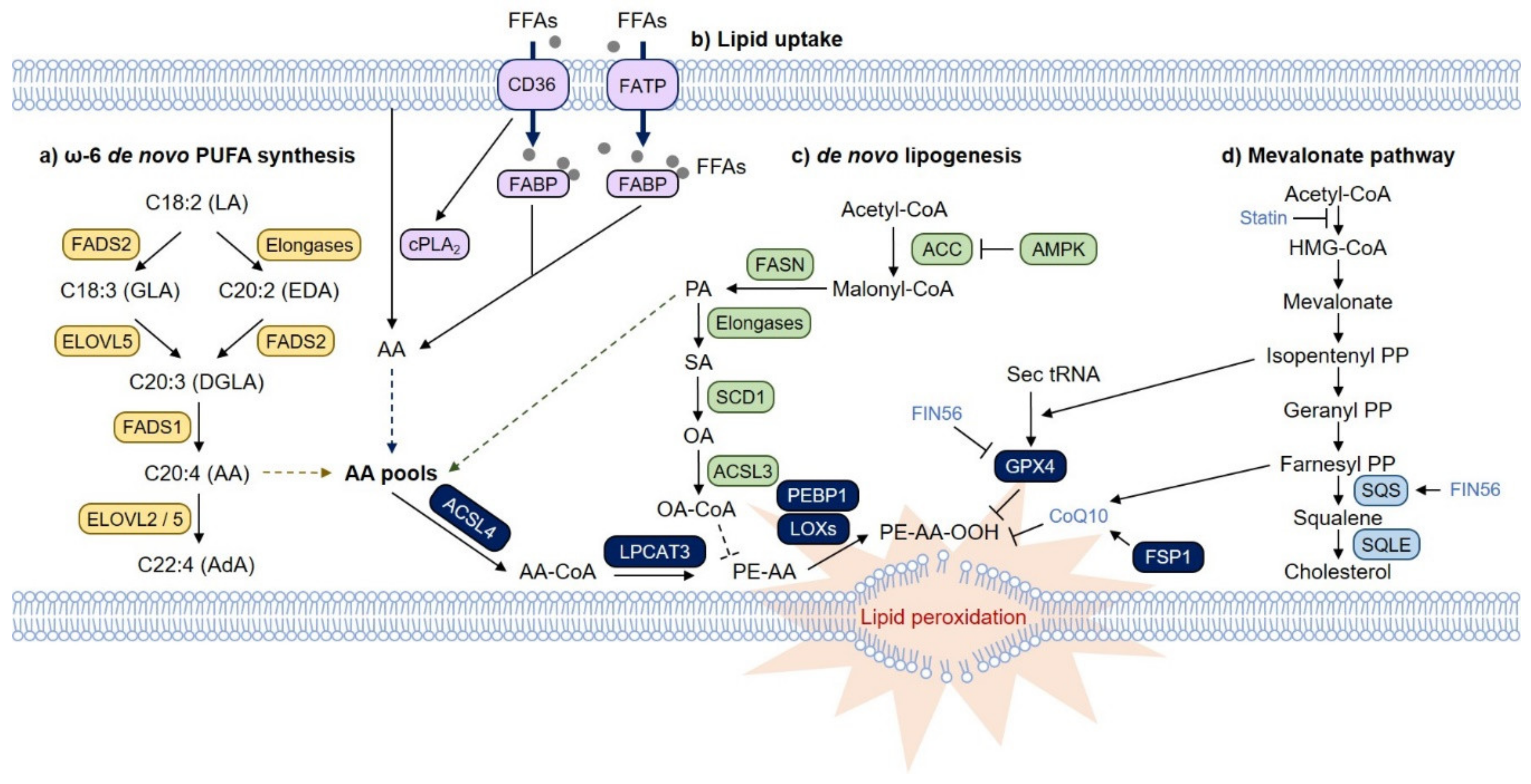
3.1. PUFA Biosynthesis Pathway and Ferroptosis
3.2. MUFAs and Ferroptosis
3.3. Fatty Acid Transport and Ferroptosis
3.4. De Novo Lipogenesis and Ferroptosis
3.5. The Mevalonate Pathway
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- D’Autréaux, B.; Toledano, M.B. ROS as Signalling Molecules: Mechanisms That Generate Specificity in ROS Homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef]
- Moloney, J.N.; Cotter, T.G. ROS Signalling in the Biology of Cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef]
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants Maintain Cellular Redox Homeostasis by Elimination of Reactive Oxygen Species. Cell. Physiol. Biochem. 2017, 44, 532–553. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Chua, C.C.; Gao, J.; Chua, K.-W.; Ho, Y.-S.; Hamdy, R.C.; Chua, B.H.L. Prevention of Ischemia/Reperfusion-Induced Cardiac Apoptosis and Injury by Melatonin Is Independent of Glutathione Peroxdiase 1. J. Pineal Res. 2009, 46, 235–241. [Google Scholar] [CrossRef] [Green Version]
- Ardanaz, N.; Yang, X.-P.; Cifuentes, M.E.; Haurani, M.J.; Jackson, K.W.; Liao, T.-D.; Carretero, O.A.; Pagano, P.J. Lack of Glutathione Peroxidase 1 Accelerates Cardiac-Specific Hypertrophy and Dysfunction in Angiotensin II Hypertension. Hypertension 2010, 55, 116–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, M.T.; Beal, M.F. Mitochondrial Dysfunction and Oxidative Stress in Neurodegenerative Diseases. Nat. Cell Biol. 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Nigam, S.; Schewe, T. Phospholipase A2s and Lipid Peroxidation. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2000, 1488, 167–181. [Google Scholar] [CrossRef]
- Adibhatla, R.M.; Hatcher, J. Phospholipase A2, Reactive Oxygen Species, and Lipid Peroxidation in Cerebral Ischemia. Free Radic. Biol. Med. 2006, 40, 376–387. [Google Scholar] [CrossRef]
- Negre-Salvayre, A.; Auge, N.; Ayala, V.; Basaga, H.; Boada, J.; Brenke, R.; Chapple, S.; Cohen, G.; Feher, J.; Grune, T.; et al. Pathological Aspects of Lipid Peroxidation. Free. Radic. Res. 2010, 44, 1125–1171. [Google Scholar] [CrossRef]
- Niki, E. Lipid Peroxidation: Physiological Levels and Dual Biological Effects. Free Radic. Biol. Med. 2009, 47, 469–484. [Google Scholar] [CrossRef] [PubMed]
- Ursini, F.; Maiorino, M.; Gregolin, C. The Selenoenzyme Phospholipid Hydroperoxide Glutathione Peroxidase. Biochim. Biophys. Acta (BBA) Gen. Subj. 1985, 839, 62–70. [Google Scholar] [CrossRef]
- Van Kuijk, F.J.; Handelman, G.J.; Dratz, E.A. Consecutive Action of Phospholipase A2 and Glutathione Peroxidase Is Required for Reduction of Phospholipid Hydroperoxides and Provides a Convenient Method to Determine Peroxide Values in Membranes. J. Free Radicals Biol. Med. 1985, 1, 421–427. [Google Scholar] [CrossRef]
- Yant, L.J.; Ran, Q.; Rao, L.; Van Remmen, H.; Shibatani, T.; Belter, J.G.; Motta, L.; Richardson, A.; Prolla, A.T. The Selenoprotein GPX4 Is Essential for Mouse Development and Protects from Radiation and Oxidative Damage Insults. Free Radic. Biol. Med. 2003, 34, 496–502. [Google Scholar] [CrossRef]
- Seiler, A.; Schneider, M.; Förster, H.; Roth, S.; Wirth, E.K.; Culmsee, C.; Plesnila, N.; Kremmer, E.; Rådmark, O.; Wurst, W.; et al. Glutathione Peroxidase 4 Senses and Translates Oxidative Stress into 12/15-Lipoxygenase Dependent-and AIF-Mediated Cell Death. Cell Metab. 2008, 8, 237–248. [Google Scholar] [CrossRef] [Green Version]
- Angeli, J.P.F.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the Ferroptosis Regulator GPX4 Triggers Acute Renal Failure in Mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of Polyunsaturated Fatty Acids by Lipoxygenases Drives Ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef] [Green Version]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ Oxidoreductase FSP1 Acts Parallel to GPX4 to Inhibit Ferroptosis. Nat. Cell Biol. 2019, 575, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; Da Silva, M.C.; Ingold, I.; Grocin, A.G.; Da Silva, T.N.X.; Panzilius, E.; Scheel, C.H.; et al. FSP1 Is a Glutathione-Independent Ferroptosis Suppressor. Nat. Cell Biol. 2019, 575, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Zhu, S.; Song, X.; Sun, X.; Fan, Y.; Liu, J.; Zhong, M.; Yuan, H.; Zhang, L.; Billiar, T.R.; et al. The Tumor Suppressor p53 Limits Ferroptosis by Blocking DPP4 Activity. Cell Rep. 2017, 20, 1692–1704. [Google Scholar] [CrossRef] [Green Version]
- Bannai, S.; Tsukeda, H.; Okumura, H. Effect of Antioxidants on Cultured Human Diploid Fibroblasts Exposed to Cystine-Free Medium. Biochem. Biophys. Res. Commun. 1977, 74, 1582–1588. [Google Scholar] [CrossRef]
- Zhou, R.-P.; Chen, Y.; Wei, X.; Yu, B.; Xiong, Z.-G.; Lu, C.; Hu, W. Novel Insights into Ferroptosis: Implications for Age-Related Diseases. Theranostics 2020, 10, 11976–11997. [Google Scholar] [CrossRef]
- Hassannia, B.; Vandenabeele, P.; Berghe, T.V. Targeting Ferroptosis to Iron Out Cancer. Cancer Cell 2019, 35, 830–849. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [Green Version]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.F.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized Arachidonic and Adrenic PEs Navigate Cells to Ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Hassannia, B.; Wiernicki, B.; Ingold, I.; Qu, F.; Van Herck, S.; Tyurina, Y.Y.; Bayır, H.; Abhari, B.A.; Angeli, J.P.F.; Choi, S.M.; et al. Nano-Targeted Induction of Dual Ferroptotic Mechanisms Eradicates High-Risk Neuroblastoma. J. Clin. Investig. 2018, 128, 3341–3355. [Google Scholar] [CrossRef]
- Wiernicki, B.; Dubois, H.; Tyurina, Y.Y.; Hassannia, B.; Bayir, H.; Kagan, V.E.; Vandenabeele, P.; Wullaert, A.; Berghe, T.V. Excessive Phospholipid Peroxidation Distinguishes Ferroptosis from Other Cell Death Modes Including Pyroptosis. Cell Death Dis. 2020, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Küch, E.-M.; Vellaramkalayil, R.; Zhang, I.; Lehnen, D.; Brügger, B.; Stremmel, W.; Ehehalt, R.; Poppelreuther, M.; Füllekrug, J. Differentially Localized Acyl-CoA Synthetase 4 Isoenzymes Mediate the Metabolic Channeling of Fatty Acids Towards Phosphatidylinositol. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2014, 1841, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Hishikawa, D.; Shindou, H.; Kobayashi, S.; Nakanishi, H.; Taguchi, R.; Shimizu, T. Discovery of a Lysophospholipid Acyltransferase Family Essential for Membrane Asymmetry and Diversity. Proc. Natl. Acad. Sci. USA 2008, 105, 2830–2835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, S.J.; Winter, G.E.; Musavi, L.S.; Lee, E.D.; Snijder, B.; Rebsamen, M.; Superti-Furga, G.; Stockwell, B.R. Human Haploid Cell Genetics Reveals Roles for Lipid Metabolism Genes in Nonapoptotic Cell Death. ACS Chem. Biol. 2015, 10, 1604–1609. [Google Scholar] [CrossRef]
- Conrad, M.; Pratt, D.A. The Chemical Basis of Ferroptosis. Nat. Chem. Biol. 2019, 15, 1137–1147. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Xu, L.; Porter, N.A. Free Radical Lipid Peroxidation: Mechanisms and Analysis. Chem. Rev. 2011, 111, 5944–5972. [Google Scholar] [CrossRef]
- Shintoku, R.; Takigawa, Y.; Yamada, K.; Kubota, C.; Yoshimoto, Y.; Takeuchi, T.; Koshiishi, I.; Torii, S. Lipoxygenase-Mediated Generation of Lipid Peroxides Enhances Ferroptosis Induced by Erastin and RSL3. Cancer Sci. 2017, 108, 2187–2194. [Google Scholar] [CrossRef]
- Wenzel, S.E.; Tyurina, Y.Y.; Zhao, J.; Croix, C.M.S.; Dar, H.H.; Mao, G.; Tyurin, V.A.; Anthonymuthu, T.S.; Kapralov, A.A.; Amoscato, A.A.; et al. PEBP1 Wardens Ferroptosis by Enabling Lipoxygenase Generation of Lipid Death Signals. Cell 2017, 171, 628–641.e26. [Google Scholar] [CrossRef] [Green Version]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid Peroxidation: Production, Metabolism, and Signaling Mechanisms of Malondialdehyde and 4-Hydroxy-2-Nonenal. Oxidative Med. Cell. Longev. 2014, 2014, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Fenton, H.J.H. LXXIII—Oxidation of Tartaric Acid in Presence of Iron. J. Chem. Soc. Trans. 1894, 65, 899–910. [Google Scholar] [CrossRef] [Green Version]
- Reis, A.; Spickett, C.M. Chemistry of Phospholipid Oxidation. Biochim. Biophys. Acta (BBA) Biomembr. 2012, 1818, 2374–2387. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, H.; Saam, J.; Eibach, S.; Holzhütter, H.-G.; Ivanov, I.; Walther, M. Structural Biology of Mammalian Lipoxygenases: Enzymatic Consequences of Targeted Alterations of the Protein Structure. Biochem. Biophys. Res. Commun. 2005, 338, 93–101. [Google Scholar] [CrossRef]
- Xu, S.; Mueser, T.C.; Marnett, L.J.; Funk, M.O. Crystal Structure of 12-Lipoxygenase Catalytic-Domain-Inhibitor Complex Identifies a Substrate-Binding Channel for Catalysis. Structure 2012, 20, 1490–1497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newcomer, M.E.; Brash, A.R. The Structural Basis for Specificity in Lipoxygenase Catalysis. Protein Sci. 2015, 24, 298–309. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.-J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-Mediated Activity during Tumour Suppression. Nat. Cell Biol. 2015, 520, 57–62. [Google Scholar] [CrossRef] [Green Version]
- Ou, Y.; Wang, S.-J.; Li, D.; Chu, B.; Gu, W. Activation of SAT1 Engages Polyamine Metabolism with p53-Mediated Ferroptotic Responses. Proc. Natl. Acad. Sci. USA 2016, 113, E6806–E6812. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.; Zhang, C.; Wu, R.; Sun, Y.; Levine, A.; Feng, Z. Glutaminase 2, a Novel p53 Target Gene Regulating Energy Metabolism and Antioxidant Function. Proc. Natl. Acad. Sci. USA 2010, 107, 7455–7460. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.; Monian, P.; Quadri, N.; Ramasamy, R.; Jiang, X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol. Cell 2015, 59, 298–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarangelo, A.; Magtanong, L.; Bieging-Rolett, K.T.; Li, Y.; Ye, J.; Attardi, L.D.; Dixon, S.J. p53 Suppresses Metabolic Stress-Induced Ferroptosis in Cancer Cells. Cell Rep. 2018, 22, 569–575. [Google Scholar] [CrossRef] [Green Version]
- Chu, B.; Kon, N.; Chen, D.; Li, T.; Liu, T.; Jiang, L.; Song, S.; Tavana, O.; Gu, W. ALOX12 is Required for p53-Mediated Tumour Suppression through a Distinct Ferroptosis Pathway. Nat. Cell Biol. 2019, 21, 579–591. [Google Scholar] [CrossRef]
- Shah, R.; Shchepinov, M.S.; Pratt, D.A. Resolving the Role of Lipoxygenases in the Initiation and Execution of Ferroptosis. ACS Central Sci. 2018, 4, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; O’Donnell, V.B.; Balzar, S.; Croix, C.M.S.; Trudeau, J.B.; Wenzel, S.E. 15-Lipoxygenase 1 Interacts with Phosphatidylethanolamine-Binding Protein to Regulate MAPK Signaling in Human Airway Epithelial Cells. Proc. Natl. Acad. Sci. USA 2011, 108, 14246–14251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeung, K.; Seitz, T.; Li, S.; Janosch, P.; McFerran, B.; Kaiser, C.; Fee, F.; Katsanakis, K.D.; Rose, D.W.; Mischak, H.; et al. Suppression of Raf-1 Kinase Activity and MAP Kinase Signalling by RKIP. Nat. Cell Biol. 1999, 401, 173–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, X.-H.; Cui, C.-C.; Shan, C.; Li, Y.-Z.; Sheng, D.-H.; Sun, B.; Chen, D.-Z. O-Phenylenediamine: A Privileged Pharmacophore of Ferrostatins for Radical-Trapping Reactivity in Blocking Ferroptosis. Org. Biomol. Chem. 2018, 16, 3952–3960. [Google Scholar] [CrossRef]
- Shah, R.; Margison, K.; Pratt, D.A. The Potency of Diarylamine Radical-Trapping Antioxidants as Inhibitors of Ferroptosis Underscores the Role of Autoxidation in the Mechanism of Cell Death. ACS Chem. Biol. 2017, 12, 2538–2545. [Google Scholar] [CrossRef] [PubMed]
- Zilka, O.; Shah, R.; Li, B.; Angeli, J.P.F.; Griesser, M.; Conrad, M.; Pratt, D.A. On the Mechanism of Cytoprotection by Ferrostatin-1 and Liproxstatin-1 and the Role of Lipid Peroxidation in Ferroptotic Cell Death. ACS Central Sci. 2017, 3, 232–243. [Google Scholar] [CrossRef]
- Anthonymuthu, T.S.; Tyurina, Y.Y.; Sun, W.-Y.; Mikulska-Ruminska, K.; Shrivastava, I.H.; Tyurin, V.A.; Cinemre, F.B.; Dar, H.H.; VanDemark, A.P.; Holman, T.R.; et al. Resolving the Paradox of Ferroptotic Cell Death: Ferrostatin-1 Binds to 15LOX/PEBP1 Complex, Suppresses Generation of per-Oxidized ETE-PE, and Protects against Ferroptosis. Redox Biol. 2021, 38, 101744. [Google Scholar] [CrossRef]
- Yang, W.-H.; Huang, Z.; Wu, J.; Ding, C.-K.C.; Murphy, S.K.; Chi, J.-T. A TAZ–ANGPTL4–NOX2 Axis Regulates Ferroptotic Cell Death and Chemoresistance in Epithelial Ovarian Cancer. Mol. Cancer Res. 2020, 18, 79–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.-H.; Ding, C.-K.C.; Sun, T.; Rupprecht, G.; Lin, C.-C.; Hsu, D.; Chi, J.-T. The Hippo Pathway Effector TAZ Regulates Ferroptosis in Renal Cell Carcinoma. Cell Rep. 2019, 28, 2501–2508.e4. [Google Scholar] [CrossRef]
- Augsburger, F.; Filippova, A.; Rasti, D.; Seredenina, T.; Lam, M.; Maghzal, G.; Mahiout, Z.; Jansen-Dürr, P.; Knaus, U.G.; Doroshow, J.; et al. Pharmacological Characterization of the Seven Human Nox Isoforms and Their Inhibitors. Redox Biol. 2019, 26, 101272. [Google Scholar] [CrossRef]
- Zou, Y.; Li, H.; Graham, E.T.; Deik, A.A.; Eaton, J.K.; Wang, W.; Sandoval-Gomez, G.; Clish, C.B.; Doench, J.G.; Schreiber, S.L. Cytochrome P450 Oxidoreductase Contributes to Phospholipid Peroxidation in Ferroptosis. Nat. Chem. Biol. 2020, 16, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Riddick, D.S.; Ding, X.; Wolf, C.R.; Porter, T.D.; Pandey, A.V.; Zhang, Q.-Y.; Gu, J.; Finn, R.D.; Ronseaux, S.; McLaughlin, L.A.; et al. NADPH–Cytochrome P450 Oxidoreductase: Roles in Physiology, Pharmacology, and Toxicology. Drug Metab. Dispos. 2012, 41, 12–23. [Google Scholar] [CrossRef] [Green Version]
- Yan, B.; Ai, Y.; Sun, Q.; Ma, Y.; Cao, Y.; Wang, J.; Zhang, Z.; Wang, X. Membrane Damage during Ferroptosis Is Caused by Oxidation of Phospholipids Catalyzed by the Oxidoreductases POR and CYB5R1. Mol. Cell 2021, 81, 355–369.e10. [Google Scholar] [CrossRef] [PubMed]
- Kapralov, A.A.; Yang, Q.; Dar, H.H.; Tyurina, Y.Y.; Anthonymuthu, T.S.; Kim, R.; Croix, C.M.S.; Mikulska-Ruminska, K.; Liu, B.; Shrivastava, I.H.; et al. Redox Lipid Reprogramming Commands Susceptibility of Macrophages and Microglia to Ferroptotic Death. Nat. Chem. Biol. 2020, 16, 278–290. [Google Scholar] [CrossRef]
- Zou, Y.; Henry, W.S.; Ricq, E.L.; Graham, E.T.; Phadnis, V.V.; Maretich, P.; Paradkar, S.; Boehnke, N.; Deik, A.A.; Reinhardt, F.; et al. Plasticity of Ether Lipids Promotes Ferroptosis Susceptibility and Evasion. Nat. Cell Biol. 2020, 585, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Guillou, H.; Zadravec, D.; Martin, P.G.; Jacobsson, A. The Key Roles of Elongases and Desaturases in Mammalian Fatty Acid Metabolism: Insights from Transgenic Mice. Prog. Lipid Res. 2010, 49, 186–199. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-Y.; Nam, M.; Son, H.Y.; Hyun, K.; Jang, S.Y.; Kim, J.W.; Kim, M.W.; Jung, Y.; Jang, E.; Yoon, S.-J.; et al. Polyunsaturated Fatty Acid Biosynthesis Pathway Determines Ferroptosis Sensitivity in Gastric Cancer. Proc. Natl. Acad. Sci. USA 2020, 117, 32433–32442. [Google Scholar] [CrossRef]
- Vriens, K.; Christen, S.; Parik, S.; Broekaert, D.; Yoshinaga, K.; Talebi, A.; Dehairs, J.; Escalona-Noguero, C.; Schmieder, R.; Cornfield, T.; et al. Evidence for an Alternative Fatty Acid Desaturation Pathway Increasing Cancer Plasticity. Nat. Cell Biol. 2019, 566, 403–406. [Google Scholar] [CrossRef] [PubMed]
- Magtanong, L.; Ko, P.-J.; To, M.; Cao, J.Y.; Forcina, G.C.; Tarangelo, A.; Ward, C.C.; Cho, K.; Patti, G.J.; Nomura, D.K.; et al. Exogenous Monounsaturated Fatty Acids Promote a Ferroptosis-Resistant Cell State. Cell Chem. Biol. 2019, 26, 420–432.e9. [Google Scholar] [CrossRef]
- Ubellacker, J.M.; Tasdogan, A.; Ramesh, V.; Shen, B.; Mitchell, E.C.; Martin-Sandoval, M.S.; Gu, Z.; McCormick, M.L.; Durham, A.B.; Spitz, D.R.; et al. Lymph Protects Metastasizing Melanoma Cells from Ferroptosis. Nat. Cell Biol. 2020, 585, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Kamphorst, J.J.; Cross, J.R.; Fan, J.; De Stanchina, E.; Mathew, R.; White, E.P.; Thompson, C.B.; Rabinowitz, J.D. Hypoxic and Ras-Transformed Cells Support Growth by Scavenging Unsaturated Fatty Acids from Lysophospholipids. Proc. Natl. Acad. Sci. USA 2013, 110, 8882–8887. [Google Scholar] [CrossRef] [Green Version]
- Abumrad, N.; Coburn, C.; Ibrahimi, A. Membrane Proteins Implicated in Long-Chain Fatty Acid Uptake by Mammalian Cells: CD36, FATP and FABPm. Biochim. et Biophys. Acta (BBA) Mol. Cell Biol. Lipids 1999, 1441, 4–13. [Google Scholar] [CrossRef]
- Glatz, J.F.C.; Luiken, J.J.F.P.; Bonen, A. Membrane Fatty Acid Transporters as Regulators of Lipid Metabolism: Implications for Metabolic Disease. Physiol. Rev. 2010, 90, 367–417. [Google Scholar] [CrossRef] [Green Version]
- Dyall, S.C.; Michael-Titus, A.T. Neurological Benefits of Omega-3 Fatty Acids. Neuro Mol. Med. 2008, 10, 219–235. [Google Scholar] [CrossRef]
- Hangauer, M.J.; Viswanathan, V.S.; Ryan, M.J.; Bole, D.; Eaton, J.K.; Matov, A.; Galeas, J.; Dhruv, H.D.; Berens, M.E.; Schreiber, S.L.; et al. Drug-Tolerant Persister Cancer Cells Are Vulnerable to GPX4 Inhibition. Nat. Cell Biol. 2017, 551, 247–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viswanathan, V.S.; Ryan, M.J.; Dhruv, H.D.; Gill, S.; Eichhoff, O.M.; Seashore-Ludlow, B.; Kaffenberger, S.D.; Eaton, J.K.; Shimada, K.; Aguirre, A.J.; et al. Dependency of a Therapy-Resistant State of Cancer Cells on a Lipid Peroxidase Pathway. Nat. Cell Biol. 2017, 547, 453–457. [Google Scholar] [CrossRef]
- Fernández, L.P.; De Cedrón, M.G.; De Molina, A.R. Alterations of Lipid Metabolism in Cancer: Implications in Prognosis and Treatment. Front. Oncol. 2020, 10, 577420. [Google Scholar] [CrossRef]
- Li, D.; Li, Y. The Interaction between Ferroptosis and Lipid Metabolism in Cancer. Signal Transduct. Target. Ther. 2020, 5, 1–10. [Google Scholar] [CrossRef]
- Nath, A.; Li, I.; Roberts, L.R.; Chan, C. Elevated Free Fatty Acid Uptake via CD36 Promotes Epithelial-Mesenchymal Transition in Hepatocellular Carcinoma. Sci. Rep. 2015, 5, 14752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascual, G.; Avgustinova, A.; Mejetta, S.; Martín, M.; Castellanos, A.; Attolini, C.S.-O.; Berenguer, A.; Prats, N.; Toll, A.; Hueto, J.A.; et al. Targeting Metastasis-Initiating Cells through the Fatty Acid Receptor CD36. Nat. Cell Biol. 2017, 541, 41–45. [Google Scholar] [CrossRef]
- Watt, M.J.; Clark, A.K.; Selth, L.A.; Haynes, V.R.; Lister, N.; Rebello, R.; Porter, L.H.; Niranjan, B.; Whitby, S.T.; Lo, J.; et al. Suppressing Fatty Acid Uptake Has Therapeutic Effects in Preclinical Models of Prostate Cancer. Sci. Transl. Med. 2019, 11, eaau5758. [Google Scholar] [CrossRef]
- Kuda, O.; Jenkins, C.M.; Skinner, J.R.; Moon, S.H.; Su, X.; Gross, R.W.; Abumrad, N.A. CD36 Protein Is Involved in Store-operated Calcium Flux, Phospholipase A2 Activation, and Production of Prostaglandin E2. J. Biol. Chem. 2011, 286, 17785–17795. [Google Scholar] [CrossRef] [Green Version]
- Al-Khami, A.A.; Zheng, L.; Del Valle, L.; Hossain, F.; Wyczechowska, D.; Zabaleta, J.; Sanchez, M.D.; Dean, M.J.; Rodriguez, P.C.; Ochoa, A.C. Exogenous Lipid Uptake Induces Metabolic and Functional Reprogramming of Tumor-Associated Myeloid-Derived Suppressor Cells. OncoImmunology 2017, 6, e1344804. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Chaudhary, O.; Rodríguez-Morales, P.; Sun, X.; Zappasodi, R.; Xu, Z.; Pinto, A.F.M.; Williams, A.; Chen, D.; Low, J.S.; et al. Oxidized Lipids and CD36-Mediated Lipid Peroxidation in CD8 T Cells Suppress Anti-Tumor Immune Responses. bioRxiv 2020. [Google Scholar] [CrossRef]
- Veglia, F.; Tyurin, V.A.; Blasi, M.; De Leo, A.; Kossenkov, A.V.; Donthireddy, L.; To, T.K.J.; Schug, Z.; Basu, S.; Wang, F.; et al. Fatty Acid Transport Protein 2 Reprograms Neutrophils in Cancer. Nat. Cell Biol. 2019, 569, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, P.C.; Hernandez, C.P.; Quiceno, D.; Dubinett, S.M.; Zabaleta, J.; Ochoa, J.B.; Gilbert, J.; Ochoa, A.C. Arginase I in Myeloid Suppressor Cells Is Induced by COX-2 in Lung Carcinoma. J. Exp. Med. 2005, 202, 931–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, B.; Chen, X.-B.; Hong, Y.-C.; Zhu, H.; He, Q.-J.; Yang, B.; Ying, M.-D.; Cao, J. Identification of PRDX6 as a Regulator of Ferroptosis. Acta Pharmacol. Sin. 2019, 40, 1334–1342. [Google Scholar] [CrossRef]
- Beharier, O.; Tyurin, V.A.; Goff, J.P.; Guerrero-Santoro, J.; Kajiwara, K.; Chu, T.; Tyurina, Y.Y.; Croix, C.M.S.; Wallace, C.T.; Parry, S.; et al. PLA2G6 Guards Placental Trophoblasts against Ferroptotic Injury. Proc. Natl. Acad. Sci. USA 2020, 117, 27319–27328. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Metabolic Control of Cell Death. Science 2014, 345, 1250256. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A Nutrient and Energy Sensor That Maintains Energy Homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.; Zandkarimi, F.; Zhang, Y.; Meena, J.K.; Kim, J.; Zhuang, L.; Tyagi, S.; Ma, L.; Westbrook, T.F.; Steinberg, G.R.; et al. Energy-Stress-Mediated AMPK Activation Inhibits Ferroptosis. Nat. Cell Biol. 2020, 22, 225–234. [Google Scholar] [CrossRef]
- Li, C.; Dong, X.; Du, W.; Shi, X.; Chen, K.; Zhang, W.; Gao, M. LKB1-AMPK Axis Negatively Regulates Ferroptosis by Inhibiting Fatty Acid Synthesis. Signal Transduct. Target. Ther. 2020, 5, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Zhu, S.; Chen, P.; Hou, W.; Wen, Q.; Liu, J.; Xie, Y.; Liu, J.; Klionsky, D.J.; Kroemer, G.; et al. AMPK-Mediated BECN1 Phosphorylation Promotes Ferroptosis by Directly Blocking System Xc–Activity. Curr. Biol. 2018, 28, 2388–2399.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forcina, G.C.; Dixon, S.J. GPX4 at the Crossroads of Lipid Homeostasis and Ferroptosis. Proteomics 2019, 19, e1800311. [Google Scholar] [CrossRef]
- Zielinski, Z.A.M.; Pratt, D.A. Cholesterol Autoxidation Revisited: Debunking the Dogma Associated with the Most Vilified of Lipids. J. Am. Chem. Soc. 2016, 138, 6932–6935. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.P.; Geiger, P.G.; Maiorino, M.; Ursini, F.; Girotti, A.W. Enzymatic Reduction of Phospholipid and Cholesterol Hydroperoxides in Artificial Bilayers and Lipoproteins. Biochim. Biophys. Acta (BBA) Lipids Lipid Metab. 1990, 1045, 252–260. [Google Scholar] [CrossRef]
- Warner, G.J.; Berry, M.J.; Moustafa, M.E.; Carlson, B.A.; Hatfield, D.L.; Faust, J.R. Inhibition of Selenoprotein Synthesis by Selenocysteine tRNA [Ser] Sec Lacking Isopentenyladenosine. J. Biol. Chem. 2000, 275, 28110–28119. [Google Scholar] [CrossRef] [Green Version]
- Clendening, J.W.; Penn, L.Z. Targeting Tumor Cell Metabolism with Statins. Oncogene 2012, 31, 4967–4978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimada, K.; Skouta, R.; Kaplan, A.; Yang, W.S.; Hayano, M.; Dixon, S.J.; Brown, L.M.; Valenzuela, C.A.; Wolpaw, A.J.; Stockwell, B.R. Global Survey of Cell Death Mechanisms Reveals Metabolic Regulation of Ferroptosis. Nat. Chem. Biol. 2016, 12, 497–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Bermudez, J.; Baudrier, L.; Bayraktar, E.C.; Shen, Y.; La, K.; Guarecuco, R.; Yucel, B.; Fiore, D.; Tavora, B.; Freinkman, E.; et al. Squalene Accumulation in Cholesterol Auxotrophic Lymphomas Prevents Oxidative Cell Death. Nat. Cell Biol. 2019, 567, 118–122. [Google Scholar] [CrossRef]
- Mao, L.; Zhao, T.; Song, Y.; Lin, L.; Fan, X.; Cui, B.; Feng, H.; Wang, X.; Yu, Q.; Zhang, J.; et al. The Emerging Role of Ferroptosis in Non-cancer Liver Diseases: Hype or Increasing Hope? Cell Death Dis. 2020, 11, 1–12. [Google Scholar] [CrossRef]
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular Mechanisms and Health Implications. Cell Res. 2021, 31, 107–125. [Google Scholar] [CrossRef] [PubMed]
- Conrad, M.; Kagan, V.E.; Bayir, H.; Pagnussat, G.C.; Head, B.; Traber, M.G.; Stockwell, B.R. Regulation of Lipid Peroxidation and Ferroptosis in Diverse Species. Genes Dev. 2018, 32, 602–619. [Google Scholar] [CrossRef] [Green Version]
- Bebber, C.M.; Müller, F.; Prieto Clemente, L.; Weber, J.; Von Karstedt, S. Ferroptosis in Cancer Cell Biology. Cancers 2020, 12, 164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, T.-J.; Park, J.H.; Lee, G.S.; Lee, J.-Y.; Shin, J.H.; Kim, M.W.; Kim, Y.S.; Kim, J.-Y.; Oh, K.-J.; Han, B.-S.; et al. Quantitative Proteomic Analyses Reveal That GPX4 Downregulation during Myocardial Infarction Contributes to Ferroptosis in Cardiomyocytes. Cell Death Dis. 2019, 10, 835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Ou, Z.; Chen, R.; Niu, X.; Chen, D.; Kang, R.; Tang, D. Activation of the p62-keap1-NRF2 Pathway Protects against Ferroptosis in Hepatocellular Carcinoma Cells. Hepatology 2016, 63, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zhang, Z.; Tsai, H.-I.; Liu, Y.; Gao, J.; Wang, M.; Song, L.; Cao, X.; Xu, Z.; Chen, H.; et al. Branched-Chain Amino Acid Aminotransferase 2 Regulates Ferroptotic Cell Death in Cancer Cells. Cell Death Differ. 2020, 1–15. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.-Y.; Kim, W.K.; Bae, K.-H.; Lee, S.C.; Lee, E.-W. Lipid Metabolism and Ferroptosis. Biology 2021, 10, 184. https://doi.org/10.3390/biology10030184
Lee J-Y, Kim WK, Bae K-H, Lee SC, Lee E-W. Lipid Metabolism and Ferroptosis. Biology. 2021; 10(3):184. https://doi.org/10.3390/biology10030184
Chicago/Turabian StyleLee, Ji-Yoon, Won Kon Kim, Kwang-Hee Bae, Sang Chul Lee, and Eun-Woo Lee. 2021. "Lipid Metabolism and Ferroptosis" Biology 10, no. 3: 184. https://doi.org/10.3390/biology10030184
APA StyleLee, J. -Y., Kim, W. K., Bae, K. -H., Lee, S. C., & Lee, E. -W. (2021). Lipid Metabolism and Ferroptosis. Biology, 10(3), 184. https://doi.org/10.3390/biology10030184