


Metabolic Reprogramming toward Aerobic Glycolysis and the Gut Microbiota Involved in the Brain Amyloid Pathology


{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Amyloid Aggregation in Alzheimer’s Disease
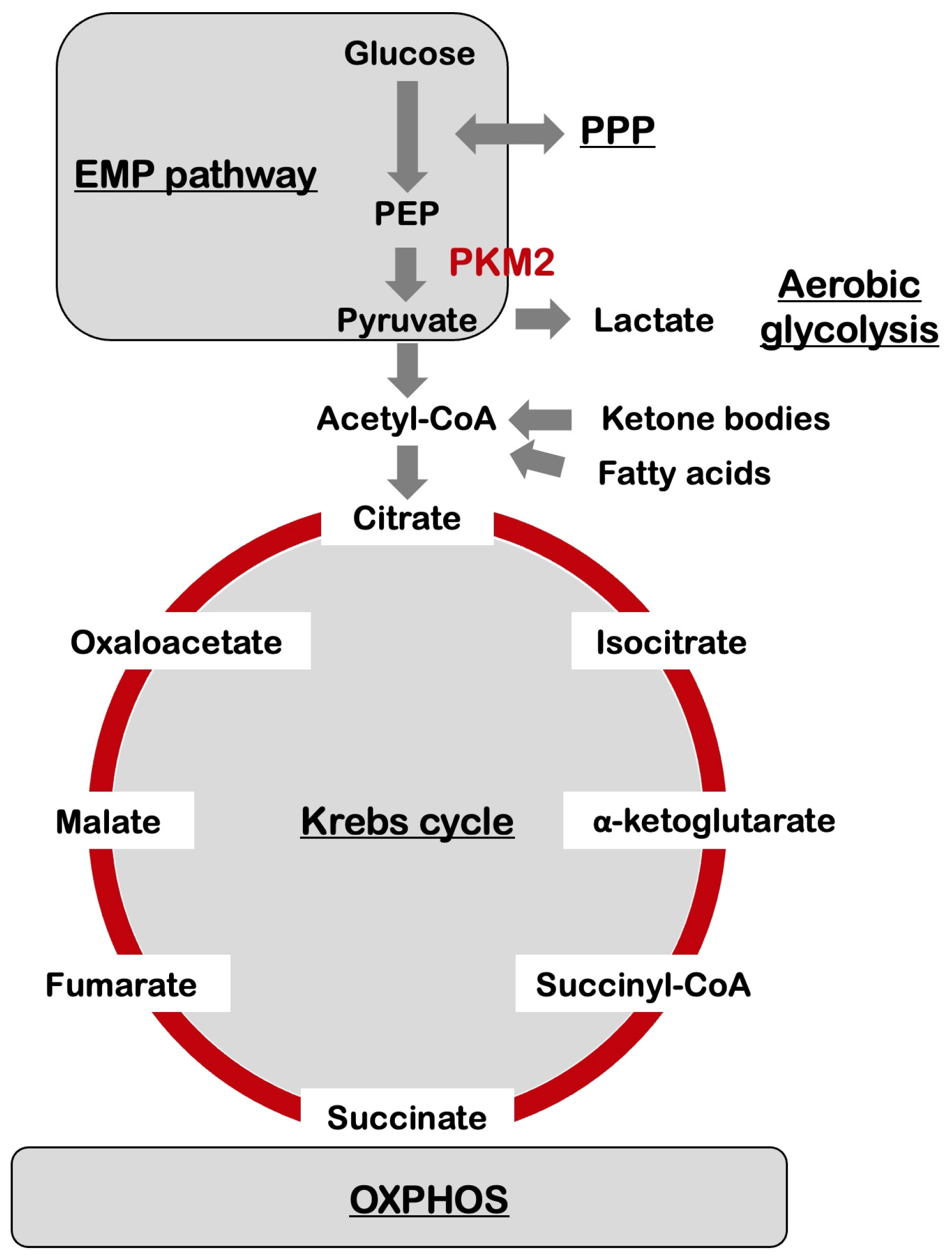
3. Metabolic Reprogramming in the Brain
4. Metabolic Reprogramming and Alzheimer’s Disease
5. Oxidative Stress and Its Connection to Alzheimer’s Disease
6. Microbiome-Microglia Interactions in Alzheimer’s Disease
7. Amyloid-β and Aerobic Glycolysis in Alzheimer’s Disease
8. Nutrition-Based Interventions for the Control of Alzheimer’s Disease and Amyloid Pathology
9. Conclusions
10. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Aβ | amyloid-β |
| AD | Alzheimer’s disease |
| ANLS | astrocyte-neuron-lactate shuttle |
| APP | amyloid precursor protein |
| ATP | adenosine 5′-triphosphate |
| CoA | coenzyme A |
| CSF | cerebrospinal fluid |
| DHAP | dihydroxyacetone phosphate |
| ETC | electron transport chain |
| FADH2 | flavin adenine dinucleotide |
| GI tract | gastrointestinal tract |
| JNK | c-Jun N-terminal kinase |
| MAPK | mitogen-activated protein kinase |
| mTOR | mammalian target of rapamycin |
| NADH | nicotinamide adenine dinucleotide |
| NADPH | nicotineamide adenine dinucleotide phosphate |
| NFT | neurofibrillary tangles |
| OXPHOS | oxidative phosphorylation |
| PEP | phosphoenolpyruvate |
| PI3K | phosphatidylinositol-3 kinase |
| PKM1 | pyruvate kinase M1 |
| PKM2 | pyruvate kinase, muscle, M2 isoform |
| PPP | pentose phosphate pathway |
| PTEN | phosphatase and tensin homolog |
| SCFA | short-chain fatty acid |
| SP | senile plaque |
| STAT3 | signal transducer and activator of tran-scription 3 |
| TCA | tricarboxylic acid |
References
- Avan, A.; Hachinski, V. Global, regional, and national trends of dementia incidence and risk factors, 1990-2019: A Global Burden of Disease study. Alzheimer’s Dement. 2023, 19, 1281–1291. [Google Scholar] [CrossRef] [PubMed]
- Balusu, S.; Praschbergerm, R.; Lauwers, E.; De Strooper, B.; Verstreken, P. Neurodegeneration cell per cell. Neuron 2023, 111, 767–786. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.M., 3rd; Cookson, M.R.; Van Den Bosch, L.; Zetterberg, H.; Holtzman, D.M.; Dewachter, I. Hallmarks of neurodegenerative diseases. Cell 2023, 186, 693–714. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.C.B.; Dammerm, E.B.; Duongm, D.M.; Pingm, L.; Zhoum, M.; Yinm, L.; Higginbothamm, L.A.; Guajardom, A.; Whitem, B.; Troncosom, J.C.; et al. Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat. Med. 2020, 26, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Dubois, B.; Feldman, H.H.; Jacova, C.; Hampel, H.; Molinuevo, J.L.; Blennow, K.; DeKosky, S.T.; Gauthier, S.; Selkoe, D.; Bateman, R.; et al. Advancing research diagnostic criteria for Alzheimer’s disease: The IWG-2 criteria. Lancet Neurol. 2014, 13, 614–629. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardie, D.G. 100 years of the Warburg effect: A historical perspective. Endocr. Relat. Cancer 2022, 29, T1–T13. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- DeBerardinis, R.J.; Chandel, N.S. We need to talk about the Warburg effect. Nat. Metab. 2020, 2, 127–129. [Google Scholar] [CrossRef]
- Bas-Orth, C.; Tan, Y.W.; Lau, D.; Bading, H. Synaptic activity drives a genomic program that promotes a neuronal Warburg effect. J. Biol. Chem. 2017, 292, 5183–5194. [Google Scholar] [CrossRef] [Green Version]
- Camandola, S.; Mattson, M.P. Brain metabolism in health, aging, and neurodegeneration. EMBO J. 2017, 36, 1474–1492. [Google Scholar] [CrossRef]
- Li, H.; Guglielmettim, C.; Seim, Y.J.; Zilberterm, M.; Le Pagem, L.M.; Shieldsm, L.; Yangm, J.; Nguyenm, K.; Tiretm, B.; Gaom, X.; et al. Neurons require glucose uptake and glycolysis in vivo. Cell Rep. 2023, 42, 112335. [Google Scholar] [CrossRef] [PubMed]
- Harper, M.E.; Patti, M.E. Metabolic terminology: What’s in a name? Nat. Metab. 2020, 2, 476–477. [Google Scholar] [CrossRef]
- Medina, M.Á. Metabolic reprogramming is a hallmark of metabolism itself. Bioessays 2020, 42, e2000058. [Google Scholar] [CrossRef]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic reprogramming and cancer progression. Science 2020, 368, eaaw5473. [Google Scholar] [CrossRef]
- Segarra-Mondejar, M.; Casellas-Díaz, S.; Ramiro-Pareta, M.; Müller-Sánchez, C.; Martorell-Riera, A.; Hermelo, I.; Reina, M.; Aragonés, J.; Martínez-Estrada, O.M.; Soriano, F.X. Synaptic activity-induced glycolysis facilitates membrane lipid provision and neurite outgrowth. EMBO J. 2018, 37, e97368. [Google Scholar] [CrossRef] [PubMed]
- Goyal, M.S.; Blazey, T.; Metcalf, N.V.; McAvoy, M.P.; Strain, J.F.; Rahmani, M.; Durbin, T.J.; Xiong, C.; Benzinger, T.L.; Morris, J.C.; et al. Brain aerobic glycolysis and resilience in Alzheimer disease. Proc. Natl. Acad. Sci. USA 2023, 120, e2212256120. [Google Scholar] [CrossRef] [PubMed]
- Traxler, L.; Herdy, J.R.; Stefanoni, D.; Eichhorner, S.; Pelucchi, S.; Szücs, A.; Santagostino, A.; Kim, Y.; Agarwal, R.K.; Schlachetzki, J.C.M.; et al. Warburg-like metabolic transformation underlies neuronal degeneration in sporadic Alzheimer’s disease. Cell Metab. 2022, 34, 1248–1263.e6. [Google Scholar] [CrossRef]
- Christofk, H.R.; Vander Heiden, M.G.; Harris, M.H.; Ramanathan, A.; Gerszten, R.E.; Wei, R.; Fleming, M.D.; Schreiber, S.L.; Cantley, L.C. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008, 452, 230–233. [Google Scholar] [CrossRef]
- Mazurek, S.; Boschek, C.B.; Hugo, F.; Eigenbrodt, E. Pyruvate kinase type M2 and its role in tumor growth and spreading. Semin. Cancer Biol. 2005, 15, 300–308. [Google Scholar] [CrossRef]
- Victor, M.B.; Tsai, L.H. Walking the high wire: How neurons maintain stability in the crossline of neurodegeneration. Cell Metab. 2022, 34, 1227–1229. [Google Scholar] [CrossRef]
- Steták, A.; Veress, R.; Ovádi, J.; Csermely, P.; Kéri, G.; Ullrich, A. Nuclear translocation of the tumor marker pyruvate kinase M2 induces programmed cell death. Cancer Res. 2007, 67, 1602–1608. [Google Scholar] [CrossRef] [Green Version]
- Murdock, M.H.; Tsai, L.H. Insights into Alzheimer’s disease from single-cell genomic approaches. Nat. Neurosci. 2023, 26, 181–195. [Google Scholar] [CrossRef]
- Butterfieldm, D.A.; Halliwellm, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef]
- Murai, T.; Matsuda, S. Pleiotropic signaling by reactive oxygen species concerted with dietary phytochemicals and microbial-derived metabolites as potent therapeutic regulators of the tumor microenvironment. Antioxidants 2023, 12, 1056. [Google Scholar] [CrossRef]
- Matsuda, S.; Nakagawa, Y.; Tsuji, A.; Kitagishi, Y.; Nakanishi, A.; Murai, T. Implications of PI3K/AKT/PTEN signaling on superoxide dismutases expression and in the pathogenesis of Alzheimer’s disease. Diseases 2018, 6, 28. [Google Scholar] [CrossRef] [Green Version]
- Mossad, O.; Erny, D. The microbiota-microglia axis in central nervous system disorders. Brain Pathol. 2020, 30, 1159–1177. [Google Scholar] [CrossRef]
- Silpe, J.E.; Balskus, E.P. Deciphering human microbiota-host chemical interactions. ACS Cent. Sci. 2021, 7, 20–29. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Sierra, A.; Stevens, B.; Tremblay, M.E.; Aguzzi, A.; Ajami, B.; Amit, I.; Audinat, E.; Bechmann, I.; Bennett, M.; et al. Microglia states and nomenclature: A field at its crossroads. Neuron 2022, 110, 3458–3483. [Google Scholar] [CrossRef]
- O’Riordan, K.J.; Collins, M.K.; Moloney, G.M.; Knox, E.G.; Aburto, M.R.; Fülling, C.; Morley, S.J.; Clarke, G.; Schellekens, H.; Cryan, J.F. Short chain fatty acids: Microbial metabolites for gut-brain axis signalling. Mol. Cell. Endocrinol. 2022, 546, 111572. [Google Scholar] [CrossRef]
- Erny, D.; Dokalis, N.; Mezo, C.; Castoldi, A.; Mossad, O.; Staszewski, O.; Frosch, M.; Villa, M.; Fuchs, V.; Mayer, A.; et al. Microbiota-derived acetate enables the metabolic fitness of the brain innate immune system during health and disease. Cell Metab. 2021, 33, 2260–2276.e7. [Google Scholar] [CrossRef] [PubMed]
- Lynch, C.M.K.; Clarke, G.; Cryan, J.F. Powering up microbiome-microglia interactions. Cell Metab. 2021, 33, 2097–2099. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.L. Glucose, glycolysis, and neurodegenerative diseases. J. Cell. Physiol. 2020, 235, 7653–7662. [Google Scholar] [CrossRef] [PubMed]
- Baik, S.H.; Kang, S.; Lee, W.; Choi, H.; Chung, S.; Kim, J.I.; Mook-Jung, I. A breakdown in metabolic reprogramming causes microglia dysfunction in Alzheimer’s disease. Cell Metab. 2019, 30, 493–507.e496. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S.; Banik, A.; Saurabh, S.; Maulik, M.; Khatri, S.N. Neuroimmunometabolism: A new pathological nexus underlying neurodegenerative disorders. J. Neurosci. 2022, 42, 1888–1907. [Google Scholar] [CrossRef] [PubMed]
- Díaz, G.; Lengele, L.; Sourdet, S.; Soriano, G.; de Souto Barreto, P. Nutrients and amyloid β status in the brain: A narrative review. Ageing Res. Rev. 2022, 81, 101728. [Google Scholar] [CrossRef]
- Murai, T.; Matsuda, S. The chemopreventive effects of chlorogenic acids, phenolic compounds in coffee, against inflammation, cancer, and neurological diseases. Molecules 2023, 28, 2381. [Google Scholar] [CrossRef]
- Murakami, M.; Ikeda, Y.; Nakagawa, Y.; Tsuji, A.; Kitagishi, Y.; Matsuda, S. Special bioactive compounds and functional foods may exhibit neuroprotective effects in patients with dementia. Biomed. Rep. 2020, 13, 1. [Google Scholar]
- Cox, P.A.; Metcalf, J.S. Traditional food items in Ogimi, Okinawa: L-Serine content and the potential for neuroprotection. Curr. Nutr. Rep. 2017, 6, 24–31. [Google Scholar] [CrossRef] [Green Version]
- Myette-Côté, É.; Soto-Mota, A.; Cunnane, S.C. Ketones: Potential to achieve brain energy rescue and sustain cognitive health during ageing. Br. J. Nutr. 2022, 128, 407–423. [Google Scholar] [CrossRef]
- Ballarini, T.; Melo van Lent, D.; Brunner, J.; Schröder, A.; Wolfsgruber, S.; Altenstein, S.; Brosseron, F.; Buerger, K.; Dechent, P.; DELCODE Study Group. Mediterranean diet, Alzheimer disease biomarkers and brain atrophy in old age. Neurology 2021, 96, e2920–e2932. [Google Scholar] [CrossRef]
- Wesselman, L.M.P.; van Lent, D.M.; Schröder, A.; van de Rest, O.; Peters, O.; Menne, F.; Fuentes, M.; Priller, J.; Spruth, E.J.; Altenstein, S.; et al. Dietary patterns are related to cognitive functioning in elderly enriched with individuals at increased risk for Alzheimer’s disease. Eur. J. Nutr. 2021, 60, 849–860. [Google Scholar] [CrossRef]
- Scarmeas, N.; Stern, Y.; Tang, M.X.; Mayeux, R.; Luchsinger, J.A. Mediterranean diet and risk for Alzheimer’s disease. Ann. Neurol. 2006, 59, 912–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiltfang, J.; Esselmann, H.; Bibl, M.; Hüll, M.; Hampel, H.; Kessler, H.; Frölich, L.; Schröder, J.; Peters, O.; Jessen, F.; et al. Amyloid β peptide ratio 42/40 but not Aβ42 correlates with phospho-Tau in patients with low- and high-CSF Aβ40 load. J. Neurochem. 2007, 101, 1053–1059. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.C.; Tangney, C.C.; Wang, Y.; Sacks, F.M.; Bennett, D.A.; Aggarwal, N.T. MIND diet associated with reduced incidence of Alzheimer’s disease. Alzheimer’s Dement. 2015, 11, 1007–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yusufov, M.; Weyandt, L.L.; Piryatinsky, I. Alzheimer’s disease and diet: A systematic review. Int. J. Neurosci. 2017, 127, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Yassine, H.N.; Self, W.; Kerman, B.E.; Santoni, G.; Navalpur Shanmugam, N.; Abdullahm, L.; Goldenm, L.R.; Fontehm, A.N.; Harringtonm, M.G.; Gräff, J.; et al. Nutritional metabolism and cerebral bioenergetics in Alzheimer’s disease and related dementias. Alzheimers Dement. 2023, 19, 1041–1066. [Google Scholar] [CrossRef] [PubMed]
- Neth, B.J.; Mintzm, A.; Whitlowm, C.; Jungm, Y.; Solingapuram Saim, K.; Registerm, T.C.; Kellarm, D.; Lockhartm, S.N.; Hoscheidtm, S.; Maldjian, J.; et al. Modified ketogenic diet is associated with improved cerebrospinal fluid biomarker profile, cerebral perfusion, and cerebral ketone body uptake in older adults at risk for Alzheimer’s disease: A pilot study. Neurobiol. Aging 2020, 86, 54–63. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murai, T.; Matsuda, S. Metabolic Reprogramming toward Aerobic Glycolysis and the Gut Microbiota Involved in the Brain Amyloid Pathology. Biology 2023, 12, 1081. https://doi.org/10.3390/biology12081081
Murai T, Matsuda S. Metabolic Reprogramming toward Aerobic Glycolysis and the Gut Microbiota Involved in the Brain Amyloid Pathology. Biology. 2023; 12(8):1081. https://doi.org/10.3390/biology12081081
Chicago/Turabian StyleMurai, Toshiyuki, and Satoru Matsuda. 2023. "Metabolic Reprogramming toward Aerobic Glycolysis and the Gut Microbiota Involved in the Brain Amyloid Pathology" Biology 12, no. 8: 1081. https://doi.org/10.3390/biology12081081
APA StyleMurai, T., & Matsuda, S. (2023). Metabolic Reprogramming toward Aerobic Glycolysis and the Gut Microbiota Involved in the Brain Amyloid Pathology. Biology, 12(8), 1081. https://doi.org/10.3390/biology12081081