A New Benchmark to Determine What Healthy Western Skin Looks Like in Terms of Biodiversity Using Standardised Methodology



Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Conditions and Sampling Procedure
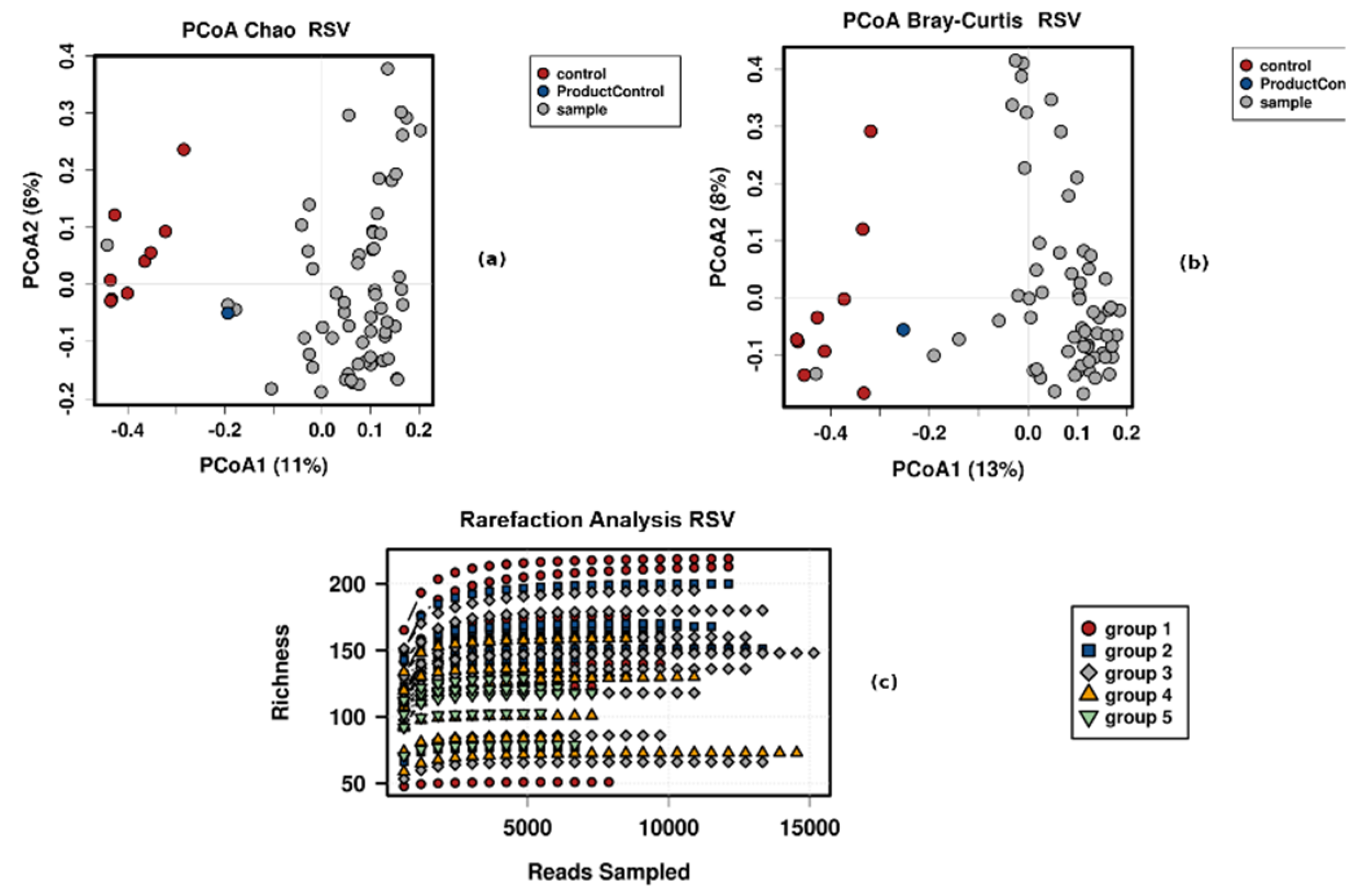
- Group 1, 18–27 years;
- Group 2, 28–37 years;
- Group 3, 38–47 years;
- Group 4, 48–57 years;
- Group 5, 58–70 years.
2.2. DNA Extraction and 16S rRNA Gene Amplification
2.3. Data Analysis
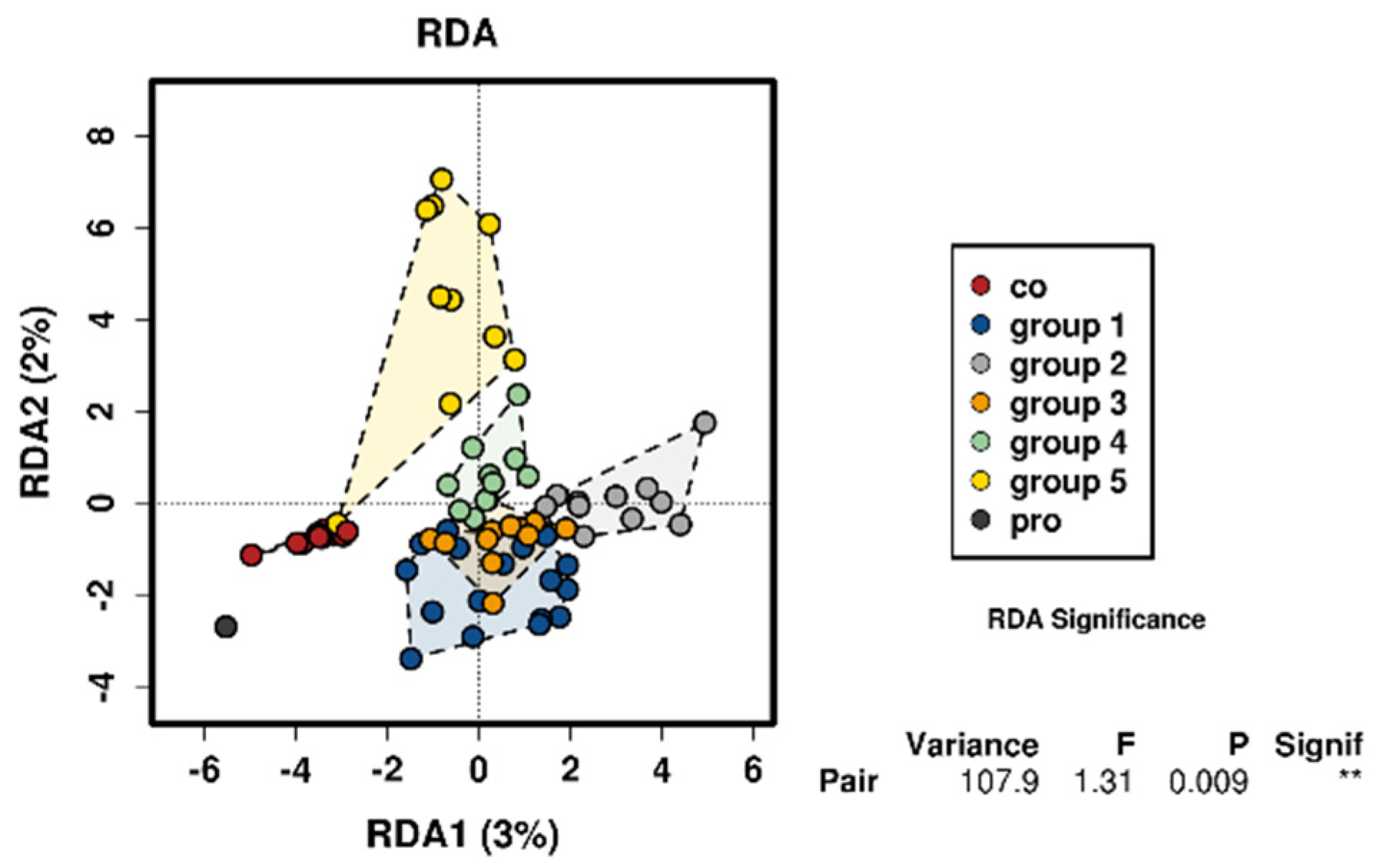
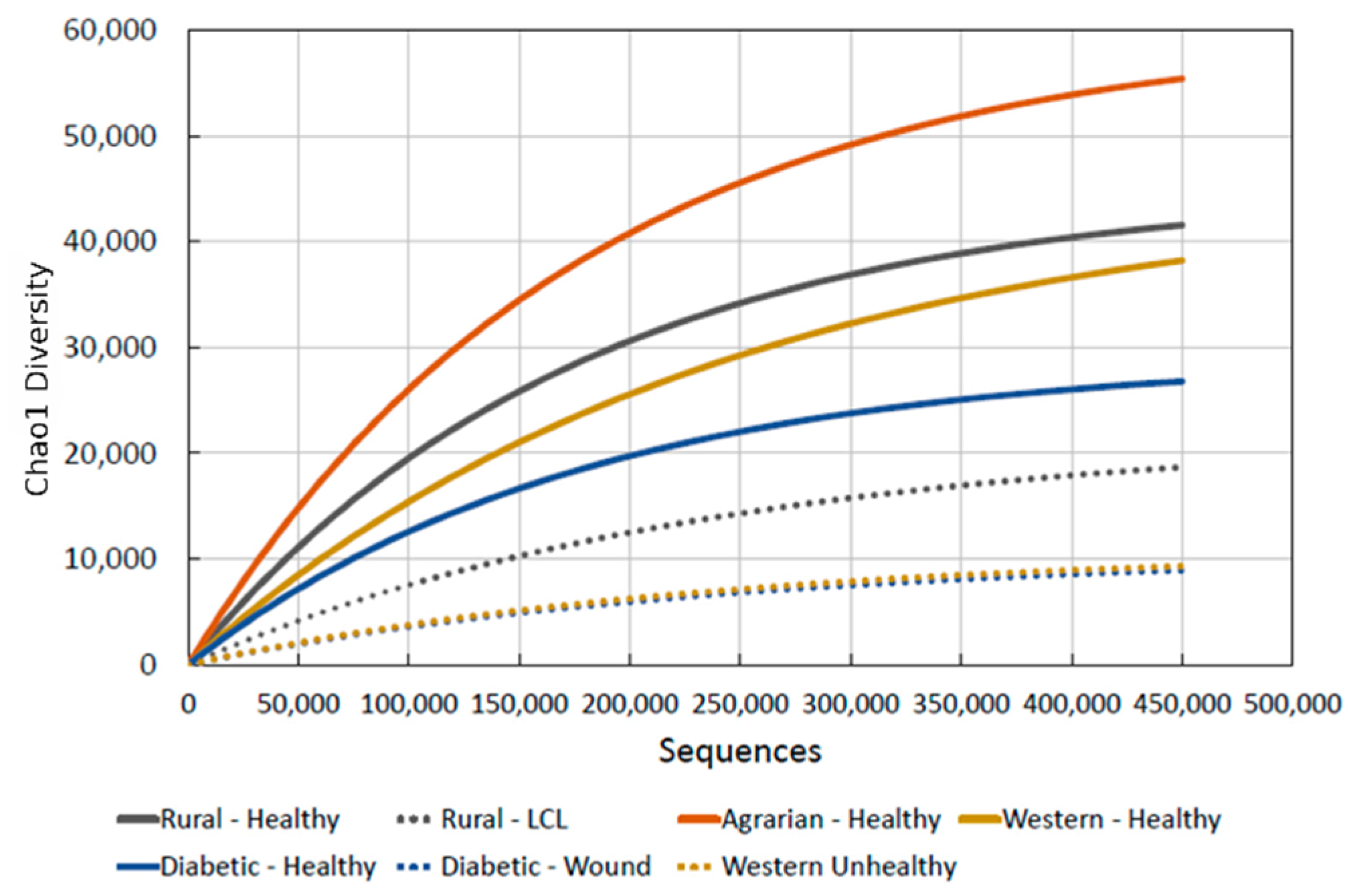
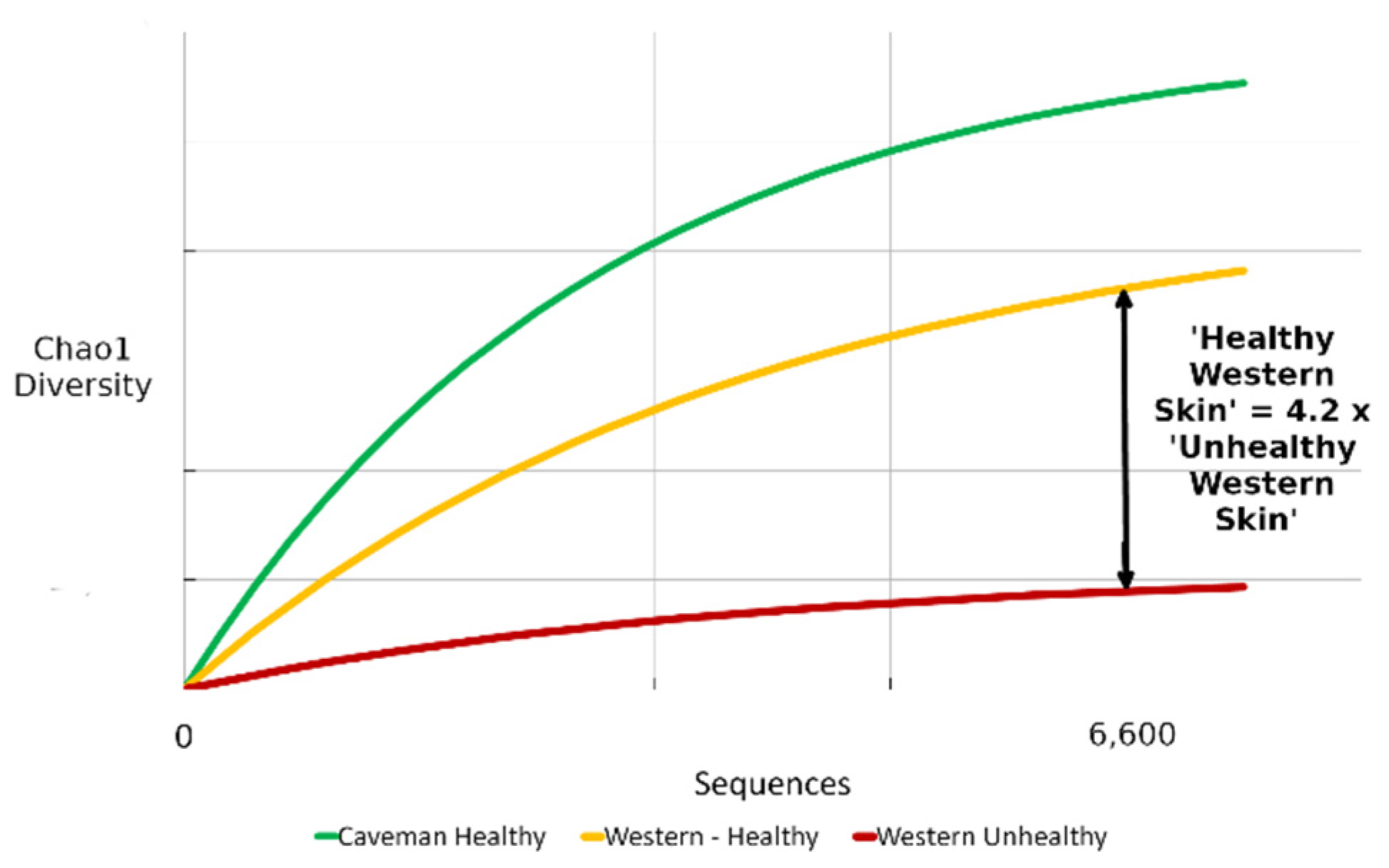
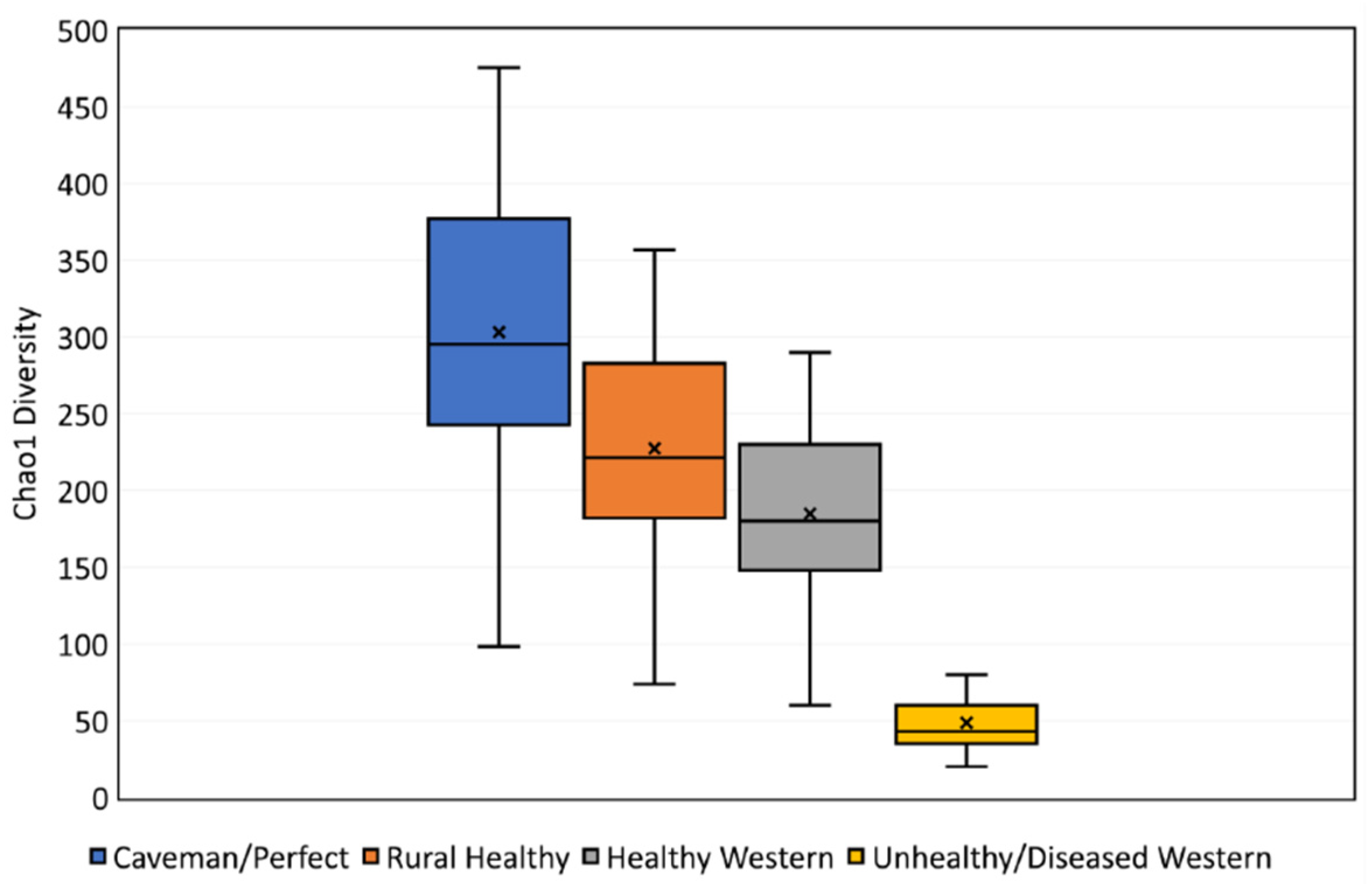
3. Results
3.1. Benchmark for Healthy Western Skin
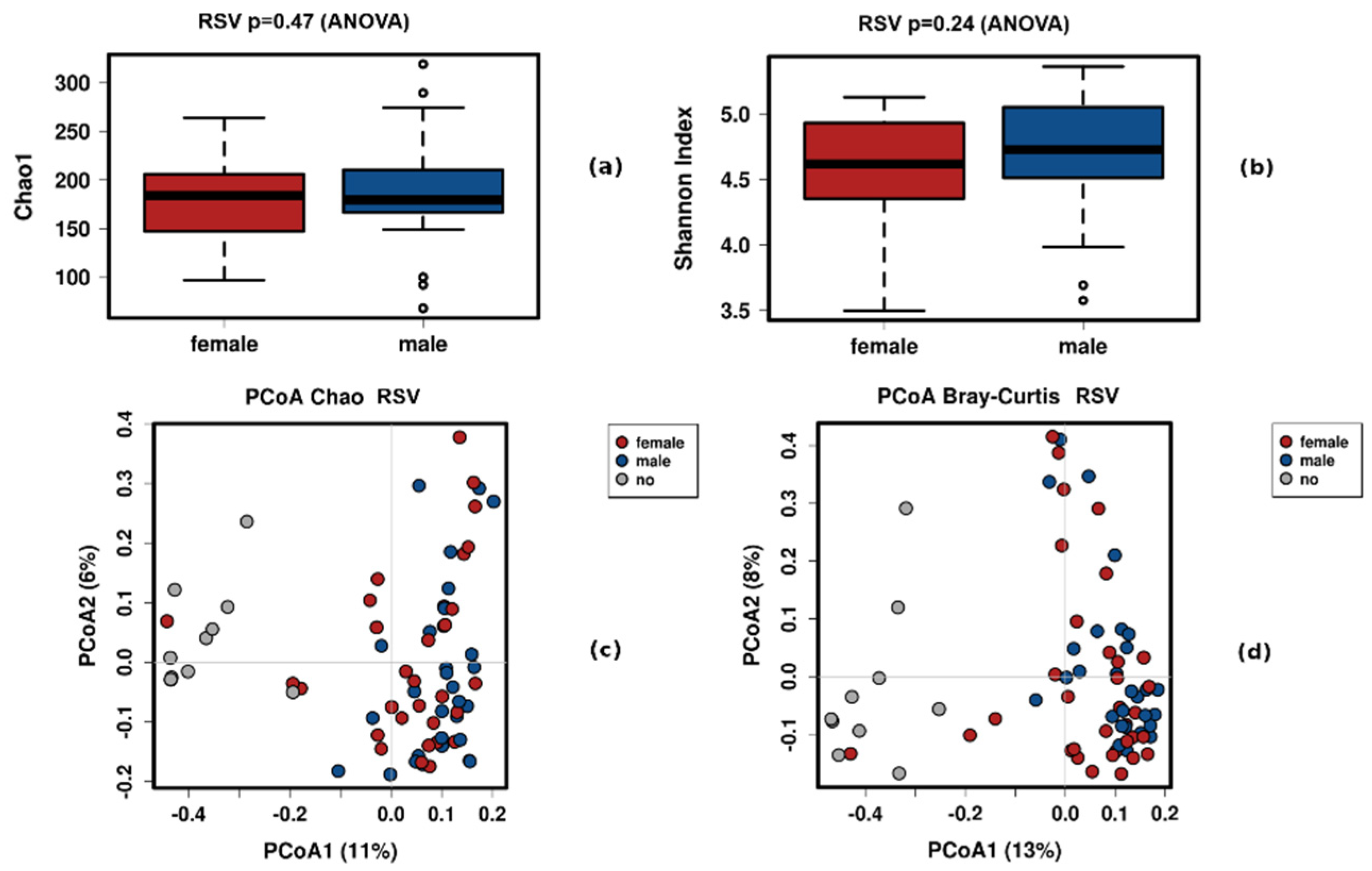
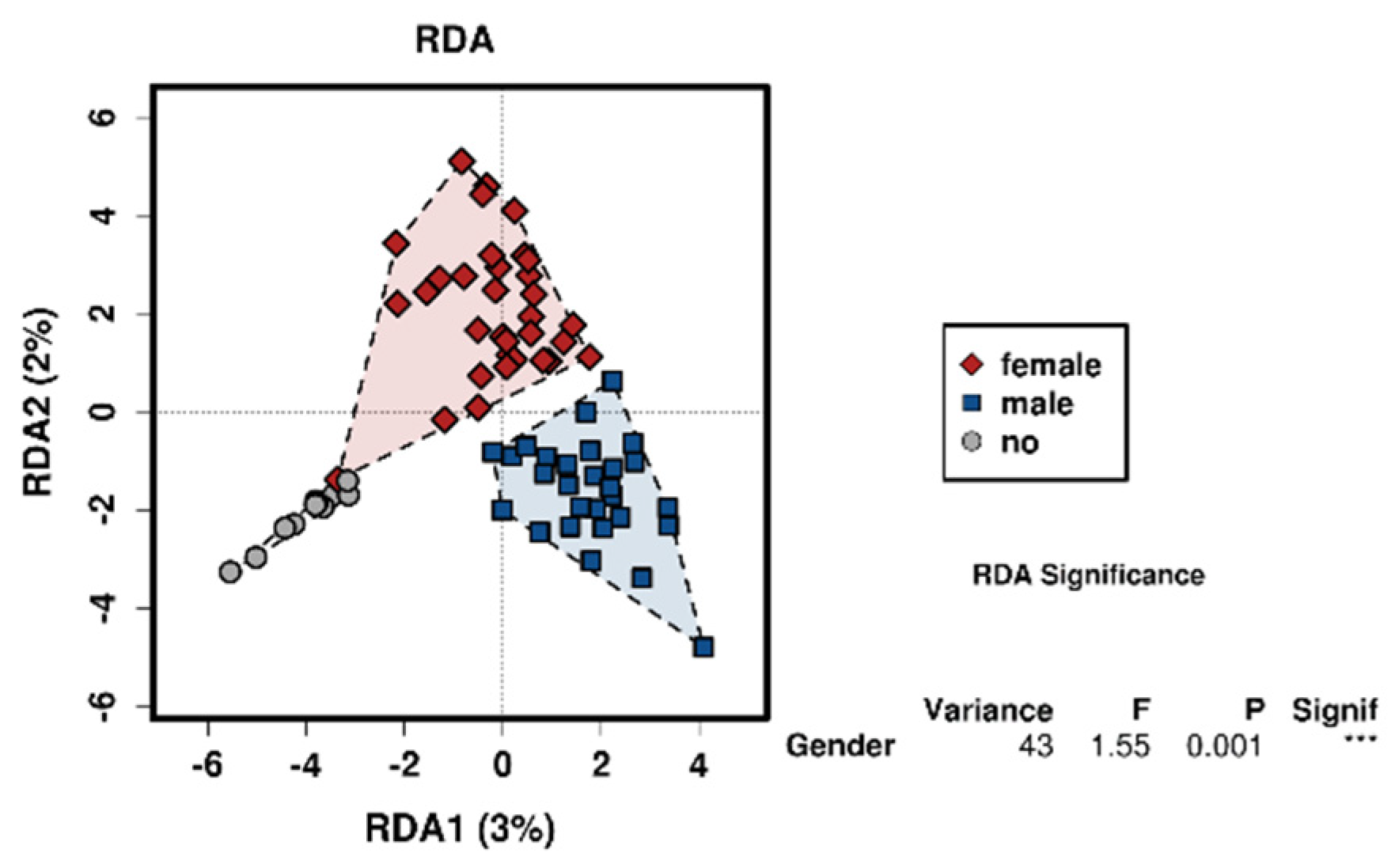
3.2. Sex
3.3. Age
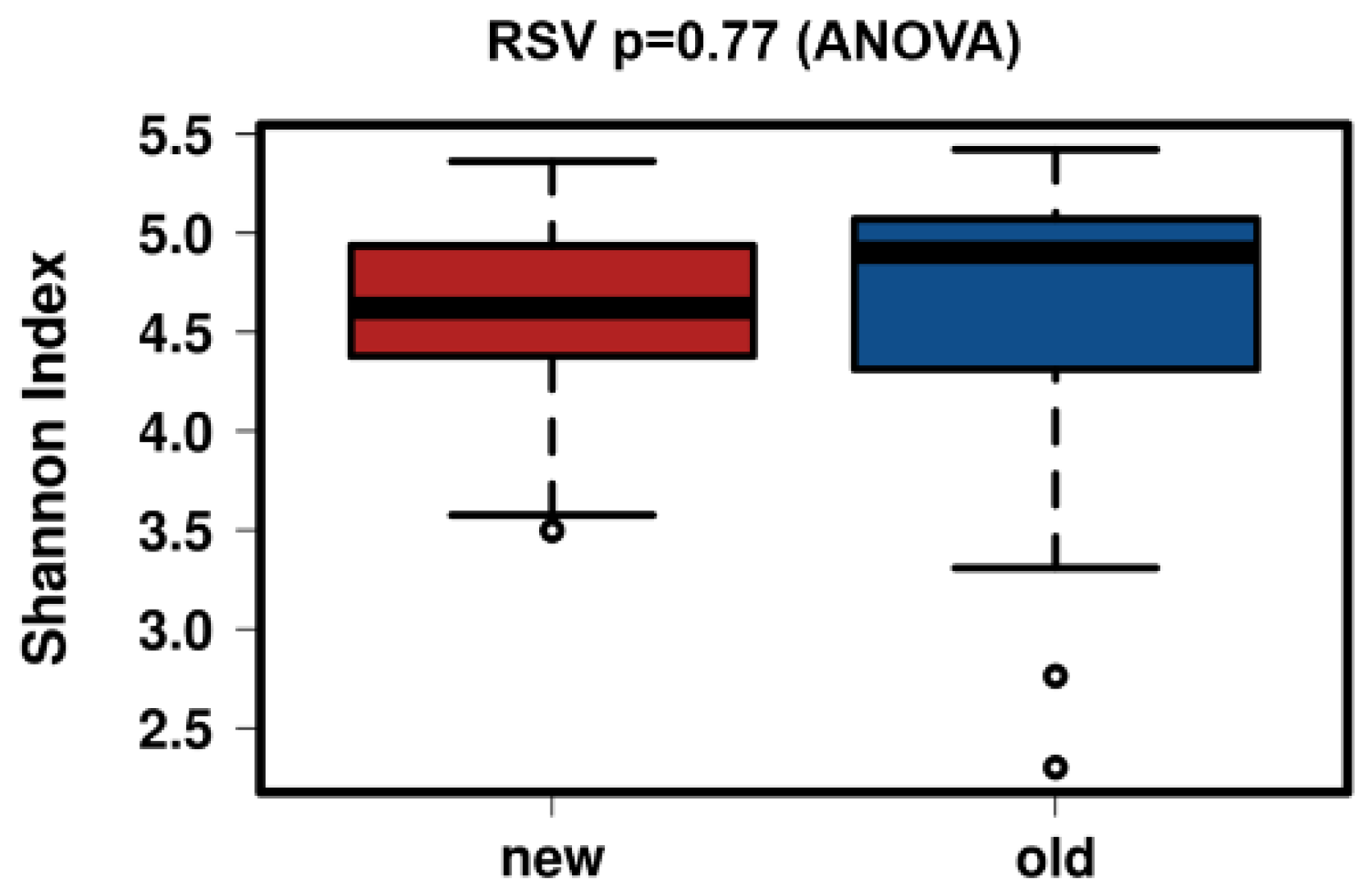
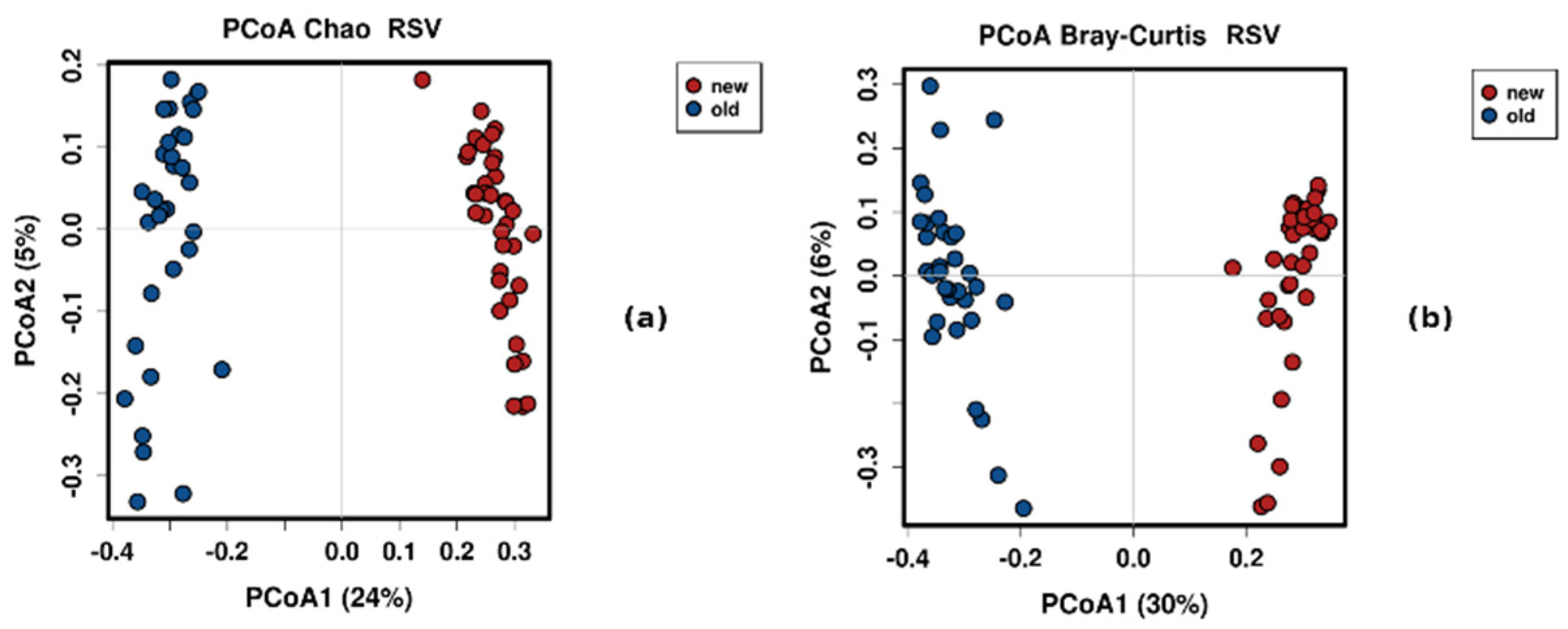
3.4. Control Testing
4. Discussion
4.1. Why the Benchmark Is Important
4.2. The Effect of Sex
4.3. The Effect of Age
4.4. Limitations and Future Work
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

References
- Hublin, J.J.; Ben-Ncer, A.; Bailey, S.E.; Freidline, S.E.; Neubauer, S.; Skinner, M.M.; Bergmann, I.; Le Cabec, A.; Benazzi, S.; Harvati, K.; et al. New fossils from Jebel Irhoud, Morocco and the pan-African origin of Homo sapiens. Nature 2017, 546, 289–292. [Google Scholar] [CrossRef] [Green Version]
- Sender, R.; Fuchs, S.; Milo, R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 2016, 14, e1002533. [Google Scholar] [CrossRef] [Green Version]
- Cogen, A.L.; Nizet, V.; Gallo, R.L. Skin microbiota: A source of disease or defence? Br. J. Dermatol. 2009, 158, 442–455. [Google Scholar] [CrossRef] [Green Version]
- Blaser, M.J.; Falkow, S. What are the consequences of the disappearing human microbiota? Nat. Rev. Microbiol. 2009, 7, 887–894. [Google Scholar] [CrossRef]
- Noble, W.C. Staphylococci on the skin. In The Skin Microflora and Microbial Skin Disease; Noble, W.C., Ed.; Cambridge University Press: London, UK, 2004; pp. 135–152. [Google Scholar]
- Holland, K.T.; Bojar, R.A. Cosmetics: What is their influence on the skin microflora? Am. J. Clin. Dermatol. 2002, 3, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Grice, E.A.; Kong, H.H.; Renaud, G.; Young, A.C.; Bouffard, G.G.; Blakesley, R.W.; Wolfsberg, T.G.; Turner, M.L.; Segre, J.A.; Program, N.C.S. A diversity profile of the human skin microbiota. Genome Res. 2008, 18, 1043–1050. [Google Scholar] [CrossRef] [Green Version]
- Sanford, J.A.; Gallo, R.L. Functions of the skin microbiota in health and disease. Semin. Immunol. 2013, 25, 370–377. [Google Scholar] [CrossRef] [Green Version]
- Barnard, E.; Li, H. Shaping of cutaneous function by encounters with commensals. J. Physiol. 2016, 595, 437–450. [Google Scholar] [CrossRef] [Green Version]
- Pamer, E.G. Immune responses to commensal and environmental microbes. Nat. Immunol. 2007, 8, 1173–1178. [Google Scholar] [CrossRef]
- Grice, E.A.; Snitkin, E.S.; Yockey, L.J.; Bermudez, D.M.; Comparative, N.; NISC Comparative Sequencing Program; Liechty, K.W.; Segre, J.A. Longitudinal shift in diabetic wound microbiota correlates with prolonged skin defense response. Proc. Natl. Acad. Sci. USA 2010, 33107, 14799–14804. [Google Scholar] [CrossRef] [Green Version]
- Kong, H.H.; Segre, J.A. Skin microbiome: Looking back to move forward. J. Investig. Dermatol. 2012, 132 Pt 2, 933–939. [Google Scholar] [CrossRef] [Green Version]
- Christensen, G.J.M.; Brüggemann, H. Bacterial skin commensals and their role as host guardians. Benef. Microbes 2014, 5, 201–215. [Google Scholar] [CrossRef] [PubMed]
- Park, B.; Iwase, T.; Liu, G.Y. Intranasal Application of S. epidermidis Prevents Colonization by Methicillin-Resistant Staphylococcus aureus in Mice. PLoS ONE 2011, 6, e25880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosebury, T. Microorganisms Indigenous to Man; McGraw Hill: New York, NY, USA, 1962. [Google Scholar]
- Mackowiak, P.A. The Normal Microbial Flora. N. Engl. J. Med. 1982, 307, 83–93. [Google Scholar] [CrossRef] [Green Version]
- Smith, K.; McCoy, K.D.; Macpherson, A.J. Use of axenic animals in studying the adaptation of mammals to their commensal intestinal microbiota. Semin. Immunol. 2007, 19, 59–69. [Google Scholar] [CrossRef]
- Gordon, H.A.; Pesti, L. The gnotobiotic animal as a tool in the study of host microbial relationships. Bacteriol. Rev. 1971, 35, 390–429. [Google Scholar] [CrossRef]
- Grice, E.A. The intersection of microbiome and host at the skin interface: Genomic- and metagenomic-based insights. Genome Res. 2015, 25, 1514–1520. [Google Scholar] [CrossRef]
- Katsuyama, M.; Ichikawa, H.; Ogawa, S.; Ikezawa, Z. A novel method to control the balance of skin microflora: Part 1. Attack on biofilm of Staphylococcus aureus without antibiotics. J. Dermatol. Sci. 2005, 38, 197–205. [Google Scholar]
- Lambers, H.; Piessens, S.; Bloem, A.; Pronk, H.; Finkel, P. Natural skin surface pH is on average below 5, which is beneficial for its resident flora. Int. J. Cosmet. Sci. 2006, 28, 359–370. [Google Scholar] [CrossRef]
- Oh, J.; Byrd, A.L.; Park, M.; Kong, H.H.; Segre, J.A.; Segre, J.A. Temporal Stability of the Human Skin Microbiome. Cell 2016, 165, 854–866. [Google Scholar] [CrossRef] [Green Version]
- Byrd, A.L.; Belkaid, Y.; Segre, J.A. The human skin microbiome. Nat. Rev. Microbiol. 2018, 16, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.; Byrd, A.L.; Deming, C.; Conlan, S.; Kong, H.H.; Segre, J.A.; NISC Comparative Sequencing Program. Biogeography and individuality shape function in the human skin metagenome. Nature 2014, 514, 59–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ley, R.E.; Lozupone, C.A.; Hamady, M.; Knight, R.; Gordon, J.I. Worlds within worlds: Evolution of the vertebrate gut microbiota. Nat. Rev. Microbiol. 2008, 6, 776–788. [Google Scholar] [CrossRef] [Green Version]
- Pei, Z.; Bini, E.J.; Yang, L.; Zhou, M.; Francois, F.; Blaser, M.J. Bacterial biota in the human distal esophagus. Proc. Natl. Acad. Sci. USA 2004, 101, 4250–4255. [Google Scholar] [CrossRef] [Green Version]
- Dessinioti, C.; Katsambas, A.D. The role of Propionibacterium acnes in acne pathogenesis: Facts and controversies. Clin. Dermatol. 2010, 28, 2–7. [Google Scholar] [CrossRef]
- Gao, Z.; Tseng, C.; Strober, B.E.; Pei, Z.; Blaser, M.J. Substantial Alterations of the Cutaneous Bacterial Biota in Psoriatic Lesions. PLoS ONE 2008, 3, e2719. [Google Scholar] [CrossRef]
- Gao, Z.; Tseng, C.; Pei, Z.; Blaser, M.J. Molecular analysis of human forearm superficial skin bacterial biota. Proc. Natl. Acad. Sci. USA 2007, 104, 2927–2932. [Google Scholar] [CrossRef] [Green Version]
- Perez, G.I.P.; Gao, Z.; Jourdain, R.; Ramirez, J.; Gany, F.; Clavaud, C.; Demaude, J.; Breton, L.; Blaser, M.J. Body Site Is a More Determinant Factor than Human Population Diversity in the Healthy Skin Microbiome. PLoS ONE 2016, 11, e0151990. [Google Scholar] [CrossRef] [Green Version]
- Kong, H.H.; Oh, J.; Deming, C.; Conlan, S.; Grice, E.A.; Beatson, M.A.; Nomicos, E.; Polley, E.C.; Komarow, H.D.; Murray, P.R.; et al. Temporal shifts in the skin microbiome associated with disease flares and treatment in children with atopic dermatitis. Genome Res. 2012, 22, 850–859. [Google Scholar] [CrossRef] [Green Version]
- Meron, E. Nonlinear Physics of Ecosystems, 1st ed.; CRC Press: Boca Raton, FL, USA, 2015. [Google Scholar]
- Goerner, S.J. Chaos and the Evolving Ecological Universe; Gordon and Breach Publishers: London, UK, 1994. [Google Scholar]
- Wallen-Russell, C.; Wallen-Russell, S. Meta Analysis of Skin Microbiome: New Link between Skin Microbiota Diversity and Skin Health with Proposal to Use This as a Future Mechanism to Determine Whether Cosmetic Products Damage the Skin. Cosmetics 2017, 4, 14. [Google Scholar] [CrossRef] [Green Version]
- Finlay, B.B.; Arrieta, M.-C. Let Them Eat Dirt. In Saving Your Child from an Oversanitized World; Windmill Books: London, UK, 2016. [Google Scholar]
- Lefcheck, J.S.; Byrnes, J.E.K.; Isbell, F.; Gamfeldt, L.; Griffin, J.N.; Eisenhauer, N.; Hensel, M.J.S.; Hector, A.; Cardinale, B.J.; Duffy, J.E. Biodiversity enhances ecosystem multifunctionality across trophic levels and habitats. Nat. Commun. 2015, 6, 6936. [Google Scholar] [CrossRef] [PubMed]
- Balvanera, P.; Pfisterer, A.B.; Buchmann, N.; He, J.-S.; Nakashizuka, T.; Raffaelli, D.; Schmid, B. Quantifying the evidence for biodiversity effects on ecosystem functioning and services. Ecol. Lett. 2006, 9, 1146–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gamfeldt, L.; Hillebrand, H.; Jonsson, P.R. Multiple functions increase the importance of biodiversity for overall ecosystem functioning. Ecology 2008, 89, 1223–1231. [Google Scholar] [CrossRef] [PubMed]
- Thrupp, L.A. The importance of biodiversity in agroecosystems. J. Crop. Improv. 2004, 12, 315–337. [Google Scholar] [CrossRef]
- Hooper, D.U.; Chapin, F.S.; Ewel, J.J.; Hector, A.; Inchausti, P.; Lavorel, S.; Lawton, J.H.; Lodge, D.M.; Loreau, M.; Naeem, S.; et al. Effects of biodiversity on ecosystem functioning: A consensus of current knowledge. Ecol. Monogr. 2005, 75, 3–35. [Google Scholar] [CrossRef]
- Srinivas, G.; Möller, S.; Wang, J.; Künzel, S.; Zillikens, D.; Baines, J.F.; Ibrahim, S.M. Genome-wide mapping of gene–microbiota interactions in susceptibility to autoimmune skin blistering. Nat. Commun. 2013, 4, 2462. [Google Scholar] [CrossRef] [Green Version]
- Clemente, J.C.; Pehrsson, E.C.; Blaser, M.J.; Sandhu, K.; Gao, Z.; Wang, B.; Magris, M.; Hidalgo, G.; Contreras, M.; Noya-Alarcón, Ó.; et al. The microbiome of uncontacted Amerindians. Sci. Adv. 2015, 1, e1500183. [Google Scholar] [CrossRef] [Green Version]
- Blaser, M.J.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Estrada, I.; Gao, Z.; Clemente, J.C.; Costello, E.K.; Knight, R. Distinct cutaneous bacterial assemblages in a sampling of South American Amerindians and US residents. ISME J. 2013, 7, 85–95. [Google Scholar] [CrossRef]
- Salgado, V.R.; de Queiroz, A.T.L.; Sanabani, S.S.; de Oliveira, C.I.; Carvalho, E.M.; Costa, J.M.L.; Barral-Netto, M.; Barral, A. The microbiological signature of human cutaneous leishmaniasis lesions exhibits restricted bacterial diversity compared to healthy skin. Memórias Inst. Oswaldo Cruz 2016, 111, 241–251. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, A.R.; Patterson, A.P.; Diesel, A.; Lawhon, S.D.; Ly, H.J.; Stephenson, C.E.; Mansell, J.; Steiner, J.M.; Dowd, S.E.; Olivry, T.; et al. The skin microbiome in healthy and allergic dogs. PLoS ONE 2014, 9, e83197. [Google Scholar]
- Gardiner, M.; Vicaretti, M.; Sparks, J.; Bansal, S.; Bush, S.; Liu, M.; Darling, A.E.; Harry, E.J.; Burke, C. A longitudinal study of the diabetic skin and wound microbiome. PeerJ 2017, 5, e3543. [Google Scholar] [CrossRef] [PubMed]
- Grice, E.A.; Segre, J.A. The skin microbiome. Nat. Rev. Microbiol. 2011, 9, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Baviera, G.; Leoni, M.C.; Capra, L.; Cipriani, F.; Longo, G.; Maiello, N.; Ricci, G.; Galli, E. Microbiota in healthy skin and in atopic eczema. BioMed Res. Int. 2014, 2014, 436921. [Google Scholar] [CrossRef]
- Manasson, J.; Reddy, S.M.; Neimann, A.L.; Segal, L.N.; Scher, J.U. Cutaneous Microbiota Features Distinguish Psoriasis from Psoriatic Arthritis. Arthritis Rheumatol. 2016, 68, 1708. [Google Scholar]
- Nakatsuji, T.; Chen, T.H.; Butcher, A.M.; Trzoss, L.L.; Nam, S.-J.; Shirakawa, K.T.; Zhou, W.; Oh, J.; Otto, M.; Fenical, W.; et al. A commensal strain of Staphylococcus epidermidis protects against skin neoplasia. Sci. Adv. 2018, 4, eaao4502. [Google Scholar] [CrossRef] [Green Version]
- Velegraki, A.; Cafarchia, C.; Gaitanis, G.; Iatta, R.; Boekhout, T. Malassezia Infections in Humans and Animals: Pathophysiology, Detection, and Treatment. PLoS Pathog. 2015, 11, e1004523. [Google Scholar] [CrossRef] [Green Version]
- Prescott, S.L.; Larcombe, D.-L.; Logan, A.C.; West, C.; Burks, W.; Caraballo, L.; Levin, M.; Van Etten, E.; Horwitz, P.; Kozyrskyj, A.; et al. The skin microbiome: Impact of modern environments on skin ecology, barrier integrity, and systemic immune programming. World Allergy Organ. J. 2017, 10, 29. [Google Scholar] [CrossRef]
- Huang, Y.J.; Marsland, B.J.; Bunyavanich, S.; O’Mahony, L.; Leung, D.Y.M.; Muraro, A.; Fleisher, T.A. The microbiome in allergic disease: Current understanding and future opportunities-2017 PRACTALL document of the American Academy of Allergy, Asthma & Immunology and the European Academy of Allergy and Clinical Immunology. J. Allergy Clin. Immunol. 2017, 139, 1099–1110. [Google Scholar]
- Sherwani, M.A.; Tufail, S.; Muzaffar, A.F.; Yusuf, N. The skin microbiome and immune system: Potential target for chemoprevention? Photodermatol. Photoimmunol. Photomed. 2018, 34, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Misic, A.M.; Gardner, S.E.; Grice, E.A. The Wound Microbiome: Modern Approaches to Examining the Role of Microorganisms in Impaired Chronic Wound Healing. Adv. Wound Care 2014, 3, 502–510. [Google Scholar] [CrossRef] [Green Version]
- Lu, L.J.; Liu, J. Human Microbiota and Ophthalmic Disease. Yale J. Biol. Med. 2016, 89, 325–330. [Google Scholar]
- Zaidi, A.K.; Spaunhurst, K.; Sprockett, D.; Thomason, Y.; Mann, M.W.; Fu, P.; Ammons, C.; Gerstenblith, M.; Tuttle, M.S.; Popkin, D.L. Characterization of the facial microbiome in twins discordant for rosacea. Exp. Dermatol. 2018, 27, 295–298. [Google Scholar] [CrossRef] [PubMed]
- Findley, K.; Oh, J.; Yang, J.; Conlan, S.; Deming, C.; Meyer, J.A.; Schoenfeld, D.; Nomicos, E.; Park, M.; NIH Intramural Sequencing Center Comparative Sequencing Program; et al. Topographic diversity of fungal and bacterial communities in human skin. Nature 2013, 498, 367–370. [Google Scholar] [CrossRef] [PubMed]
- SanMiguel, A.; Grice, E.A. Interactions between host factors and the skin microbiome. Cell. Mol. Life Sci. 2015, 72, 1499–1515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giacomoni, P.U.; Mammone, T.; Teri, M. Gender-linked differences in human skin. J. Dermatol. Sci. 2009, 55, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Ying, S.; Zeng, D.-N.; Chi, L.; Tan, Y.; Galzote, C.; Cardona, C.; Lax, S.; Gilbert, J.; Quan, Z.-X. The Influence of Age and Gender on Skin-Associated Microbial Communities in Urban and Rural Human Populations. PLoS ONE 2015, 10, e0141842. [Google Scholar] [CrossRef] [PubMed]
- Fierer, N.; Hamady, M.; Lauber, C.L.; Knight, R. The influence of sex, handedness, and washing on the diversity of hand surface bacteria. Proc. Natl. Acad. Sci. USA 2008, 105, 17994–17999. [Google Scholar] [CrossRef] [Green Version]
- Shibagaki, N.; Suda, W.; Clavaud, C.; Bastien, P.; Takayasu, L.; Iioka, E.; Kurokawa, R.; Yamashita, N.; Hattori, Y.; Shindo, C.; et al. Aging-related changes in the diversity of women’s skin microbiomes associated with oral bacteria. Sci. Rep. 2017, 7, 10567. [Google Scholar] [CrossRef]
- Taylor, B.; Wadsworth, M.; Wadsworth, J.; Peckham, C. Changes in the reported prevalence of childhood eczema since the 1939-45 war. Lancet 1984, 324, 1255–1257. [Google Scholar] [CrossRef]
- Simpson, C.R.; Newton, J.; Hippisley-Cox, J.; Sheikh, A. Trends in the epidemiology and prescribing of medication for eczema in England. J. R. Soc. Med. 2009, 102, 108–117. [Google Scholar] [CrossRef] [Green Version]
- Burd, R.M. Psoriasis: A general overview. Br. J. Hosp. Med. 2006, 67, 259–262. [Google Scholar] [CrossRef] [PubMed]
- Prescott, S.L.; Tang, M.L.K. The Australasian Society of Clinical Immunology and Allergy position statement: Summary of allergy prevention in children. Med. J. Aust. 2005, 182, 464–467. [Google Scholar] [CrossRef] [PubMed]
- Asher, M.I.; Montefort, S.; Björkstén, B.; Lai, C.K.; Strachan, D.P.; Weiland, S.K.; Williams, H. Worldwide time trends in the prevalence of symptoms of asthma, allergic rhinoconjunctivitis, and eczema in childhood: ISAAC Phases One and Three repeat multicountry cross-sectional surveys. Lancet 2006, 368, 733–743. [Google Scholar] [CrossRef]
- Shaw, T.E.; Currie, G.P.; Koudelka, C.W.; Simpson, E.L. Eczema Prevalence in the United States: Data from the 2003 National Survey of Children’s Health. J. Investig. Dermatol. 2011, 131, 67–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- NIH HMP Working Group TNHW; Peterson, J.; Garges, S.; Giovanni, M.; McInnes, P.; Wang, L.; Schloss, J.A.; Bonazzi, V.; McEwen, J.E.; Wetterstrand, K.A.; et al. The NIH Human Microbiome Project. Genome Res. 2009, 19, 2317–2323. [Google Scholar]
- Grabchak, M.; Marcon, E.; Lang, G.; Zhang, Z. The generalized Simpson’s entropy is a measure of biodiversity. PLoS ONE 2017, 12, e0173305. [Google Scholar] [CrossRef]
- Mouillot, D.; Bellwood, D.R.; Baraloto, C.; Chave, J.; Galzin, R.; Harmelin-Vivien, M.; Kulbicki, M.; Lavergne, S.; Lavorel, S.; Mouquet, N.; et al. Rare Species Support Vulnerable Functions in High-Diversity Ecosystems. PLoS Biol. 2013, 11, e1001569. [Google Scholar] [CrossRef] [Green Version]
- Leitão, R.P.; Zuanon, J.; Villéger, S.; Williams, S.E.; Baraloto, C.; Fortune, C.; Mendonça, F.P.; Mouillot, D. Rare species contribute disproportionately to the functional structure of species assemblages. Proc. R. Soc. B Biol. Sci. 2016, 283, 20160084. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. 1), 4516–4522. [Google Scholar] [CrossRef] [Green Version]
- Zakrzewski, M.; Proietti, C.; Ellis, J.J.; Hasan, S.; Brion, M.J.; Berger, B.; Krause, L. Calypso: A user-friendly web-server for mining and visualizing microbiome-environment interactions. Bioinformatics 2017, 33, 782–783. [Google Scholar] [CrossRef] [Green Version]
- Bouslimani, A.; Porto, C.; Rath, C.M.; Wang, M.; Guo, Y.; Gonzalez, A.; Berg-Lyon, D.; Ackermann, G.; Christensen, G.J.M.; Nakatsuji, T.; et al. Molecular cartography of the human skin surface in 3D. Proc. Natl. Acad. Sci. USA 2015, 112, E2120–E2129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, A. Nonparametric Estimation of the Number of Classes in a Population Nonparametric Estimation of the Number of Classes in a Population. Scand. J. Stat. 1984, 11, 265–270. [Google Scholar]
- Colwell, R.K.; Coddington, J.A. Estimating terrestrial biodiversity through extrapolation. Philos. Trans. R. Soc. B Biol. Sci. 1994, 345, 101–118. [Google Scholar]
- Wallen-Russell, C. The Role of Every-Day Cosmetics in Altering the Skin Microbiome: A Study Using Biodiversity. Cosmetics 2018, 6, 2. [Google Scholar] [CrossRef] [Green Version]
- Kuczynski, J.; Liu, Z.; Lozupone, C.; McDonald, D.; Fierer, N.; Knight, R. Microbial community resemblance methods differ in their ability to detect biologically relevant patterns. Nat. Methods 2010, 7, 813–819. [Google Scholar] [CrossRef]
- Van den Wollenberg, A.L. Redundancy analysis an alternative for canonical correlation analysis. Psychometrika 1977, 42, 207–219. [Google Scholar] [CrossRef]
- Rao, C.R. The Use and Interpretation of Principal Component Analysis in Applied Research. Sankhyā Indian J. Stat. Ser. A 1964, 26, 329–358. [Google Scholar]
- Paliy, O.; Shankar, V. Application of multivariate statistical techniques in microbial ecology. Mol. Ecol. 2016, 25, 1032–1057. [Google Scholar] [CrossRef] [Green Version]
- Ramette, A. Multivariate analyses in microbial ecology. FEMS Microbiol. Ecol. 2007, 62, 142–160. [Google Scholar] [CrossRef] [Green Version]
- Concato, J.; Hartigan, J.A. P values: From suggestion to superstition. J. Investig. Med. 2016, 64, 1166–1171. [Google Scholar] [CrossRef] [Green Version]
- Johnson, V.E. Revised standards for statistical evidence. Proc. Natl. Acad. Sci. USA 2013, 110, 19313–19317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stehlikova, Z.; Kostovcik, M.; Kostovcikova, K.; Kverka, M.; Juzlova, K.; Rob, F.; Hercogova, J.; Bohac, P.; Pinto, Y.; Uzan, A.; et al. Dysbiosis of Skin Microbiota in Psoriatic Patients: Co-occurrence of Fungal and Bacterial Communities. Front. Microbiol. 2019, 10. [Google Scholar] [CrossRef]
- Steglińska, A.; Jachowicz, A.; Szulc, J.; Adamiak, J.; Otlewska, A.; Pielech-Przybylska, K.; Gutarowska, B. Factors Influencing Microbiological Biodiversity of Human Foot Skin. Int. J. Environ. Res. Public Health 2019, 16, 3503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grice, E.A.; Kong, H.H.; Conlan, S.; Deming, C.B.; Davis, J.; Young, A.C.; Bouffard, G.G.; Blakesley, R.W.; Murray, P.R.; Green, E.D.; et al. Topographical and temporal diversity of the human skin microbiome. Science 2009, 324, 1190–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costello, E.K.; Lauber, C.L.; Hamady, M.; Fierer, N.; Gordon, J.I.; Knight, R. Bacterial Community Variation in Human Body Habitats Across Space and Time. Science 2009, 326, 1694–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.K.; Choi, S.Y.; Byun, H.J.; Huh, C.H.; Park, K.C.; Patel, R.A.; Shinn, A.H.; Youn, S.W. Evaluation of gender difference in skin type and pH. J. Dermatol. Sci. 2006, 41, 153–156. [Google Scholar] [CrossRef] [PubMed]
- Dao, H.; Kazin, R.A. Gender differences in skin: A review of the literature. Gend. Med. 2007, 4, 308–328. [Google Scholar] [CrossRef]
- Fierer, N.; Jackson, R.B. The diversity and biogeography of soil bacterial communities. Proc. Natl. Acad. Sci. USA 2006, 103, 626–631. [Google Scholar] [CrossRef] [Green Version]
- Roth, R.R.; James, W.D. Microbial Ecology of the Skin. Annu. Rev. Microbiol. 1988, 42, 441–464. [Google Scholar] [CrossRef]
- Marples, R.R. Sex, constancy, and skin bacteria. Arch. Dermatol. Res. 1982, 272, 317–320. [Google Scholar] [CrossRef]
- Dimitriu, P.A.; Iker, B.; Malik, K.; Leung, H.; Mohn, W.W.; Hillebrand, G.G. New insights into the intrinsic and extrinsic factors that shape the human skin microbiome. MBio 2019, 10, e00839-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, M.H.Y.; Wilkins, D.; Lee, P.K.H. Insights into the pan-microbiome: Skin microbial communities of Chinese individuals differ from other racial groups. Sci. Rep. 2015, 5, srep11845. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kim, J.J.; Myeong, N.R.; Kim, T.; Kim, D.A.; An, S.; Kim, H.; Park, T.; Jang, S.I.; Yeon, J.H.; et al. Segregation of age-related skin microbiome characteristics by functionality. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.O.; Kim, H.S.; Youn, J.C.; Shin, E.C.; Park, S. Serum cytokine profiles in healthy young and elderly population assessed using multiplexed bead-based immunoassays. J. Transl. Med. 2011, 9, 113. [Google Scholar] [CrossRef] [Green Version]
- Luebberding, S.; Krueger, N.; Kerscher, M. Skin physiology in men and women: In vivo evaluation of 300 people including TEWL, SC hydration, sebum content and skin surface pH. Int. J. Cosmet. Sci. 2013, 35, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Wallen-Russell, C. Is There a Relationship between Transepidermal Water Loss and Microbial Biodiversity on the Skin? Cosmetics 2019, 6, 18. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Lauber, C.L.; Costello, E.K.; Berg-Lyons, D.; Gonzalez, A.; Stombaugh, J.; Knights, D.; Gajer, P.; Ravel, J.; Fierer, N.; et al. Moving pictures of the human microbiome. Genome Biol. 2011, 12, R50. [Google Scholar] [CrossRef] [Green Version]
- Huttenhower, C.; Gevers, D.; Knight, R.; Abubucker, S.; Badger, J.H.; Chinwalla, A.T. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar]
- Park, S.-C.; Won, S. Evaluation of 16S rRNA Databases for Taxonomic Assignments Using a Mock Community. Genom. Inform. 2018, 16, e24. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.R.; Shin, J.; Guevarra, R.B.; Lee, J.H.; Kim, D.W.; Seol, K.H.; Lee, J.-H.; Kim, H.B.; Isaacson, R.E. Deciphering diversity indices for a better understanding of microbial communities. J. Microbiol. Biotechnol. 2017, 27, 2089–2093. [Google Scholar] [CrossRef] [Green Version]
- Zhan, A.; Xiong, W.; He, S.; MacIsaac, H.J. Influence of Artifact Removal on Rare Species Recovery in Natural Complex Communities Using High-Throughput Sequencing. PLoS ONE 2014, 9, e96928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betensky, R.A. The p-Value Requires Context, Not a Threshold. Am. Stat. 2019, 73 (Suppl. 1), 115–117. [Google Scholar] [CrossRef] [Green Version]
- Di Leo, G.; Sardanelli, F. Statistical significance: P value, 0.05 threshold, and applications to radiomics—reasons for a conservative approach. Eur. Radiol. Exp. 2020, 4, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Skin Diversity Benchmark | Multiplication Factor |
|---|---|
| Unhealthy/diseased western | 0.24 (1/4.2) |
| Healthy western | - |
| Rural healthy | 1.23 |
| Caveman/perfect | 1.64 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wallen-Russell, C.; Wallen-Russell, S. A New Benchmark to Determine What Healthy Western Skin Looks Like in Terms of Biodiversity Using Standardised Methodology. Cosmetics 2020, 7, 79. https://doi.org/10.3390/cosmetics7040079
Wallen-Russell C, Wallen-Russell S. A New Benchmark to Determine What Healthy Western Skin Looks Like in Terms of Biodiversity Using Standardised Methodology. Cosmetics. 2020; 7(4):79. https://doi.org/10.3390/cosmetics7040079
Chicago/Turabian StyleWallen-Russell, Christopher, and Sam Wallen-Russell. 2020. "A New Benchmark to Determine What Healthy Western Skin Looks Like in Terms of Biodiversity Using Standardised Methodology" Cosmetics 7, no. 4: 79. https://doi.org/10.3390/cosmetics7040079
APA StyleWallen-Russell, C., & Wallen-Russell, S. (2020). A New Benchmark to Determine What Healthy Western Skin Looks Like in Terms of Biodiversity Using Standardised Methodology. Cosmetics, 7(4), 79. https://doi.org/10.3390/cosmetics7040079