
Tracking Down of a Selected Panel of Parabens: A Validated Method to Evaluate Their Occurrence in Skin Layers

 ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Skin Sample Preparation and Extraction Method
2.3. Liquid Chromatography Method
2.4. Chromatographic Method Validation
2.5. Calibration Curve and Linearity
2.6. Limits of Detection and Quantification
2.7. Precision and Accuracy
2.8. Selectivity
2.9. Carry-Over
2.10. Matrix Effect
2.11. Robustness
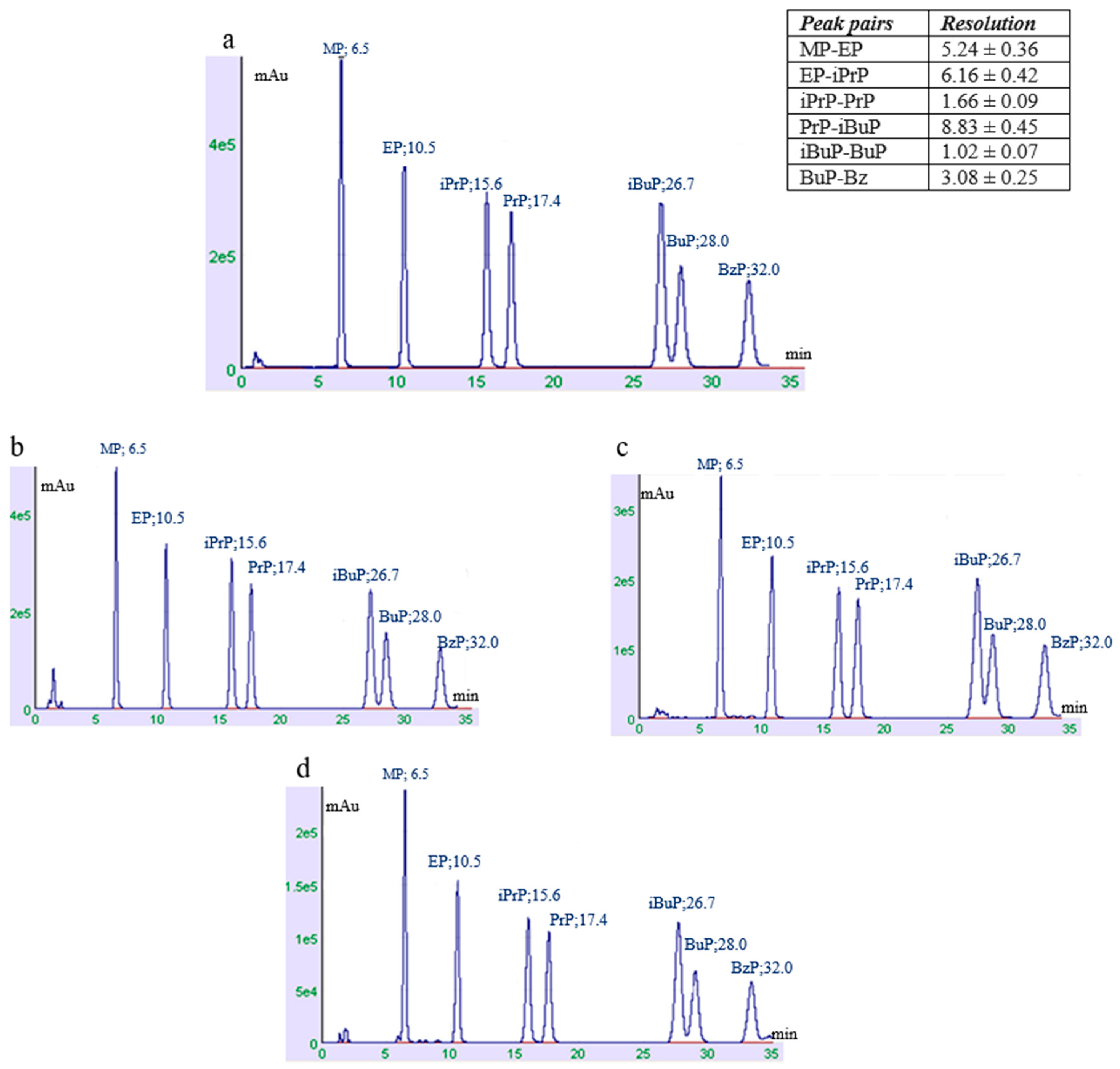
3. Results and Discussion
3.1. Optimization of Extraction and Chromatographic Method
3.2. Method Validation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Al-Halaseh, L.K.; Al-Adaileh, S.; Mbaideen, A.; Hajleh, M.N.A.; Al-Samydai, A.; Zakaraya, Z.Z.; Dayyih, W.A. Implication of parabens in cosmetics and cosmeceuticals: Advantages and limitations. J. Cosmet. Dermatol. 2022, 21, 3265–3271. [Google Scholar] [CrossRef] [PubMed]
- Błędzka, D.; Gromadzińska, J.; Wąsowicz, W. Parabens. From environmental studies to human health. Environ. Int. 2014, 67, 27–42. [Google Scholar] [CrossRef]
- Ma, X.; Wang, H.; Song, Y.; Pan, Y. Skin irritation potential of cosmetic preservatives: An exposure-relevant study. J. Cosmet. Dermatol. 2021, 20, 195–203. [Google Scholar] [CrossRef]
- Soni, M.G.; Carabin, I.G.; Burdock, G.A. Safety assessment of esters of p-hydroxybenzoic acid (parabens). Food Chem. Toxicol. 2005, 43, 985–1015. [Google Scholar] [CrossRef] [PubMed]
- Pollock, T.; Weaver, R.E.; Ghasemi, R.; deCatanzaro, D. Butyl paraben and propyl paraben modulate bisphenol A and estradiol concentrations in female and male mice. Toxicol. Appl. Pharmacol. 2017, 325, 18–24. [Google Scholar] [CrossRef] [PubMed]
- EU. Regulation (EC) No 1004/2014 of the European Parliament and of the Council on Cosmetic Products; Official Journal of the European Union: Luxembourg, 2014. [Google Scholar]
- Bodin, L.; Rogiers, V.; Bernauer, U.; Chaudhry, Q.; Coenraads, P.J.; Dusinska, M.; Ezendam, J.; Gaffet, E.; Galli, C.L.; Granum, B.; et al. Opinion of the Scientific Committee on Consumer Safety (SCCS)—Final Opinion on propylparaben (CAS No 94-13-3, EC No 202-307-7). Regul. Toxicol. Pharmacol. 2021, 125, 105005. [Google Scholar] [CrossRef]
- Neupane, R.; Boddu, S.H.S.; Renukuntla, J.; Babu, R.J.; Tiwari, A.K. Alternatives to Biological Skin in Permeation Studies: Current Trends and Possibilities. Pharmaceutics 2020, 12, 152. [Google Scholar] [CrossRef] [Green Version]
- Neri, I.; Laneri, S.; Di Lorenzo, R.; Dini, I.; Russo, G.; Grumetto, L. Parabens Permeation through Biological Membranes: A Comparative Study Using Franz Cell Diffusion System and Biomimetic Liquid Chromatography. Molecules 2022, 27, 4263. [Google Scholar] [CrossRef]
- Van der Schyff, V.; Suchánková, L.; Kademoglou, K.; Melymuk, L.; Klánová, J. Parabens and antimicrobial compounds in conventional and “green” personal care products. Chemosphere 2022, 297, 134019. [Google Scholar] [CrossRef]
- Piao, C.; Chen, L.; Wang, Y. A review of the extraction and chromatographic determination methods for the analysis of parabens. J. Chromatogr. B 2014, 969, 139–148. [Google Scholar] [CrossRef]
- Ferreira-Nunes, R.; Angelo, T.; da Silva, S.M.M.; Magalhaes, P.O.; Gratieri, T.; da Cunha, M.S.S.; Gelfuso, G.M. Versatile chromatographic method for catechin determination in development of topical formulations containing natural extracts. Biomed. Chromatogr. 2018, 32, e4062. [Google Scholar] [CrossRef] [PubMed]
- Pereira, M.N.; Matos, B.N.; Gratieri, T.; Cunha-Filho, M.; Gelfuso, G.M. Development and validation of a simple chromatographic method for simultaneous determination of clindamycin phosphate and rifampicin in skin permeation studies. J. Pharm. Biomed. Anal. 2018, 159, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Özcan, S.; Levent, S.; Can, N.; Kozanli, M. A Novel HPLC Method for Simultaneous Determination of Methyl, Ethyl, n-propyl, Isopropyl, n-butyl, Isobutyl and Benzyl Paraben in Pharmaceuticals and Cosmetics. Comb. Chem. High Throughput Screen. 2021, 24, 352–365. [Google Scholar] [CrossRef] [PubMed]
- European Union Guidelines. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/ich-guideline-q2r2-validation-analytical-procedures-step-2b_en.pdf (accessed on 20 September 2022).
- Shen, H.-Y.; Jiang, H.-L.; Mao, H.-L.; Pan, G.; Zhou, L.; Cao, Y.-F. Simultaneous determination of seven phthalates and four parabens in cosmetic products using HPLC-DAD and GC-MS methods. J. Sep. Sci. 2007, 30, 48–54. [Google Scholar] [CrossRef]
- Khesina, Z.B.; Iartsev, S.D.; Revelsky, A.I.; Buryak, A.K. Microextraction by packed sorbent optimized by statistical design of experiment as an approach to increase the sensitivity and selectivity of HPLC-UV determination of parabens in cosmetics. J. Pharm. Biomed. Anal. 2021, 195, 113843. [Google Scholar] [CrossRef] [PubMed]
- Katakam, L.N.R.; Ettaboina, S.K.; Dongala, T. A simple and rapid HPLC method for determination of parabens and their degradation products in pharmaceutical dosage forms. Biomed. Chromatogr. 2021, 35, e5152. [Google Scholar] [CrossRef]
- Dewani, A.P.; Bakal, R.L.; Kokate, P.G.; Chandewar, A.V.; Patra, S. Development of a Single Ion Pair HPLC Method for Analysis of Terbinafine, Ofloxacin, Ornidazole, Clobetasol, and Two Preservatives in a Cream Formulation: Application to In Vitro Drug Release in Topical Simulated Media-Phosphate Buffer Through Rat Skin. J. AOAC Int. 2015, 98, 913–920. [Google Scholar] [CrossRef]
- Qadir, M.A.; Ahmed, M.; Shafiq, M.I.; Ali, A.; Sadiq, A. Analytical Method for the Identification and Assay of Kojic Acid, Methylparaben, and Propylparaben in Cosmetic Products Using UPLC: Application of ISO 12787:2011 Standard. J. AOAC Int. 2016, 99, 1191–1196. [Google Scholar] [CrossRef]
- Abd El-Hay, S.S.; Mohram, M.S. Development and Validation of New RP-HPLC Method for Simultaneous Determination of Methyl and Propyl Parabens with Levetiracetam in Pure Form and Pharmaceutical Formulation. Chromatogr. Res. Int. 2016, 2016, 4909547. [Google Scholar] [CrossRef] [Green Version]
- Vela-Soria, F.; Ballesteros, O.; Rodríguez, I.; Zafra-Gómez, A.; Ballesteros, L.; Cela, R.; Navalón, A. A new treatment by dispersive liquid-liquid microextraction for the determination of parabens in human serum samples. Anal. Bioanal. Chem. 2013, 405, 7259–7267. [Google Scholar] [CrossRef]
- Lecce, R.; Regazzoni, L.; Mustazza, C.; Incarnato, G.; Porrà, R.; Panusa, A. Screening of preservatives by HPLC-PDA-ESI/MS: A focus on both allowed and recently forbidden compounds in the new EU cosmetics regulation. J. Pharm. Biomed. Anal. 2016, 125, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Mansour, R.S.H.; Hamdan, I.I.; Salem, M.S.H.; Khalil, E.A.; Sallam, A.A. HPLC method development/validation and skin diffusion study of caffeine, methyl paraben and butyl paraben as skin-diffusing model drugs. PLoS ONE 2021, 16, e0247879. [Google Scholar] [CrossRef] [PubMed]
- Andersen, A. Final Amended Report on the Safety Assessment of Methylparaben, Ethylparaben, Propylparaben, Isopropylparaben, Butylparaben, Isobutylparaben, and Benzylparaben as used in Cosmetic Products. Int. J. Toxicol. 2008, 27, 1–82. [Google Scholar] [CrossRef]


{kind=link}
| Calibration Parameters | MP | EP | iPrP | PrP | iBuP | BuP | BzP |
|---|---|---|---|---|---|---|---|
| Linear range µg mL−1 | 50.0–0.5 | 50.0–0.5 | 50.0–0.5 | 50.0–0.5 | 100.0–1.0 | 50.0–0.5 | 50.0–0.5 |
| Slope | 4836.6 | 4256.8 | 4657.3 | 4512.7 | 2133.7 | 6501.9 | 3177.8 |
| Intercept | 7668.8 | 5554.6 | 2590.7 | 1816.1 | 4877.7 | 450.06 | 2853.4 |
| R2 | 0.986 | 0.990 | 0.997 | 0.998 | 0.997 | 0.995 | 0.997 |
| Repeatability (n = 5) RSD% | 4.908 | 7.480 | 6.480 | 5.518 | 8.977 | 3.351 | 20.155 |
| Intermediate precision (n = 10) RSD% | 4.492 | 7.581 | 7.518 | 8.173 | 10.193 | 7.919 | 30.041 |
| LOQ µg mL−1 | 0.087 | 0.161 | 0.244 | 0.197 | 0.301 | 0.121 | 0.216 |
| LOD µg mL−1 | 0.026 | 0.048 | 0.073 | 0.059 | 0.090 | 0.036 | 0.065 |
| Matrix effect SC | 0.92 | 0.83 | 0.85 | 0.86 | 1.01 | 0.81 | 0.80 |
| Matrix effect Epidermis | 0.92 | 0.84 | 0.87 | 0.88 | 0.98 | 0.80 | 0.89 |
| Matrix effect Dermis | 0.89 | 0.81 | 0.83 | 0.84 | 0.98 | 0.82 | 0.78 |
| Recovery µg mL−1 | MP | EP | iPrP | PrP | iBuP | BuP | BzP |
|---|---|---|---|---|---|---|---|
| Stratum Corneum * | 90.45 ± 1.75 | 82.19 ± 1.74 | 84.21 ± 1.52 | 84.91 ± 1.42 | 109.90 ± 3.34 | 62.27 ± 0.59 | 78.77 ± 1.59 |
| Stratum Corneum ** | 100.08 ± 5.06 | 92.10 ± 4.70 | 74.93 ± 4.86 | 72.19 ± 5.33 | 76.05 ± 2.88 | 61.80 ± 2.96 | 76.60 ± 2.90 |
| Epidermis * | 90.92 ± 0.47 | 83.53 ± 0.39 | 86.09 ± 0.90 | 87.37 ± 1.03 | 106.85 ± 0.50 | 63.69 ± 0.62 | 87.53 ± 3.05 |
| Epidermis ** | 100.72 ± 4.43 | 99.26 ± 1.31 | 82.52 ± 2.50 | 77.15 ± 2.92 | 79.44 ± 1.61 | 64.94 ± 2.78 | 76.97 ± 1.85 |
| Dermis * | 79.78 ± 4.78 | 73.37 ± 3.37 | 76.02 ± 4.11 | 76.00 ± 5.01 | 109.52 ± 2.32 | 68.89 ± 0.42 | 66.07 ± 3.68 |
| Dermis ** | 105.73 ± 1.65 | 99.46 ± 0.28 | 82.72 ± 0.28 | 77.08 ± 0.10 | 79.66 ± 0.57 | 65.34 ± 0.57 | 70.98 ± 1.47 |
| Analytes | Vehicle | Method | LOD µg mL−1 | LOQ µg mL−1 | Ref. |
|---|---|---|---|---|---|
| MP, PrP | Ethanol | LC_UV | 0.003–0.053 | 0.087–0.197 | Present study |
| MP, PrP | Water: acetonitrile (50:50) | LC_UV | 0.026–0.026 | n.r. # | [18] |
| MP, PrP | Pharmaceutical formulation | LC_UV | 0.110–0.090 | 1.85–0.06 | * [19] |
| MP, and PP | Cosmetic products | UPLC_DAD | 0.068–0.064 | 0.02 and 0.04 | * [20] |
| MP and PP | Pharmaceutical formulation | LC_UV | 0.025–0.029 | 0.076–0.088 | * [21] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Lorenzo, R.; Neri, I.; Russo, G.; Laneri, S.; Grumetto, L. Tracking Down of a Selected Panel of Parabens: A Validated Method to Evaluate Their Occurrence in Skin Layers. Cosmetics 2022, 9, 102. https://doi.org/10.3390/cosmetics9050102
Di Lorenzo R, Neri I, Russo G, Laneri S, Grumetto L. Tracking Down of a Selected Panel of Parabens: A Validated Method to Evaluate Their Occurrence in Skin Layers. Cosmetics. 2022; 9(5):102. https://doi.org/10.3390/cosmetics9050102
Chicago/Turabian StyleDi Lorenzo, Ritamaria, Ilaria Neri, Giacomo Russo, Sonia Laneri, and Lucia Grumetto. 2022. "Tracking Down of a Selected Panel of Parabens: A Validated Method to Evaluate Their Occurrence in Skin Layers" Cosmetics 9, no. 5: 102. https://doi.org/10.3390/cosmetics9050102
APA StyleDi Lorenzo, R., Neri, I., Russo, G., Laneri, S., & Grumetto, L. (2022). Tracking Down of a Selected Panel of Parabens: A Validated Method to Evaluate Their Occurrence in Skin Layers. Cosmetics, 9(5), 102. https://doi.org/10.3390/cosmetics9050102