Isotope Fractionation during Gas Chromatography Can Enhance Mass Spectrometry-Based Measures of 2H-Labeling of Small Molecules

 ,
, {kind=link}
{kind=link}
{kind=link}
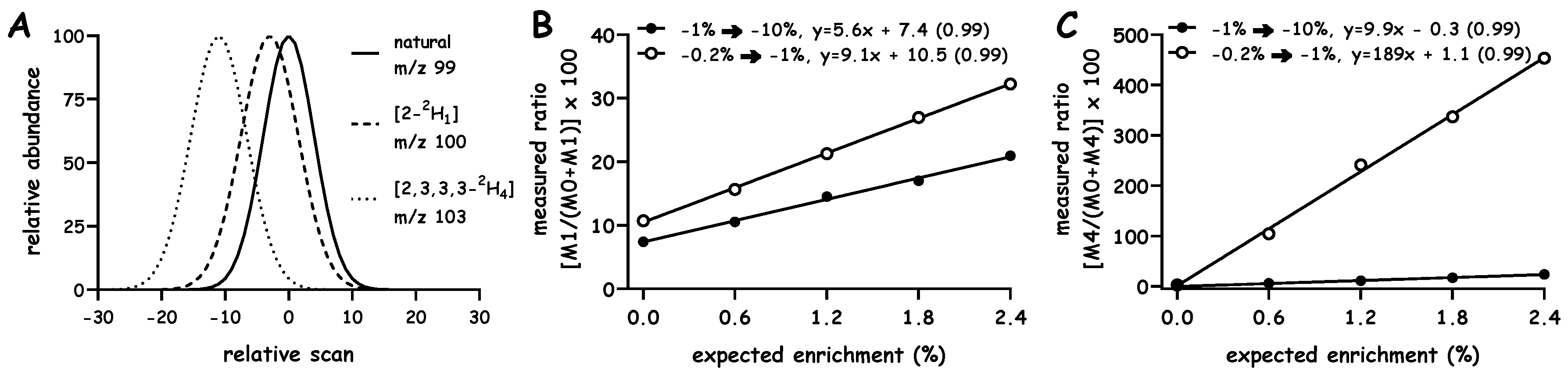{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Simulating and Modeling the Isotope Fractionation
2.2. Using GC-MS Analyses to Test the Model Predictions
2.3. Enhancing Isotope Ratio Analyses in Cases of Poor Chromatography and/or Low Signal Intensity
2.4. Additional Considerations of the Theory
3. Materials and Methods
3.1. Chemicals and Supplies
3.2. Biological
3.3. Analytical
3.4. Simulation and Mathematical Modeling of Isotope Fractionation
3.5. Processing GC-MS Data Following the Analyses of Alanine Standards
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Patterson, B.W. Use of stable isotopically labeled tracers for studies of metabolic kinetics: An overview. Metabolism 1997, 46, 322–329. [Google Scholar] [CrossRef]
- Daurio, N.A.; Wang, Y.; Chen, Y.; Zhou, H.; Carballo-Jane, E.; Mane, J.; Rodriguez, C.G.; Zafian, P.; Houghton, A.; Addona, G.; et al. Spatial and temporal studies of metabolic activity: Contrasting biochemical kinetics in tissues and pathways during fasted and fed states. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E1105–E1117. [Google Scholar] [CrossRef] [PubMed]
- Brook, M.S.; Wilkinson, D.J.; Atherton, P.J.; Smith, K. Recent developments in deuterium oxide tracer approaches to measure rates of substrate turnover: Implications for protein, lipid, and nucleic acid research. Curr. Opin. Clin. Nutr. Metab. Care 2017, 20, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Foletta, V.C.; Palmieri, M.; Kloehn, J.; Mason, S.; Previs, S.F.; McConville, M.J.; Sieber, O.M.; Bruce, C.R.; Kowalski, G.M. Analysis of Mammalian Cell Proliferation and Macromolecule Synthesis Using Deuterated Water and Gas Chromatography-Mass Spectrometry. Metabolites 2016, 6, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busch, R.; Kim, Y.K.; Neese, R.A.; Schade-Serin, V.; Collins, M.; Awada, M.; Gardner, J.L.; Beysen, C.; Marino, M.E.; Misell, L.M.; et al. Measurement of protein turnover rates by heavy water labeling of nonessential amino acids. Biochim. Biophys. Acta 2006, 1760, 730–744. [Google Scholar] [CrossRef]
- Turner, S.M.; Murphy, E.J.; Neese, R.A.; Antelo, F.; Thomas, T.; Agarwal, A.; Go, C.; Hellerstein, M.K. Measurement of TG synthesis and turnover in vivo by 2H2O incorporation into the glycerol moiety and application of MIDA. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E790–E803. [Google Scholar] [CrossRef] [Green Version]
- Neese, R.A.; Misell, L.M.; Turner, S.; Chu, A.; Kim, J.; Cesar, D.; Hoh, R.; Antelo, F.; Strawford, A.; McCune, J.M.; et al. Measurement in vivo of proliferation rates of slow turnover cells by 2H2O labeling of the deoxyribose moiety of DNA. Proc. Natl. Acad. Sci. USA 2002, 99, 15345–15350. [Google Scholar] [CrossRef] [Green Version]
- Brenna, J.T.; Corso, T.N.; Tobias, H.J.; Caimi, R.J. High-precision continuous-flow isotope ratio mass spectrometry. Mass Spectrom. Rev. 1997, 16, 227–258. [Google Scholar] [CrossRef]
- Wolfe, R.R.; Chinkes, D.L. Isotope Tracers in Metabolic Research: Principles and Practice of Kinetic Analyses; Wiley-Liss: Hoboken, NJ, USA, 2005. [Google Scholar]
- Matthews, D.E.; Hayes, J.M. Systematic errors in gas chromatography-mass spectrometry isotope ratio measurements. Anal. Chem. 1975, 48, 1375–1382. [Google Scholar] [CrossRef]
- Antoniewicz, M.R.; Kelleher, J.K.; Stephanopoulos, G. Accurate assessment of amino acid mass isotopomer distributions for metabolic flux analysis. Anal. Chem. 2007, 79, 7554–7559. [Google Scholar] [CrossRef]
- Katanik, J.; Mccabe, B.J.; Brunengraber, D.Z.; Chandramouli, V.; Nishiyama, F.J.; Anderson, V.E.; Previs, S.F. Measuring gluconeogenesis using a low dose of (H2O)-H-2: Advantage of isotope fractionation during gas chromatography. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E1043–E1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patterson, B.W.; Zhang, X.J.; Chen, Y.; Klein, S.; Wolfe, R.R. Measurement of very low stable isotope enrichments by gas chromatography/mass spectrometry: Application to measurement of muscle protein synthesis. Metabolism 1997, 46, 943–948. [Google Scholar] [CrossRef]
- Slater, C.; Preston, T.; Mcmillan, D.C.; Falconer, J.S.; Fearon, K.C.H. Gc/Ms Analysis of [H-2(5)]Phenylalanine at Very-Low Enrichment—Measurement of Protein-Synthesis in Health and Disease. J. Mass Spectrom. 1995, 30, 1325–1332. [Google Scholar] [CrossRef]
- Yang, D.; Diraison, F.; Beylot, M.; Brunengraber, D.Z.; Samols, M.A.; Anderson, V.E.; Brunengraber, H. Assay of low deuterium enrichment of water by isotopic exchange with [U-13C3]acetone and gas chromatography-mass spectrometry. Anal. Biochem. 1998, 258, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Hazey, J.W.; Yang, D.; Powers, L.; Previs, S.F.; David, F.; Beaulieu, A.D.; Puchowicz, M.A.; Potter, J.L.; Palmquist, D.L.; Brunengraber, H. Tracing gluconeogenesis with deuterated water: Measurement of low deuterium enrichments on carbons 6 and 2 of glucose. Anal. Biochem. 1997, 248, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Shah, V.; Herath, K.; Previs, S.F.; Hubbard, B.K.; Roddy, T.P. Headspace analyses of acetone: A rapid method for measuring the 2H-labeling of water. Anal. Biochem. 2010, 404, 235–237. [Google Scholar] [CrossRef]
- Bluck, L.J.C.; Coward, W.A. Peak measurement in gas chromatographic mass spectrometric isotope studies. J. Mass Spectrom. 1997, 32, 1212–1218. [Google Scholar] [CrossRef]
- Thenot, J.P.; Horning, E.C. Amino acid N-dimethylaminomethylene alkyl esters. New derivatives for gas chromatographic and gas chromatographic-mass spectrometric studies. Anal. Lett. 1972, 5, 519–529. [Google Scholar] [CrossRef]
- Gasier, H.G.; Fluckey, J.D.; Previs, S.F. The application of (H2O)-H-2 to measure skeletal muscle protein synthesis. Nutr. Metab. 2010, 7, 31. [Google Scholar] [CrossRef] [Green Version]
- O’Leary, T.S.; Snyder, J.; Sadayappan, S.; Day, S.M.; Previs, M.J. MYBPC3 truncation mutations enhance actomyosin contractile mechanics in human hypertrophic cardiomyopathy. J. Mol. Cell. Cardiol. 2019, 127, 165–173. [Google Scholar] [CrossRef]
- Dyson, N. Chromatographic Integration Methods, 2nd ed.; Royal Society of Chemistry: Cambridge, UK, 1998. [Google Scholar]
- Herath, K.; Yang, J.; Zhong, W.; Kulick, A.; Rohm, R.J.; Lassman, M.E.; Castro-Perez, J.M.; Mahsut, A.; Dunn, K.; Johns, D.G.; et al. Determination of low levels of 2H-labeling using high-resolution mass spectrometry (HR-MS): Application in studies of lipid flux and beyond. J. Am. Soc. Mass Spectrom. 2011, 22, 154. [Google Scholar]
- Trotzmuller, M.; Triebl, A.; Ajsic, A.; Hartler, J.; Kofeler, H.; Regittnig, W. Determination of the Isotopic Enrichment of (13)C- and (2)H-Labeled Tracers of Glucose Using High-Resolution Mass Spectrometry: Application to Dual- and Triple-Tracer Studies. Anal. Chem. 2017, 89, 12252–12260. [Google Scholar] [CrossRef] [PubMed]
- Sadygov, R.G.; Avva, J.; Rahman, M.; Lee, K.; Ilchenko, S.; Kasumov, T.; Borzou, A. d2ome, Software for in Vivo Protein Turnover Analysis Using Heavy Water Labeling and LC-MS, Reveals Alterations of Hepatic Proteome Dynamics in a Mouse Model of NAFLD. J. Proteome Res. 2018, 17, 3740–3748. [Google Scholar] [CrossRef] [PubMed]
- Borzou, A.; Sadygov, V.R.; Zhang, W.; Sadygov, R.G. Proteome Dynamics from Heavy Water Metabolic Labeling and Peptide Tandem Mass Spectrometry. Int. J. Mass Spectrom. 2019, 445, 116194. [Google Scholar] [CrossRef]
- Naylor, B.C.; Porter, M.T.; Wilson, E.; Herring, A.; Lofthouse, S.; Hannemann, A.; Piccolo, S.R.; Rockwood, A.L.; Price, J.C. DeuteRater: A tool for quantifying peptide isotope precision and kinetic proteomics. Bioinformatics 2017, 33, 1514–1520. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Downes, D.P.; Kasumov, T.; Daurio, N.A.; Wood, N.B.; Previs, M.J.; Sheth, P.R.; McLaren, D.G.; Previs, S.F. Isotope Fractionation during Gas Chromatography Can Enhance Mass Spectrometry-Based Measures of 2H-Labeling of Small Molecules. Metabolites 2020, 10, 474. https://doi.org/10.3390/metabo10110474
Downes DP, Kasumov T, Daurio NA, Wood NB, Previs MJ, Sheth PR, McLaren DG, Previs SF. Isotope Fractionation during Gas Chromatography Can Enhance Mass Spectrometry-Based Measures of 2H-Labeling of Small Molecules. Metabolites. 2020; 10(11):474. https://doi.org/10.3390/metabo10110474
Chicago/Turabian StyleDownes, Daniel P., Takhar Kasumov, Natalie A. Daurio, Neil B. Wood, Michael J. Previs, Payal R. Sheth, David G. McLaren, and Stephen F. Previs. 2020. "Isotope Fractionation during Gas Chromatography Can Enhance Mass Spectrometry-Based Measures of 2H-Labeling of Small Molecules" Metabolites 10, no. 11: 474. https://doi.org/10.3390/metabo10110474
APA StyleDownes, D. P., Kasumov, T., Daurio, N. A., Wood, N. B., Previs, M. J., Sheth, P. R., McLaren, D. G., & Previs, S. F. (2020). Isotope Fractionation during Gas Chromatography Can Enhance Mass Spectrometry-Based Measures of 2H-Labeling of Small Molecules. Metabolites, 10(11), 474. https://doi.org/10.3390/metabo10110474