Oral Gavage Delivery of Stable Isotope Tracer for In Vivo Metabolomics

 , and
, and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
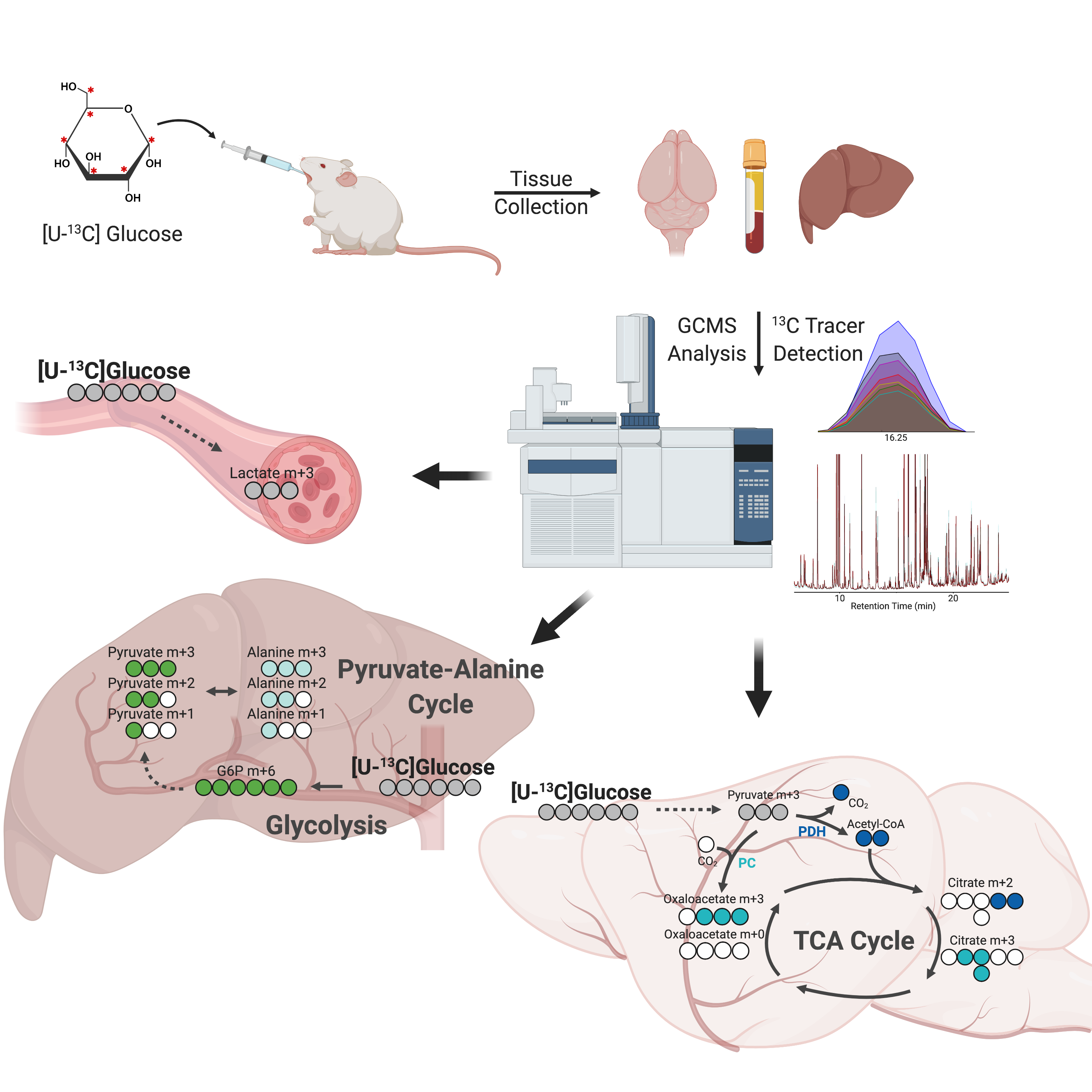
2.1. Experimental Workflow and Stable Isotope Tissue Distribution
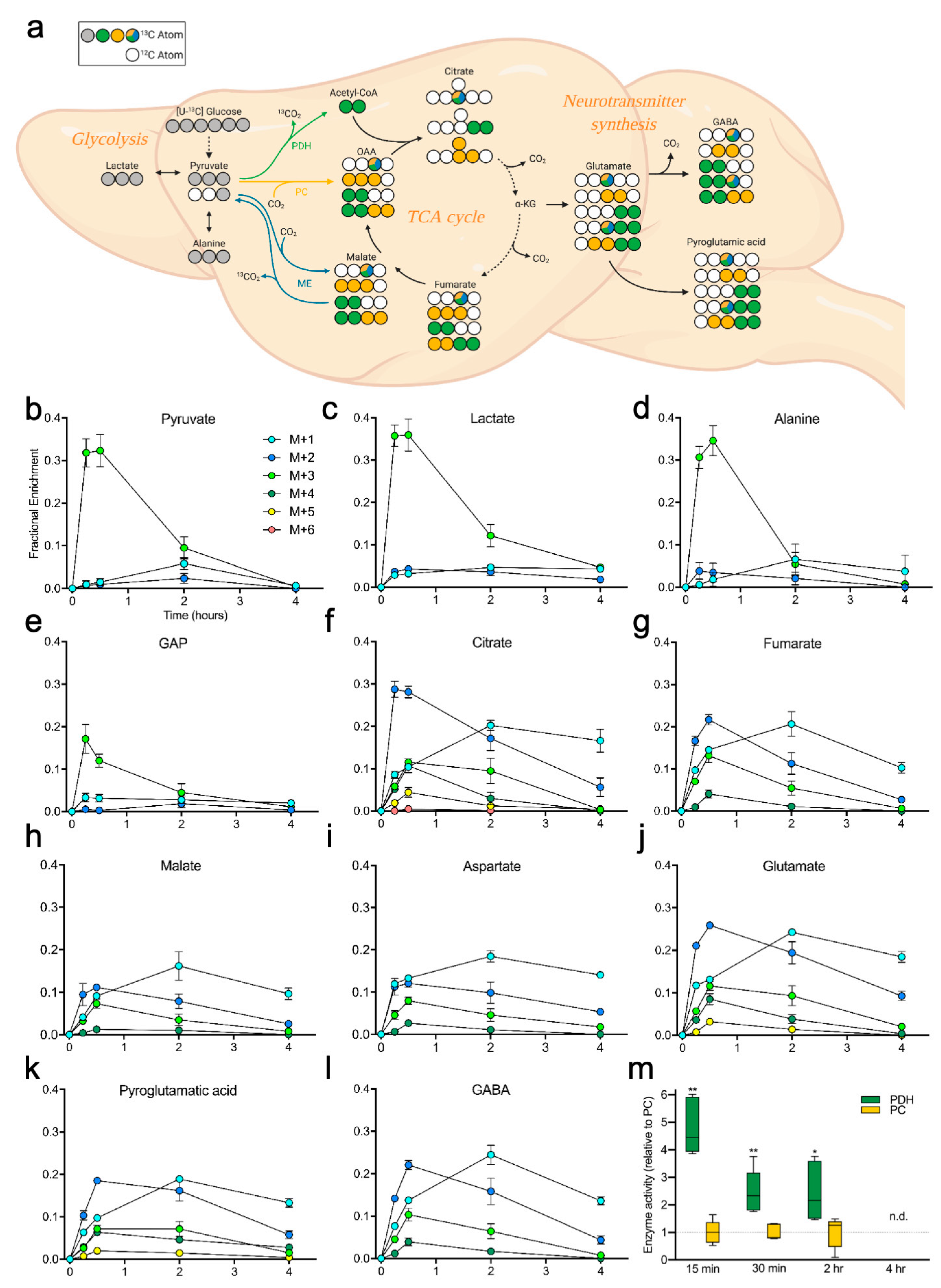
2.2. Brain Metabolites Display Varying Patterns of 13C Labeling
2.3. Measuring Glucose Metabolism in an Alzheimer’s Disease Model
2.4. Liver Metabolites Display Varying Patterns of 13C Labeling
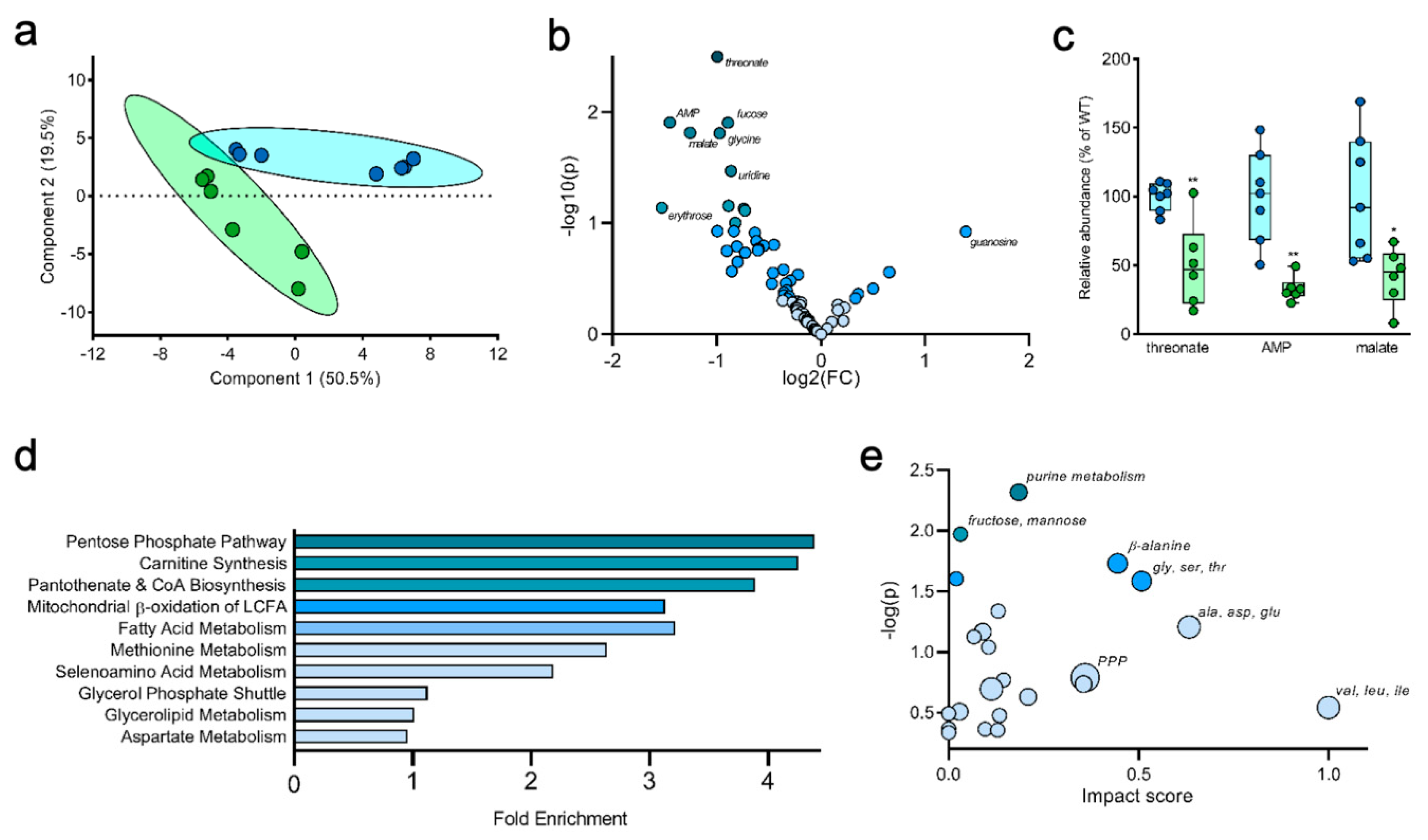
2.5. Tracing Glucose Metabolism in a Mouse Model of Type II Diabetes
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Gavage of [U-13C] Glucose Solution
4.3. Plasma and Tissue Collection
4.4. Glucose Colorimetric Assay
4.5. Triglyceride Assay
4.6. Sample Preparation for GCMS Analysis
4.7. Glycogen Preparation for GCMS Analysis
4.8. GCMS Quantitation
4.9. Metabolomics Data Analysis
4.10. Animal Cognitive Testing
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fan, T.W.; Lorkiewicz, P.K.; Sellers, K.; Moseley, H.N.; Higashi, R.M.; Lane, A.N. Stable isotope-resolved metabolomics and applications for drug development. Pharmacol. Ther. 2012, 133, 366–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lane, A.N.; Higashi, R.M.; Fan, T.W.-M. Preclinical models for interrogating drug action in human cancers using stable isotope resolved metabolomics (sirm). Metabolomics 2016, 12, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, T.W.-M.; Tan, J.; McKinney, M.M.; Lane, A.N. Stable isotope resolved metabolomics analysis of ribonucleotide and rna metabolism in human lung cancer cells. Metabolomics 2012, 8, 517–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higashi, R.M.; Fan, T.W.-M.; Lorkiewicz, P.K.; Moseley, H.N.B.; Lane, A.N. Stable isotope-labeled tracers for metabolic pathway elucidation by gc-ms and ft-ms. In Mass Spectrometry in Metabolomics: Methods and Protocols; Raftery, D., Ed.; Springer: New York, NY, USA, 2014; pp. 147–167. [Google Scholar]
- Hiller, K.; Metallo, C.M. Profiling metabolic networks to study cancer metabolism. Curr. Opin. Biotechnol. 2013, 24, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ahn, W.S.; Gameiro, P.A.; Keibler, M.A.; Zhang, Z.; Stephanopoulos, G. 13C isotope-assisted methods for quantifying glutamine metabolism in cancer cells. Methods Enzymol. 2014, 542, 369–389. [Google Scholar] [PubMed] [Green Version]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by idh1 mediates lipogenesis under hypoxia. Nature 2012, 481, 380–384. [Google Scholar] [CrossRef] [Green Version]
- Ternette, N.; Yang, M.; Laroyia, M.; Kitagawa, M.; O’Flaherty, L.; Wolhulter, K.; Igarashi, K.; Saito, K.; Kato, K.; Fischer, R.; et al. Inhibition of mitochondrial aconitase by succination in fumarate hydratase deficiency. Cell Rep. 2013, 3, 689–700. [Google Scholar] [CrossRef] [Green Version]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Lane, A.N.; Ricketts, C.J.; Sourbier, C.; Wei, M.H.; Shuch, B.; Pike, L.; Wu, M.; Rouault, T.A.; Boros, L.G.; et al. Metabolic reprogramming for producing energy and reducing power in fumarate hydratase null cells from hereditary leiomyomatosis renal cell carcinoma. PLoS ONE 2013, 8, e72179. [Google Scholar] [CrossRef]
- Hoekstra, A.S.; de Graaff, M.A.; Briaire-de Bruijn, I.H.; Ras, C.; Seifar, R.M.; van Minderhout, I.; Cornelisse, C.J.; Hogendoorn, P.C.W.; Breuning, M.H.; Suijker, J.; et al. Inactivation of sdh and fh cause loss of 5hmc and increased h3k9me3 in paraganglioma/pheochromocytoma and smooth muscle tumors. Oncotarget 2015, 6, 38777–38788. [Google Scholar] [CrossRef] [Green Version]
- Saxena, N.; Maio, N.; Crooks, D.R.; Ricketts, C.J.; Yang, Y.; Wei, M.-H.; Fan, T.W.-M.; Lane, A.N.; Sourbier, C.; Singh, A.; et al. Sdhb-deficient cancers: The role of mutations that impair iron sulfur cluster delivery. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiehn, O. Metabolomics by gas chromatography-mass spectrometry: Combined targeted and untargeted profiling. Curr. Protoc. Mol. Biol. 2016, 114, 30.4.1–30.4.32. [Google Scholar] [CrossRef] [PubMed]
- Jang, C.; Chen, L.; Rabinowitz, J.D. Metabolomics and isotope tracing. Cell 2018, 173, 822–837. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Zheng, H.; Chen, J.; Li, C.; Du, Y.; Xia, H.; Gao, H. Metabolic fate of glucose in the brain of app/ps1 transgenic mice at 10 months of age: A 13C nmr metabolomic study. Metab. Brain Dis. 2018, 33, 1661–1668. [Google Scholar] [CrossRef]
- Sugiura, Y.; Honda, K.; Kajimura, M.; Suematsu, M. Visualization and quantification of cerebral metabolic fluxes of glucose in awake mice. Proteomics 2014, 14, 829–838. [Google Scholar] [CrossRef]
- Lane, A.N.; Yan, J.; Fan, T.W. 13C tracer studies of metabolism in mouse tumor xenografts. Bio Protoc. 2015, 5, e1650. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Zhang, Q.; Li, P.; Hong, G.; Wang, D.; Liu, J.; Zhou, H.; Chai, G.; Lu, B.; He, S.; et al. Nicotine pharmacokinetics in rat brain and blood by simultaneous microdialysis, stable-isotope labeling, and uhplc-hrms: Determination of nicotine metabolites. Anal. Chem. 2019, 91, 2916–2922. [Google Scholar] [CrossRef]
- Broekaert, D.; Fendt, S.M. Measuring in vivo tissue metabolism using 13C glucose infusions in mice. Methods Mol. Biol. (Clifton N.J.) 2019, 1862, 67–82. [Google Scholar]
- Sun, R.C.; Fan, T.W.; Deng, P.; Higashi, R.M.; Lane, A.N.; Le, A.T.; Scott, T.L.; Sun, Q.; Warmoes, M.O.; Yang, Y. Noninvasive liquid diet delivery of stable isotopes into mouse models for deep metabolic network tracing. Nat. Commun. 2017, 8, 1646. [Google Scholar] [CrossRef] [PubMed]
- Courtney, K.D.; Bezwada, D.; Mashimo, T.; Pichumani, K.; Vemireddy, V.; Funk, A.M.; Wimberly, J.; McNeil, S.S.; Kapur, P.; Lotan, Y.; et al. Isotope tracing of human clear cell renal cell carcinomas demonstrates suppressed glucose oxidation in vivo. Cell Metab. 2018, 28, 793–800.e2. [Google Scholar] [CrossRef] [Green Version]
- Kindt, A.; Liebisch, G.; Clavel, T.; Haller, D.; Hörmannsperger, G.; Yoon, H.; Kolmeder, D.; Sigruener, A.; Krautbauer, S.; Seeliger, C.; et al. The gut microbiota promotes hepatic fatty acid desaturation and elongation in mice. Nat. Commun. 2018, 9, 3760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLaren, D.G.; He, T.; Wang, S.P.; Mendoza, V.; Rosa, R.; Gagen, K.; Bhat, G.; Herath, K.; Miller, P.L.; Stribling, S.; et al. The use of stable-isotopically labeled oleic acid to interrogate lipid assembly in vivo: Assessing pharmacological effects in preclinical species. J. Lipid Res. 2011, 52, 1150–1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glick, G.D.; Rossignol, R.; Lyssiotis, C.A.; Wahl, D.; Lesch, C.; Sanchez, B.; Liu, X.; Hao, L.Y.; Taylor, C.; Hurd, A.; et al. Anaplerotic metabolism of alloreactive t cells provides a metabolic approach to treat graft-versus-host disease. J. Pharmacol. Exp. Ther. 2014, 351, 298–307. [Google Scholar] [CrossRef] [PubMed]
- O’Doherty, R.M.; Jensen, P.B.; Anderson, P.; Jones, J.G.; Berman, H.K.; Kearney, D.; Newgard, C.B. Activation of direct and indirect pathways of glycogen synthesis by hepatic overexpression of protein targeting to glycogen. Clin. Investig. 2000, 105, 479–488. [Google Scholar] [CrossRef] [Green Version]
- Shulman, G.I.; Rothman, D.L.; Smith, D.; Johnson, C.M.; Blair, J.B.; Shulman, R.G.; DeFronzo, R.A. Mechanism of liver glycogen repletion in vivo by nuclear magnetic resonance spectroscopy. J. Clin. Investig. 1985, 76, 1229–1236. [Google Scholar] [CrossRef]
- Fernández-Calleja, J.M.S.; Bouwman, L.M.S.; Swarts, H.J.M.; Oosting, A.; Keijer, J.; van Schothorst, E.M. Extended indirect calorimetry with isotopic CO2 sensors for prolonged and continuous quantification of exogenous vs. Total substrate oxidation in mice. Sci. Rep. 2019, 9, 11507. [Google Scholar] [CrossRef] [Green Version]
- Jang, C.; Hui, S.; Lu, W.; Cowan, A.J.; Morscher, R.J.; Lee, G.; Liu, W.; Tesz, G.J.; Birnbaum, M.J.; Rabinowitz, J.D. The small intestine converts dietary fructose into glucose and organic acids. Cell Metab. 2018, 27, 351–361.e3. [Google Scholar] [CrossRef]
- Nissen, J.D.; Pajęcka, K.; Stridh, M.H.; Skytt, D.M.; Waagepetersen, H.S. Dysfunctional tca-cycle metabolism in glutamate dehydrogenase deficient astrocytes. Glia 2015, 63, 2313–2326. [Google Scholar] [CrossRef]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. Off. J. Soc. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef]
- Xia, J.; Wishart, D.S. Metpa: A web-based metabolomics tool for pathway analysis and visualization. Bioinformatics 2010, 26, 2342–2344. [Google Scholar] [CrossRef] [Green Version]
- Hasenour, C.M.; Rahim, M.; Young, J.D. In vivo estimates of liver metabolic flux assessed by 13C-propionate and 13C-lactate are impacted by tracer recycling and equilibrium assumptions. Cell Rep. 2020, 32, 107986. [Google Scholar] [CrossRef] [PubMed]
- Andres, D.A.; Young, L.E.A.; Veeranki, S.; Hawkinson, T.R.; Levitan, B.M.; He, D.; Wang, C.; Satin, J.; Sun, R.C. Improved workflow for mass spectrometry-based metabolomics analysis of the heart. J. Biol. Chem. 2020, 295, 2676–2686. [Google Scholar] [CrossRef] [PubMed]
- Young, L.E.A.; Brizzee, C.O.; Macedo, J.K.A.; Murphy, R.D.; Contreras, C.J.; DePaoli-Roach, A.A.; Roach, P.J.; Gentry, M.S.; Sun, R.C. Accurate and sensitive quantitation of glucose and glucose phosphates derived from storage carbohydrates by mass spectrometry. Carbohydr. Polym. 2020, 230, 115651. [Google Scholar] [CrossRef] [PubMed]
- Coleman, D.L.; Hummel, K.P. The influence of genetic background on the expression of the obese (ob) gene in the mouse. Diabetologia 1973, 9, 287–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buescher, J.M.; Antoniewicz, M.R.; Boros, L.G.; Burgess, S.C.; Brunengraber, H.; Clish, C.B.; DeBerardinis, R.J.; Feron, O.; Frezza, C.; Ghesquiere, B.; et al. A roadmap for interpreting 13C metabolite labeling patterns from cells. Curr. Opin. Biotechnol. 2015, 34, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Fan, T.W.; Lane, A.N.; Higashi, R.M. Stable isotope resolved metabolomics studies in ex vivo tissue slices. Bio Protoc. 2016, 6, e1730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández-García, J.; Altea-Manzano, P.; Pranzini, E.; Fendt, S.-M. Stable isotopes for tracing mammalian-cell metabolism in vivo. Trends Biochem. Sci. 2020, 45, 185–201. [Google Scholar] [CrossRef]
- Phillips, P.J. Oral glucose tolerance testing. Aust. Fam. Physician 2012, 41, 391–393. [Google Scholar]
- Ayala, J.E.; Samuel, V.T.; Morton, G.J.; Obici, S.; Croniger, C.M.; Shulman, G.I.; Wasserman, D.H.; McGuinness, O.P. Standard operating procedures for describing and performing metabolic tests of glucose homeostasis in mice. Dis. Models Mech. 2010, 3, 525–534. [Google Scholar] [CrossRef] [Green Version]
- Hoggatt, A.F.; Hoggatt, J.; Honerlaw, M.; Pelus, L.M. A spoonful of sugar helps the medicine go down: A novel technique to improve oral gavage in mice. J. Am. Assoc. Lab. Anim. Sci. 2010, 49, 329–334. [Google Scholar]
- Macdonald, I.R.; DeBay, D.R.; Reid, G.A.; O’Leary, T.P.; Jollymore, C.T.; Mawko, G.; Burrell, S.; Martin, E.; Bowen, C.V.; Brown, R.E.; et al. Early detection of cerebral glucose uptake changes in the 5xfad mouse. Curr. Alzheimer Res. 2014, 11, 450–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verfaillie, S.C.; Adriaanse, S.M.; Binnewijzend, M.A.; Benedictus, M.R.; Ossenkoppele, R.; Wattjes, M.P.; Pijnenburg, Y.A.; van der Flier, W.M.; Lammertsma, A.A.; Kuijer, J.P.; et al. Cerebral perfusion and glucose metabolism in alzheimer’s disease and frontotemporal dementia: Two sides of the same coin? Eur. Radiol. 2015, 25, 3050–3059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Guo, L.; Lu, L.; Sun, H.; Shao, M.; Beck, S.J.; Li, L.; Ramachandran, J.; Du, Y.; Du, H. Synaptosomal mitochondrial dysfunction in 5xfad mouse model of alzheimer’s disease. PLoS ONE 2016, 11, e0150441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devi, L.; Ohno, M. Mitochondrial dysfunction and accumulation of the β-secretase-cleaved c-terminal fragment of app in alzheimer’s disease transgenic mice. Neurobiol. Dis. 2012, 45, 417–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.; Wirtz, M.; Parker, N.; Hogan, M.; Strahler, J.; Michailidis, G.; Schmidt, S.; Vidal-Puig, A.; Diano, S.; Andrews, P.; et al. Leptin-mediated changes in hepatic mitochondrial metabolism, structure, and protein levels. Proc. Natl. Acad. Sci. USA 2009, 106, 13100–13105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llufrio, E.M.; Wang, L.; Naser, F.J.; Patti, G.J. Sorting cells alters their redox state and cellular metabolome. Redox. Biol. 2018, 16, 381–387. [Google Scholar] [CrossRef]
- Sun, R.C.; Dukhande, V.V.; Zhou, Z.; Young, L.E.A.; Emanuelle, S.; Brainson, C.F.; Gentry, M.S. Nuclear glycogenolysis modulates histone acetylation in human non-small cell lung cancers. Cell Metab. 2019, 30, 903–916.e7. [Google Scholar] [CrossRef]
- Brewer, M.K.; Uittenbogaard, A.; Austin, G.L.; Segvich, D.M.; DePaoli-Roach, A.; Roach, P.J.; McCarthy, J.J.; Simmons, Z.R.; Brandon, J.A.; Zhou, Z.; et al. Targeting pathogenic lafora bodies in lafora disease using an antibody-enzyme fusion. Cell Metab. 2019, 30, 689–705.e6. [Google Scholar] [CrossRef]
- Kind, T.; Tolstikov, V.; Fiehn, O.; Weiss, R.H. A comprehensive urinary metabolomic approach for identifying kidney cancerr. Anal. Biochem. 2007, 363, 185–195. [Google Scholar] [CrossRef]
- Fiehn, O.; Kopka, J.; Dörmann, P.; Altmann, T.; Trethewey, R.N.; Willmitzer, L. Metabolite profiling for plant functional genomics. Nat. Biotechnol. 2000, 18, 1157–1161. [Google Scholar] [CrossRef]
- Kind, T.; Wohlgemuth, G.; Lee, D.Y.; Lu, Y.; Palazoglu, M.; Shahbaz, S.; Fiehn, O. Fiehnlib: Mass spectral and retention index libraries for metabolomics based on quadrupole and time-of-flight gas chromatography/mass spectrometry. Anal. Chem. 2009, 81, 10038–10048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, L.A.; Zuloaga, K.L.; Kugelman, T.L.; Mader, K.S.; Morre, J.T.; Zuloaga, D.G.; Weber, S.; Marzulla, T.; Mulford, A.; Button, D.; et al. Amelioration of metabolic syndrome-associated cognitive impairments in mice via a reduction in dietary fat content or infusion of non-diabetic plasma. EBioMedicine 2016, 3, 26–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Williams, H.C.; Piron, M.A.; Nation, G.K.; Walsh, A.E.; Young, L.E.A.; Sun, R.C.; Johnson, L.A. Oral Gavage Delivery of Stable Isotope Tracer for In Vivo Metabolomics. Metabolites 2020, 10, 501. https://doi.org/10.3390/metabo10120501
Williams HC, Piron MA, Nation GK, Walsh AE, Young LEA, Sun RC, Johnson LA. Oral Gavage Delivery of Stable Isotope Tracer for In Vivo Metabolomics. Metabolites. 2020; 10(12):501. https://doi.org/10.3390/metabo10120501
Chicago/Turabian StyleWilliams, Holden C., Margaret A. Piron, Grant K. Nation, Adeline E. Walsh, Lyndsay E. A. Young, Ramon C. Sun, and Lance A. Johnson. 2020. "Oral Gavage Delivery of Stable Isotope Tracer for In Vivo Metabolomics" Metabolites 10, no. 12: 501. https://doi.org/10.3390/metabo10120501
APA StyleWilliams, H. C., Piron, M. A., Nation, G. K., Walsh, A. E., Young, L. E. A., Sun, R. C., & Johnson, L. A. (2020). Oral Gavage Delivery of Stable Isotope Tracer for In Vivo Metabolomics. Metabolites, 10(12), 501. https://doi.org/10.3390/metabo10120501