Enhancing Glycolysis Protects against Ischemia-Reperfusion Injury by Reducing ROS Production

 ,
,
Abstract
:1. Introduction
2. Results
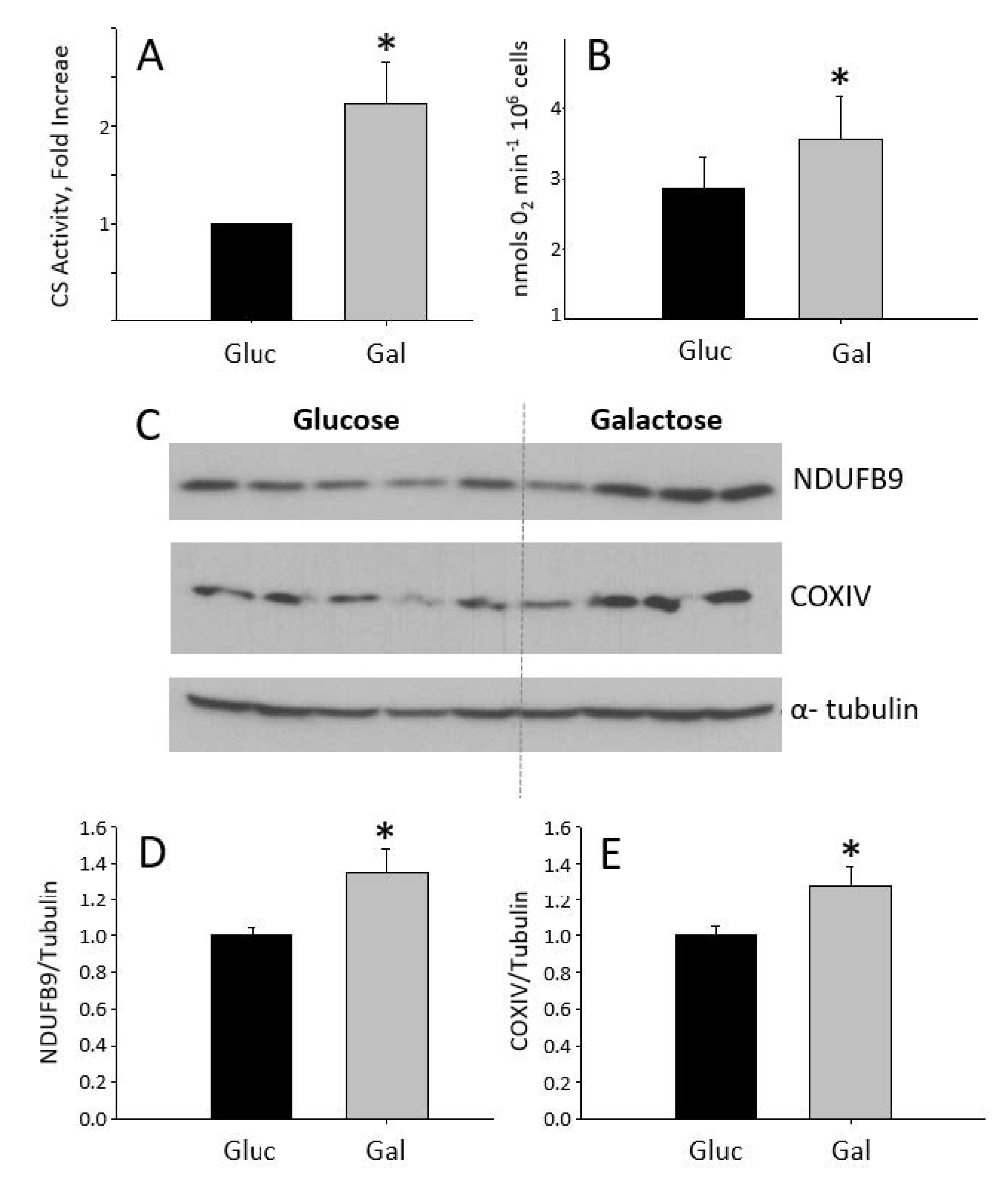
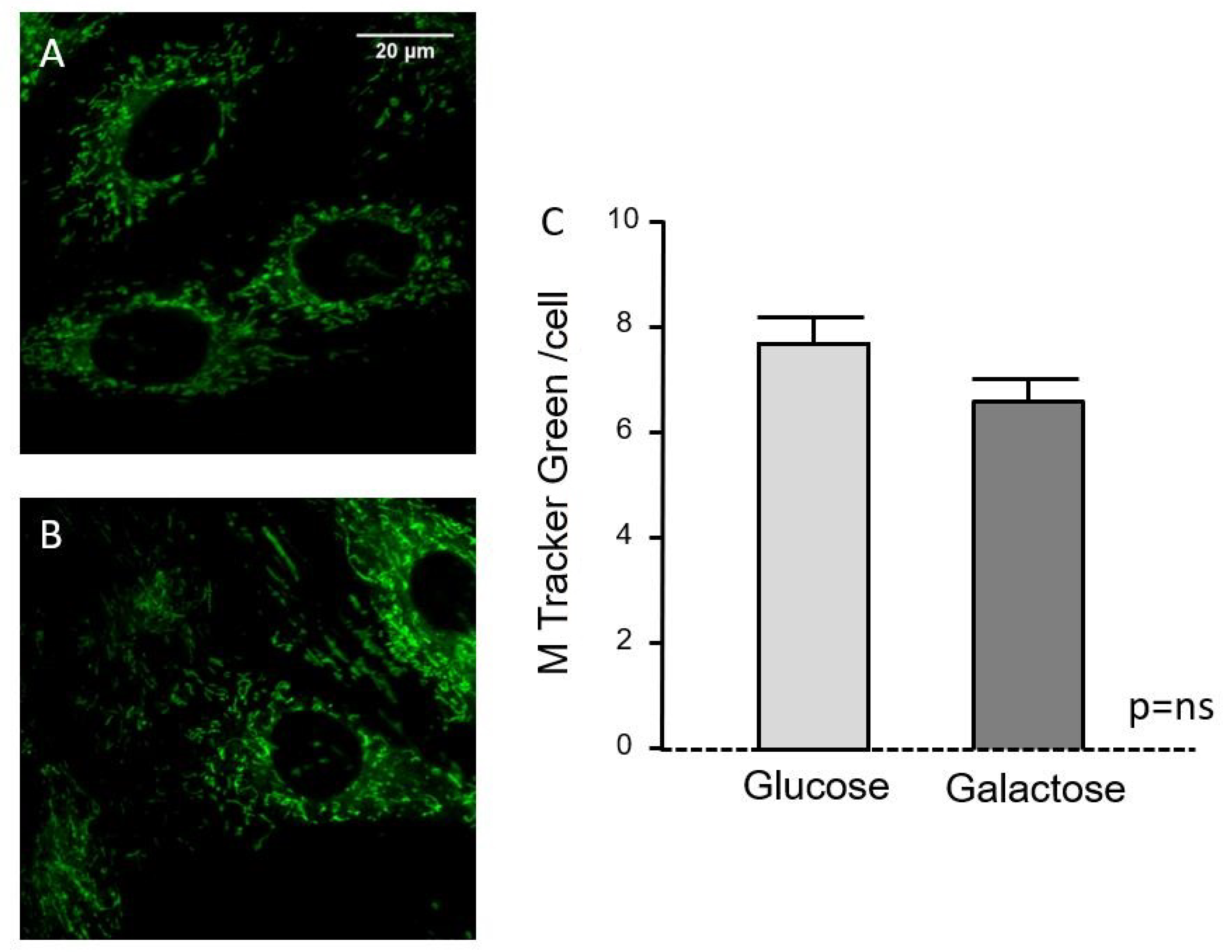
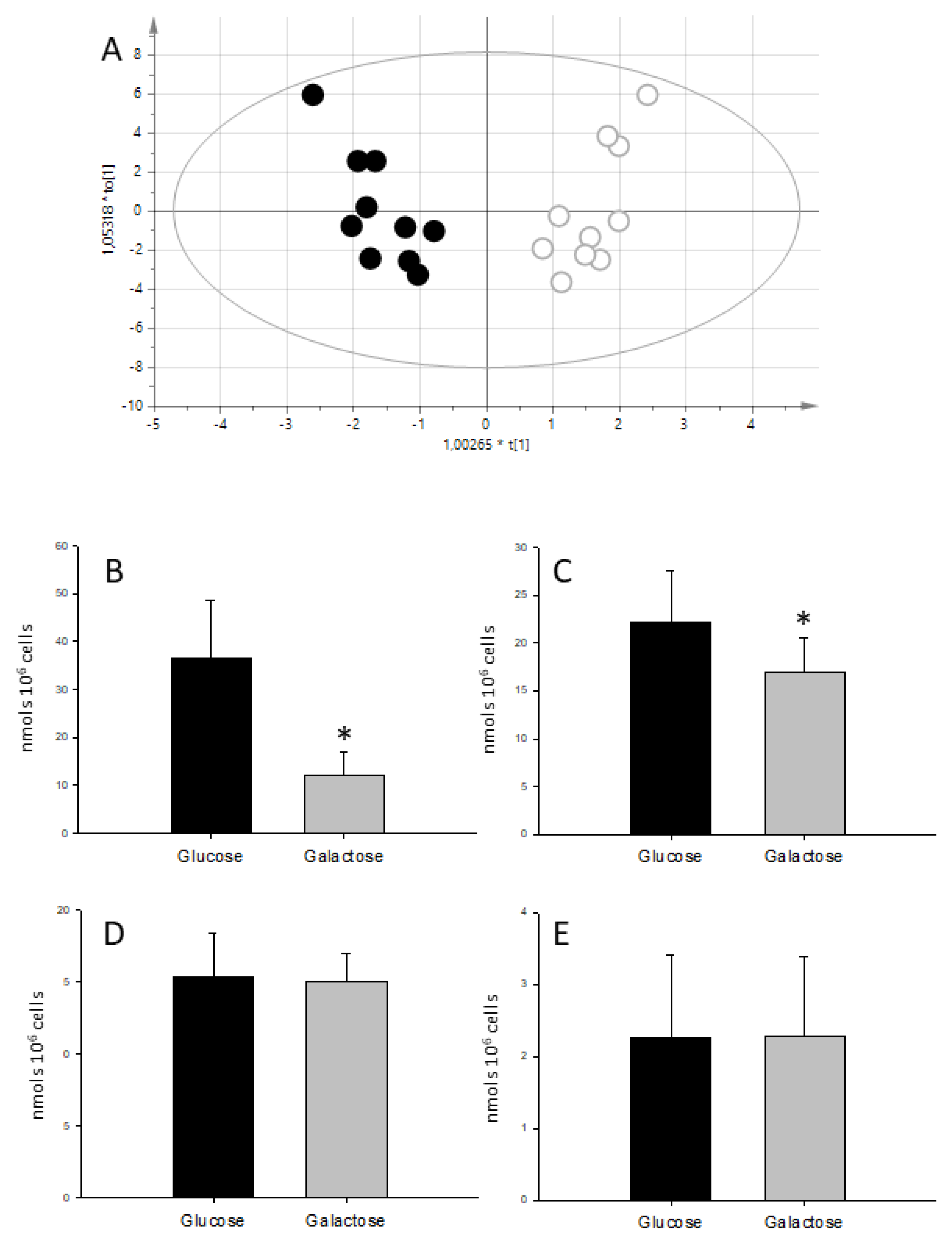
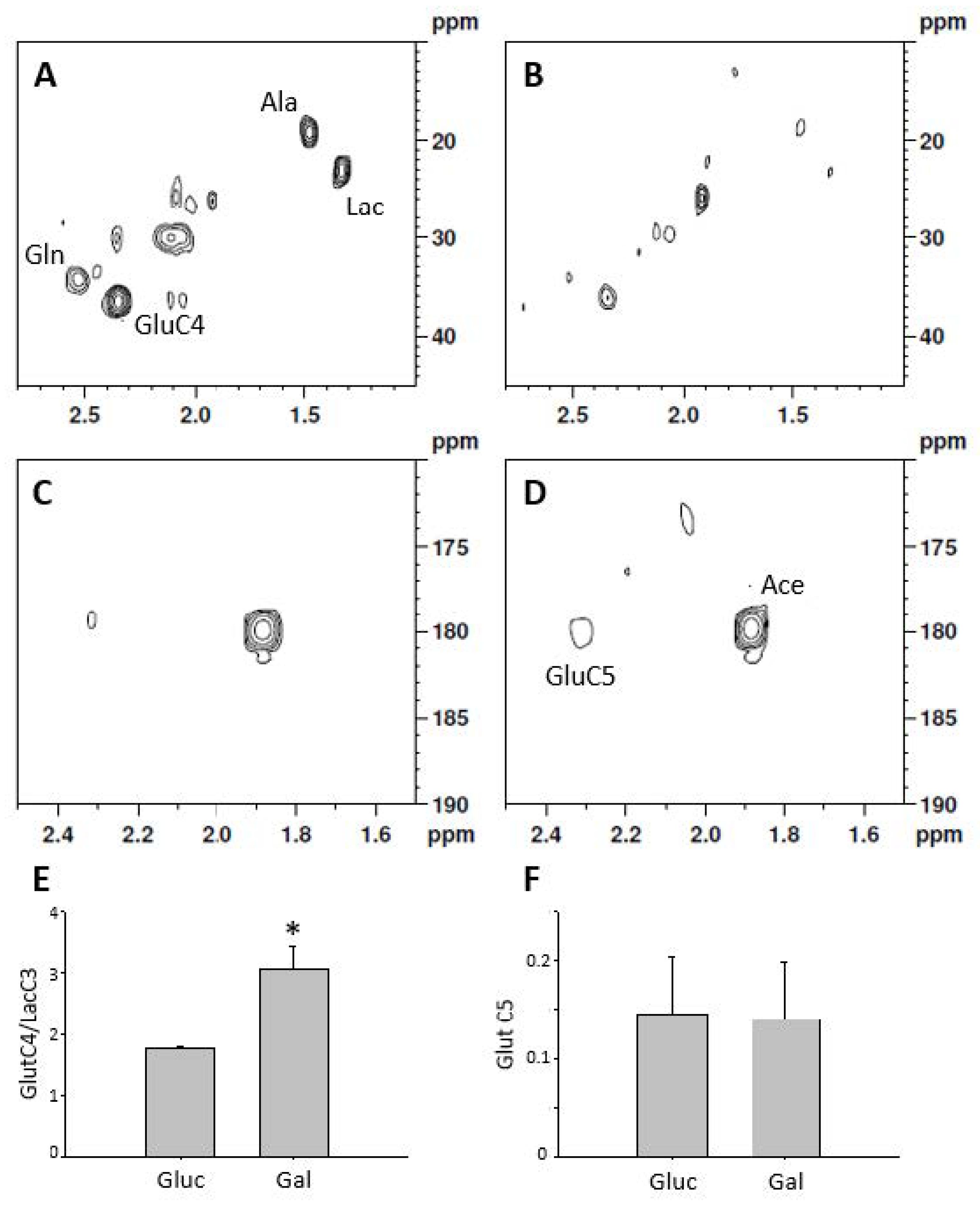
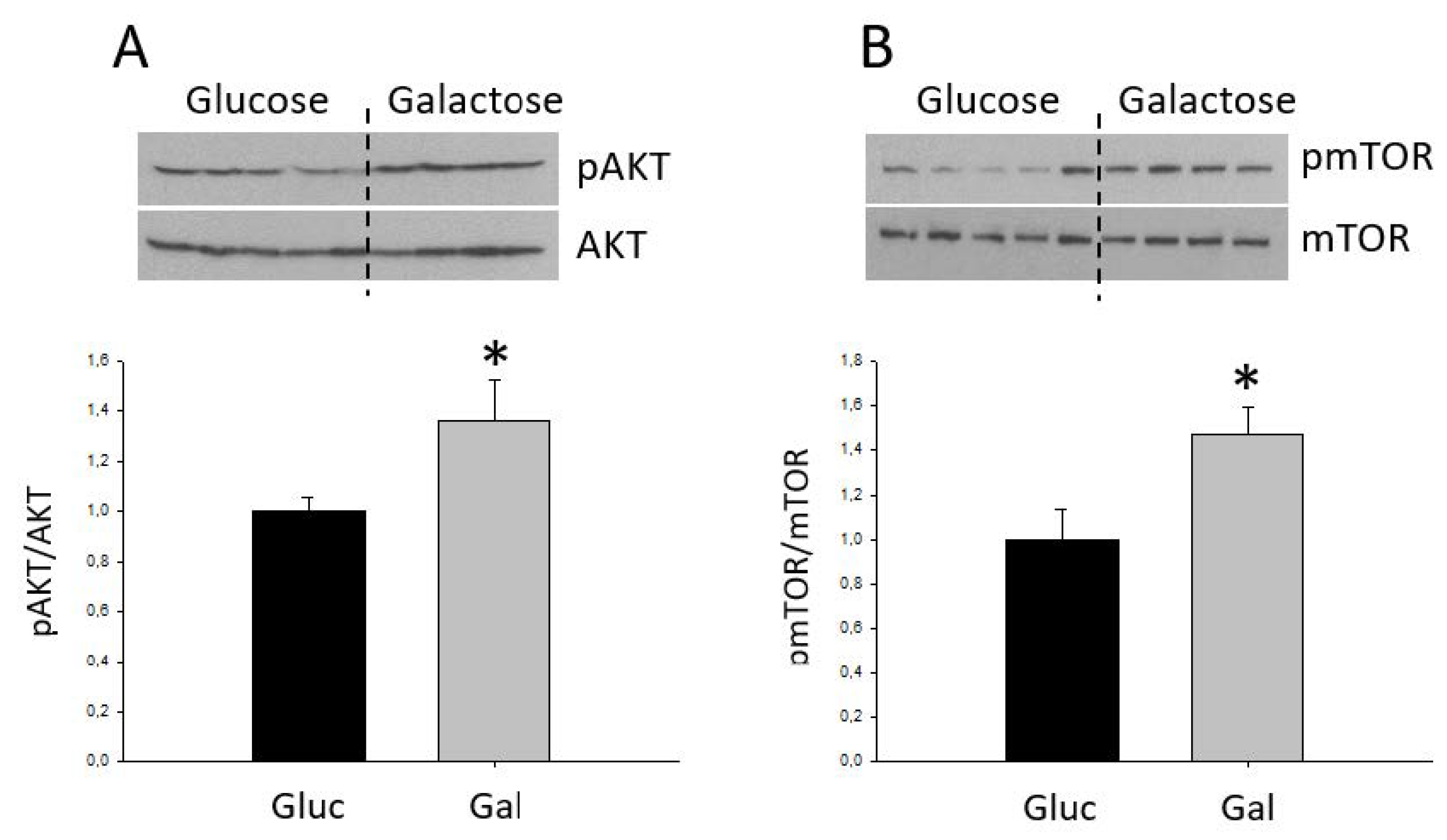
2.1. Characterization of the Galactose-Induced Metabolic Modulation
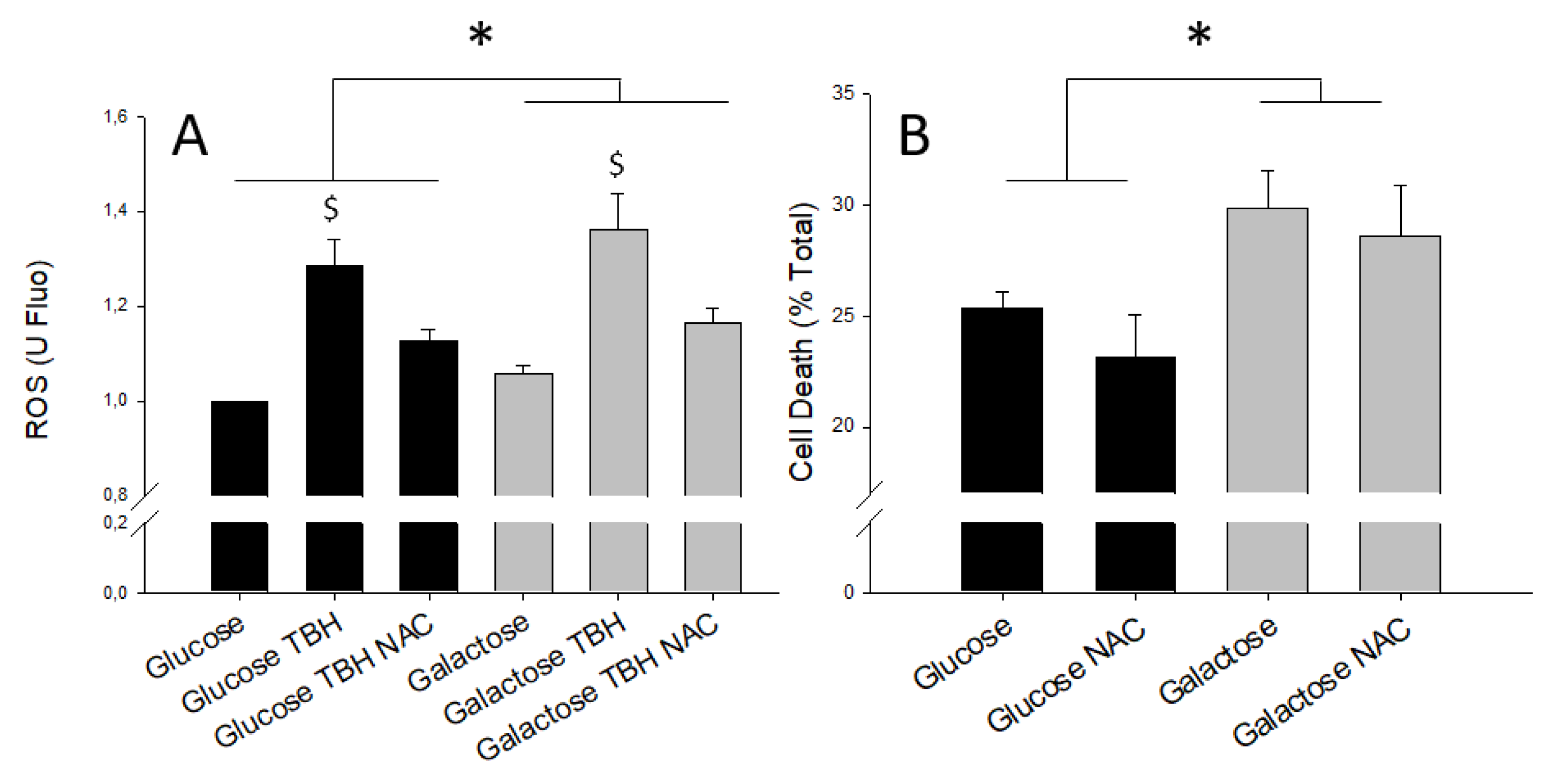
2.2. Glucose Oxidation and Protection against Ischemia-Reperfusion Injury
3. Discussion
4. Methods
4.1. Cell Culture
4.2. Oxygen Consumption
4.3. Simulated Iischemia-Reperfusion
4.4. ROS
4.5. Western Blot
4.6. Mitochondrial Pool
4.7. Gene Expression
4.8. Nuclear Magnetic Resonance
4.9. Pattern Recognition
4.10. Statistics
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mathers, C.D.; Loncar, D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006, 3, 2011–2030. [Google Scholar] [CrossRef] [Green Version]
- Ibáñez, B.; Heusch, G.; Ovize, M.; Van De Werf, F. Evolving therapies for myocardial ischemia/reperfusion injury. J. Am. Coll. Cardiol. 2015, 65, 1454–1471. [Google Scholar] [CrossRef] [Green Version]
- Piper, H.M.; García-Dorado, D.; Ovize, M. A fresh look at reperfusion injury. Cardiovasc. Res. 1998, 38, 291–300. [Google Scholar] [CrossRef] [Green Version]
- Yellon, D.M.; Hausenloy, D.J. Myocardial reperfusion injury. N. Engl. J. Med. 2007, 357, 1121–1135. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Botker, H.E.; Engstrom, T.; Erlinge, D.; Heusch, G.; Ibáñez, B.; Kloner, R.A.; Ovize, M.; Yellon, D.M.; Garcia-Dorado, D. Targeting reperfusion injury in patients with ST-segment elevation myocardial infarction: Trials and tribulations. Eur. Heart J. 2016, ehw145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopaschuk, G.D.; Ussher, J.R.; Folmes, C.D.L.; Jaswal, J.S.; Stanley, W.C. Myocardial Fatty Acid Metabolism in Health and Disease. Physiol. Rev. 2010, 90, 207–258. [Google Scholar] [CrossRef] [PubMed]
- Lionetti, V.; Stanley, W.C.; Recchia, F.A. Modulating fatty acid oxidation in heart failure. Cardiovasc. Res. 2011, 90, 202–209. [Google Scholar] [CrossRef]
- Liu, B.; Clanachan, A.S.; Schulz, R.; Lopaschuk, G.D. Cardiac Efficiency Is Improved After Ischemia by Altering Both the Source and Fate of Protons. Circ. Res. 1996, 79, 940–948. [Google Scholar] [CrossRef]
- Folmes, C.D.L.; Sowah, D.; Clanachan, A.S.; Lopaschuk, G.D. High rates of residual fatty acid oxidation during mild ischemia decrease cardiac work and ef fi ciency. J. Mol. Cell. Cardiol. 2009, 47, 142–148. [Google Scholar] [CrossRef]
- Malmberg, K.; Norhammar, A.; Wedel, H.; Rydén, L. Glycometabolic State at Admission: Important Risk Marker of Mortality in Conventionally Treated Patients With Diabetes Mellitus and Acute Myocardial Infarction. Circulation 1999, 99, 2626–2632. [Google Scholar] [CrossRef]
- Opie, L.L.; Lopaschuk, G.D. Fuels, aerobic and anaerobic metabolism. In Heart Physiology, from Cell to Circulation; Opie, L.L., Ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2004; p. 306. [Google Scholar]
- Nadtochiy, S.M.; Wang, Y.T.; Nehrke, K.; Munger, J.; Brookes, P.S. Cardioprotection by nicotinamide mononucleotide (NMN): Involvement of glycolysis and acidic pH. J. Mol. Cell. Cardiol. 2018, 121, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D. Metabolic Modulators in Heart Disease: Past, Present, and Future. Can. J. Cardiol. 2017, 33, 838–849. [Google Scholar] [CrossRef] [PubMed]
- Selker, H.P.; Beshanksy, J.R.; Sheehan, P.R.; Massaro, J.M.; Griffith, J.L.; D’Agostino, R.B.; Ruthazer, R.; Atkins, J.M.; Sayah, A.J.; Levy, M.K.; et al. Out-of-Hospital Administration of Intravenous Glucose-Insulin-Potassium in Patients With Suspected Acute Coronary Syndromes. Jama 2012, 307, 1925–1933. [Google Scholar] [PubMed] [Green Version]
- Van Der Horst, I.C.C.; De Luca, G.; Ottervanger, J.P.; De Boer, M.-J.; Hoorntje, J.C.A.; Suryapranata, H.; Dambrink, J.-H.E.; Gosselink, A.T.M.; Zijlstra, F.; Van’t Hof, A.W.J. ST-segment elevation resolution and outcome in patients treated with primary angioplasty and glucose-insulin-potassium infusion. Am. Heart, J. 2005, 149, 1135.e1–1135.e9. [Google Scholar] [CrossRef]
- Wiggers, H.; Nørrelund, H.; Nielsen, S.S.; Andersen, N.H.; Nielsen-kudsk, J.E.; Christiansen, J.S.; Nielsen, T.T.; Møller, N.; Bøtker, H.E.; Nørrelund, H.; et al. Influence of insulin and free fatty acids on contractile function in patients with chronically stunned and hibernating myocardium. Am. J. Physiol. Heart Circ. Physiol. 2005, 938–946. [Google Scholar] [CrossRef] [Green Version]
- Andrienko, T.N.; Pasdois, P.; Pereira, G.C.; Ovens, M.J.; Halestrap, A.P. The role of succinate and ROS in reperfusion injury—A critical appraisal. J. Mol. Cell. Cardiol. 2017, 110, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Meana, M.; Inserte, J.; Fernandez-Sanz, C.; Hernando, V.; Miro-Casas, E.; Barba, I.; Garcia-Dorado, D. The role of mitochondrial permeability transition in reperfusion-induced cardiomyocyte death depends on the duration of ischemia. Basic Res. Cardiol. 2011, 106, 1259–1268. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; James, A.M.; Work, L.M.; Saeb-Parsy, K.; Frezza, C.; Krieg, T.; Murphy, M.P. A unifying mechanism for mitochondrial superoxide production during ischemia-reperfusion injury. Cell Metab. 2016, 23, 254–263. [Google Scholar] [CrossRef] [Green Version]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijević, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef] [Green Version]
- Valls-Lacalle, L.; Barba, I.; Miro-Casas, E.; Alburquerque-Béjar, J.J.; Ruiz-Meana, M.; Fuertes-Agudo, M.; Rodríguez-Sinovas, A.; García-Dorado, D. Succinate dehydrogenase inhibition with malonate during reperfusion reduces infarct size by preventing mitochondrial permeability transition. Cardiovasc. Res. 2016, 109, 374–384. [Google Scholar] [CrossRef] [Green Version]
- Eagle, H.; Barban, S.; Levy, M.; Schulze, H.O. The utilization of carbohydrates by human cell cultures. J. Biol. Chem. 1958, 233, 551–558. [Google Scholar] [PubMed]
- Aguer, C.; Gambarotta, D.; Mailloux, R.J.; Moffat, C.; Dent, R.; McPherson, R.; Harper, M.E. Galactose enhances oxidative metabolism and reveals mitochondrial dysfunction in human primary muscle cells. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marroquin, L.D.; Hynes, J.; Dykens, J.A.; Jamieson, J.D.; Will, Y. Circumventing the crabtree effect: Replacing media glucose with galactose increases susceptibility of hepG2 cells to mitochondrial toxicants. Toxicol. Sci. 2007, 97, 539–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mot, A.I.; Liddell, J.R.; White, A.R.; Crouch, P.J. Circumventing the Crabtree Effect: A method to induce lactate consumption and increase oxidative phosphorylation in cell culture. Int. J. Biochem. Cell Biol. 2016, 79, 128–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deus, C.M.; Zehowski, C.; Nordgren, K.; Wallace, K.B.; Skildum, A.; Oliveira, P.J. Stimulating basal mitochondrial respiration decreases doxorubicin apoptotic signaling in H9c2 cardiomyoblasts. Toxicology 2015, 334, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Dott, W.; Mistry, P.; Wright, J.; Cain, K.; Herbert, K.E. Modulation of mitochondrial bioenergetics in a skeletal muscle cell line model of mitochondrial toxicity. Redox Biol. 2014, 2, 224–233. [Google Scholar] [CrossRef] [Green Version]
- Kuznetsov, A.V.; Javadov, S.; Sickinger, S.; Frotschnig, S.; Grimm, M. H9c2 and HL-1 cells demonstrate distinct features of energy metabolism, mitochondrial function and sensitivity to hypoxia-reoxygenation. Biochim. Biophys. Acta-Mol. Cell Res. 2015, 1853, 276–284. [Google Scholar] [CrossRef] [Green Version]
- Pereira, S.L.; Ramalho-Santos, J.; Branco, A.F.; Sardão, V.A.; Oliveira, P.J.; Carvalho, R.A. Metabolic remodeling during H9c2 myoblast differentiation: Relevance for in vitro toxicity studies. Cardiovasc. Toxicol. 2011, 11, 180–190. [Google Scholar] [CrossRef]
- Mashimo, T.; Pichumani, K.; Vemireddy, V.; Hatanpaa, K.J.; Singh, D.K.; Sirasanagandla, S.; Nannepaga, S.; Piccirillo, S.G.; Kovacs, Z.; Foong, C.; et al. Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell 2014, 159, 1603–1614. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Yan, B.; Yu, S.; Zhang, C.; Wang, B.; Wang, Y.; Wang, J.; Yuan, Z.; Zhang, L.; Pan, J. Coenzyme Q10 inhibits the aging of mesenchymal stem cells induced by D-galactose through Akt/mTOR signaling. Oxid. Med. Cell. Longev. 2015, 2015. [Google Scholar] [CrossRef]
- Govindaraju, V.; Young, K.; Maudsley, A.A. Proton NMR chemical shifts and coupling constants for brain metabolites. NMR Biomed. 2000, 13, 129–153. [Google Scholar] [CrossRef]
- Villena, J.A. New insights into PGC-1 coactivators : Redefining their role in the regulation of mitochondrial function and beyond. FEBS J. 2014, 282, 647–672. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.J.; Kahl, A.; Qian, L.; Stepanova, A.; Starkov, A.; Manfredi, G.; Iadecola, C.; Zhou, P. Prohibitin is a positive modulator of mitochondrial function in PC12 cells under oxidative stress. J. Neurochem. 2018, 146, 235–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, J.A.; Robles-Escajeda, E.; Oaxaca, D.M.; Padilla, D.L.; Kirken, R.A. The prohibitin protein complex promotes mitochondrial stabilization and cell survival in hematologic malignancies. Oncotarget 2017, 8, 65445–65456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kase, E.T.; Bakke, S.S.; Aas, V.; Nikolić, N.; Rustan, A.C.; Bogen, K.K.; Thoresen, G.H. Remodeling of Oxidative Energy Metabolism by Galactose Improves Glucose Handling and Metabolic Switching in Human Skeletal Muscle Cells. PLoS ONE 2013, 8, e59972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Yao, H.; Chen, X.; Wang, Z.; Xiang, Y.; Xia, J.; Liu, Y.; Wang, Y. Ginsenoside Rg1 Decreases Oxidative Stress and Down-Regulates Akt/mTOR Signalling to Attenuate Cognitive Impairment in Mice and Senescence of Neural Stem Cells Induced by d-Galactose. Neurochem. Res. 2018, 43, 430–440. [Google Scholar] [CrossRef]
- Nozari, Y.; Eshraghi, A.; Talasaz, A.H.; Bahremand, M.; Salamzadeh, J.; Salarifar, M.; Pourhosseini, H.; Jalali, A.; Mortazavi, S.H. Protection from Reperfusion Injury with Intracoronary N-Acetylcysteine in Patients with STEMI Undergoing Primary Percutaneous Coronary Intervention in a Cardiac Tertiary Center. Am. J. Cardiovasc. Drugs 2018, 18, 213–221. [Google Scholar] [CrossRef]
- Zweier, J.L.; Flaherty, J.T.; Weisfeldt, M.L. Direct measurement of free radical generation following reperfusion of ischemic myocardium. Proc. Natl. Acad. Sci. USA 1987, 84, 1404–1407. [Google Scholar] [CrossRef] [Green Version]
- Fujio, Y.; Nguyen, T.; Wencker, D.; Kitsis, R.N.; Walsh, K. Akt Promotes Survival of Cardiomyocytes In Vitro and Protects Against Ischemia-Reperfusion Injury in Mouse Heart. Circ. 2000, 101, 660–667. [Google Scholar] [CrossRef]
- Bai, J.; Lyden, P.D. Revisiting cerebral postischemic reperfusion injury: New insights in understanding reperfusion failure, hemorrhage, and edema. Int. J. Stroke 2015, 10, 143–152. [Google Scholar] [CrossRef]
- Jaeschke, H. Reperfusion injury after warm ischemia or cold storage of the liver: Role of apoptotic cell death. Transplant. Proc. 2002, 34, 2656–2658. [Google Scholar] [CrossRef]
- Enguix, N.; Pardo, R.; González, A.; López, V.; Simó, R.; Kralli, A.; Villena, J. Mice lacking PGC-1β in adipose tissues reveal a dissociation between mitochondrial dysfunction and insulin resistance. Mol. Metab. 2013, 2, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Barba, I.; Chavarria, L.; Ruiz-Meana, M.; Mirabet, M.; Agulló, E.; Garcia-Dorado, D. Effect of intracellular lipid droplets on cytosolic Ca2+ and cell death during ischaemia-reperfusion injury in cardiomyocytes. J. Physiol. 2009, 587, 1331–1341. [Google Scholar] [CrossRef] [PubMed]
- Pardo, R.; Vilà, M.; Cervela, L.; de Marco, M.; Gama-Pérez, P.; González-Franquesa, A.; Statuto, L.; Vilallonga, R.; Simó, R.; Garcia-Roves, P.; et al. Calorie restriction prevents diet-induced insulin resistance independently of PGC-1-driven mitochondrial biogenesis in white adipose tissue. FASEB J. 2019, 33, 2343–2358. [Google Scholar] [CrossRef] [Green Version]
- Castillo, A.; Vilà, M.; Pedriza, I.; Pardo, R.; Cámara, Y.; Martín, E.; Beiroa, D.; Torres-Torronteras, J.; Oteo, M.; Morcillo, M.A.; et al. Adipocyte MTERF4 regulates non-shivering adaptive thermogenesis and sympathetic-dependent glucose homeostasis. Biochim. Biophys. Acta-Mol. Basis Dis. 2019, 1865, 1298–1312. [Google Scholar] [CrossRef]
- Barba, I.; Miró-Casas, E.; Torrecilla, J.L.; Pladevall, E.; Tejedor, S.; Sebastián-Pérez, R.; Ruiz-Meana, M.; Berrendero, J.R.; Cuevas, A.; García-Dorado, D. High Fat Diet Induces Metabolic Changes and Reduces Oxidative Stress in Female Mouse Hearts. J. Nutr. Biochem. 2016, 40, 187–193. [Google Scholar] [CrossRef]
- Barba, I.; de León, G.; Martín, E.; Cuevas, A.; Aguade, S.; Candell-Riera, J.; Barrabés, J.A.; Garcia-Dorado, D. Nuclear magnetic resonance-based metabolomics predicts exercise-induced ischemia in patients with suspected coronary artery disease. Magn. Reson. Med. 2008, 60, 27–32. [Google Scholar] [CrossRef]
- Barba, I.; Andrés, M.; Picón, I.; Aguade, S.; Garcia-Dorado, D. Sex differences in the 1H NMR metabolic profile of serum in cardiovascular risk patients. Sci. Rep. 2019, 9, 2380. [Google Scholar] [CrossRef] [Green Version]
- Frick, M.A.; Barba, I.; Fenoy-alejandre, M.; Lopez-Lopez, P.; Baquero-artigao, F.; Rodriguez-Molino, P.; Noguera-julian, A.; Nicolás-López, M.; de la Fuente-Juárez, A.; Codina-Grau, M.G.; et al. 1H-NMR Urinary Metabolic Profile, A Promising Tool for the Management of Infants with Human Cytomegalovirus-Infection. Metabolites 2019, 9, 288. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolite | Galactoe | Glucose | p |
|---|---|---|---|
| ATP | 15.6 ± 1.88 | 15.31 ± 3.06 | 0.832 |
| Acetate | 16.3 ± 19.67 | 15.43 ± 24.83 | 0.932 |
| Alanine | 16.93 ± 3.69 | 22.14 ± 5.44 | 0.022 |
| Creatine | 11.74 ± 3.57 | 11.01 ± 3.39 | 0.642 |
| Creatine phosphate | 2.28 ± 1.10 | 2.26 ± 1.16 | 0.969 |
| Galactose | 39.68 ± 13.54 | n.d. | |
| Glucose | 17.75 ± 4.80 | 56.82 ± 42.60 | 0.099 |
| Glutamate | 184.52 ± 38.07 | 170.01 ± 32.73 | 0.373 |
| Glutamine | 46.51 ± 11.12 | 41.10 ± 13.64 | 0.344 |
| Glycine | 56.16 ± 25.97 | 50.07 ± 18.46 | 0.554 |
| Isoleucine | 12.72 ± 4.00 | 9.54 ± 2.48 | 0.047 |
| Lactate | 12.25 ± 4.75 | 36.55 ± 12.11 | <0.001 |
| Leucine | 12.17 ± 2.52 | 9.82 ± 2.74 | 0.061 |
| Succinate | 2.28 ± 0.90 | 3.08 ± 1.12 | 0.095 |
| Taurine | 65.26 ± 19.41 | 69.72 ± 27.78 | 0.687 |
| Threonine | 31.29 ± 12.37 | 26.45 ± 8.21 | 0.351 |
| Valine | 13.06 ± 13.22 | 10.60 ± 4.04 | 0.149 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beltran, C.; Pardo, R.; Bou-Teen, D.; Ruiz-Meana, M.; Villena, J.A.; Ferreira-González, I.; Barba, I. Enhancing Glycolysis Protects against Ischemia-Reperfusion Injury by Reducing ROS Production. Metabolites 2020, 10, 132. https://doi.org/10.3390/metabo10040132
Beltran C, Pardo R, Bou-Teen D, Ruiz-Meana M, Villena JA, Ferreira-González I, Barba I. Enhancing Glycolysis Protects against Ischemia-Reperfusion Injury by Reducing ROS Production. Metabolites. 2020; 10(4):132. https://doi.org/10.3390/metabo10040132
Chicago/Turabian StyleBeltran, Claudia, Rosario Pardo, Diana Bou-Teen, Marisol Ruiz-Meana, Josep A. Villena, Ignacio Ferreira-González, and Ignasi Barba. 2020. "Enhancing Glycolysis Protects against Ischemia-Reperfusion Injury by Reducing ROS Production" Metabolites 10, no. 4: 132. https://doi.org/10.3390/metabo10040132
APA StyleBeltran, C., Pardo, R., Bou-Teen, D., Ruiz-Meana, M., Villena, J. A., Ferreira-González, I., & Barba, I. (2020). Enhancing Glycolysis Protects against Ischemia-Reperfusion Injury by Reducing ROS Production. Metabolites, 10(4), 132. https://doi.org/10.3390/metabo10040132