QSRR Automator: A Tool for Automating Retention Time Prediction in Lipidomics and Metabolomics

Abstract
:1. Introduction
2. Results
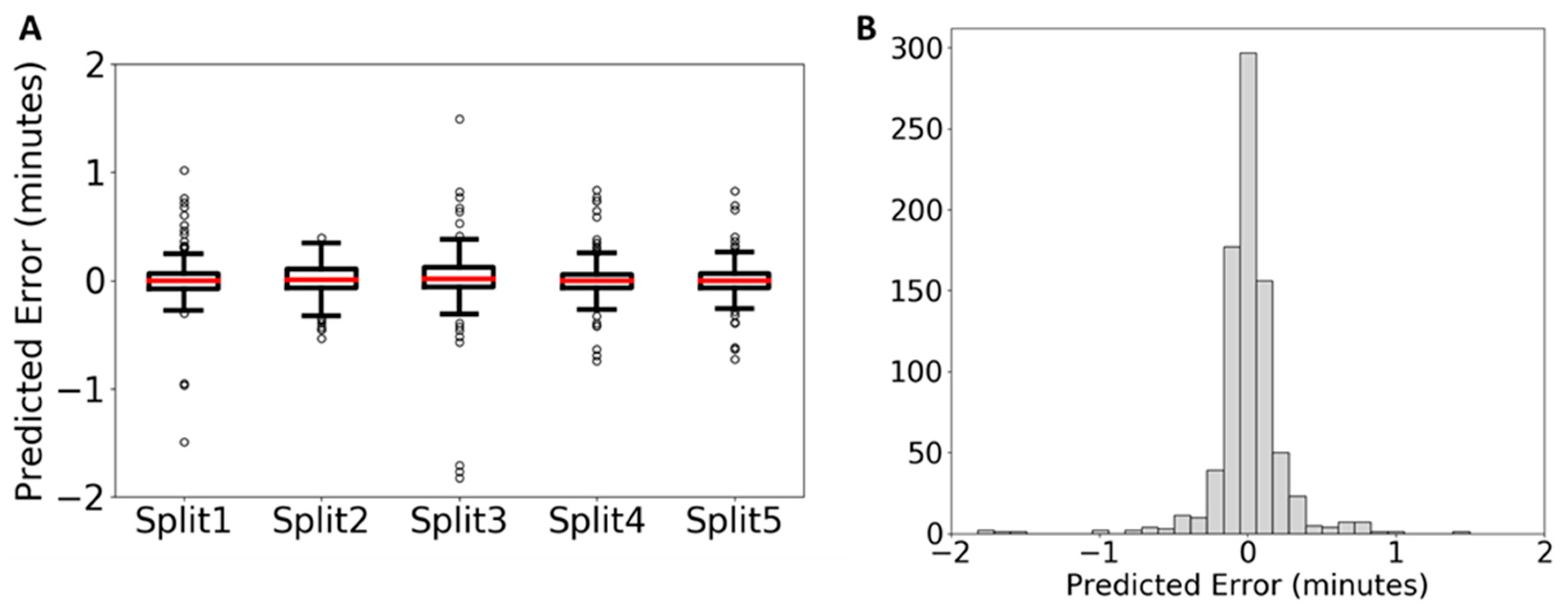
2.1. Comparison to Previously Published Data
2.1.1. Direct Comparison of QSRR Automator to Published Data-Sets
2.1.2. Use of Published Test and Training Data-Sets
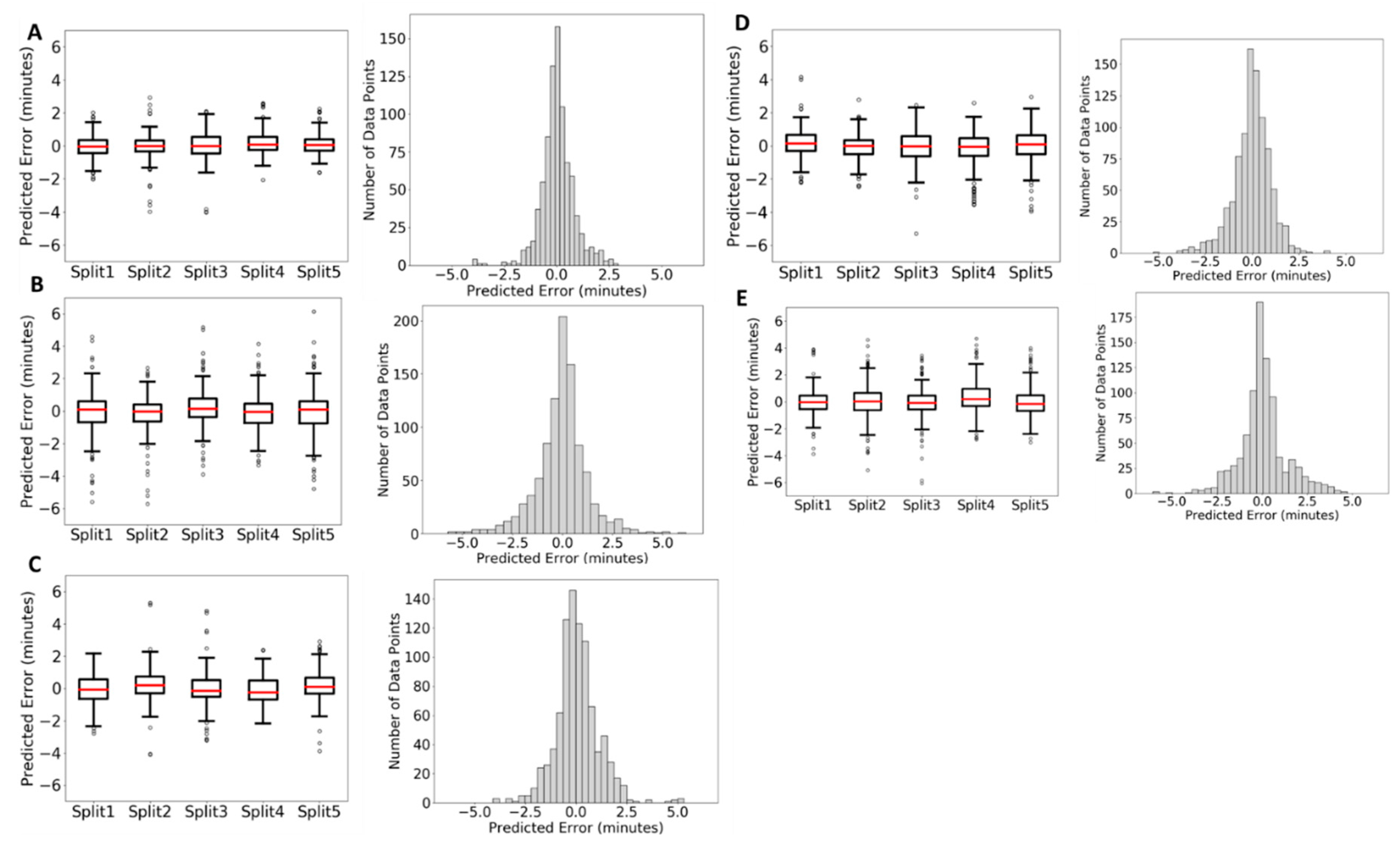
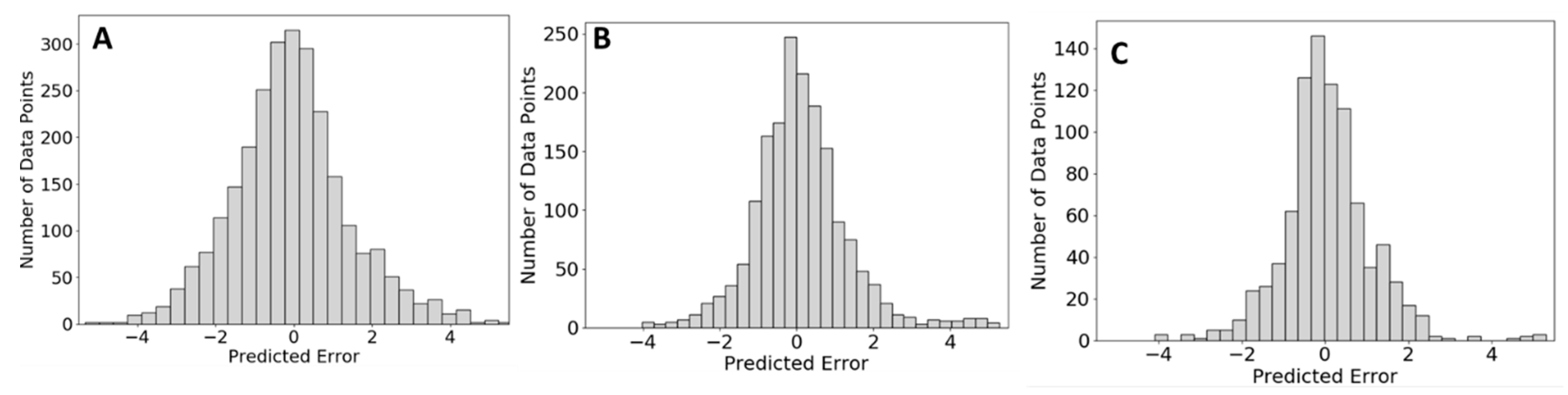
2.2. Tests on In-House Data
2.2.1. Lipidomics Data
2.2.2. Metabolomics Data
2.2.3. Comparison of QSRR Automator Models on HILIC Columns
3. Discussion
4. Materials and Methods
4.1. QSRR Automator Software
4.1.1. Software Used in the Creation of QSRR Automator
4.1.2. QSRR Automator Workflow
4.2. Comparison to Previously Published Data
4.3. Tests on in-House Data
4.3.1. Lipid Extraction
4.3.2. Metabolomics Data
4.3.3. Limited Feature Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Program Availability
References
- Aicheler, F.; Li, J.; Hoene, M.; Lehmann, R.; Xu, G.; Kohlbacher, O. Retention Time Prediction Improves Identification in Nontargeted Lipidomics Approaches. Anal. Chem. 2015, 87, 7698–7704. [Google Scholar] [CrossRef] [PubMed]
- Creek, D.J.; Jankevics, A.; Breitling, R.; Watson, D.G.; Barrett, M.P.; Burgess, K.E. Toward global metabolomics analysis with hydrophilic interaction liquid chromatography-mass spectrometry: Improved metabolite identification by retention time prediction. Anal. Chem. 2011, 83, 8703–8710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanstrup, J.; Neumann, S.; Vrhovšek, U. PredRet: Prediction of retention time by direct mapping between multiple chromatographic systems. Anal. Chem. 2015, 87, 9421–9428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bach, E.; Szedmak, S.; Brouard, C.; Böcker, S.; Rousu, J. Liquid-chromatography retention order prediction for metabolite identification. Bioinformatics 2018, 34, i875–i883. [Google Scholar] [CrossRef] [PubMed]
- Mahieu, N.G.; Patti, G.J. Systems-Level Annotation of a Metabolomics Data Set Reduces 25 000 Features to Fewer than 1000 Unique Metabolites. Anal. Chem. 2017, 89, 10397–10406. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.; Fraser, K.; Huege, J.; Featonby, T.; Rasmussen, S.; Jones, C. Predicting retention time in hydrophilic interaction liquid chromatography mass spectrometry and its use for peak annotation in metabolomics. Metabolomics 2015, 11, 696–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goryński, K.; Bojko, B.; Nowaczyk, A.; Buciński, A.; Pawliszyn, J.; Kaliszan, R. Quantitative structure-retention relationships models for prediction of high performance liquid chromatography retention time of small molecules: Endogenous metabolites and banned compounds. Anal. Chim. Acta 2013, 797, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Bouwmeester, R.; Martens, L.; Degroeve, S. Comprehensive and Empirical Evaluation of Machine Learning Algorithms for Small Molecule LC Retention Time Prediction. Anal. Chem. 2019, 91, 3694–3703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsugawa, H.; Cajka, T.; Kind, T.; Ma, Y.; Higgins, B.; Ikeda, K.; Kanazawa, M.; VanderGheynst, J.; Fiehn, O.; Arita, M. MS-DIAL: Data-independent MS/MS deconvolution for comprehensive metabolome analysis. Nat. Methods 2015, 12, 523–526. [Google Scholar] [CrossRef] [PubMed]
- Kaliszan, R. QSRR: Quantitative structure-(chromatographic) retention relationships. Chem. Rev. 2007, 107, 3212–3246. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Uchikata, T.; Sakamoto, S.; Yokoi, Y.; Fukusaki, E.; Bamba, T. Development of a lipid profiling system using reverse-phase liquid chromatography coupled to high-resolution mass spectrometry with rapid polarity switching and an automated lipid identification software. J. Chromatogr. A 2013, 1292, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Sandra, K.; Pereira, A.O.S.; Vanhoenacker, G.; David, F.; Sandra, P. Comprehensive blood plasma lipidomics by liquid chromatography/quadrupole time-of-flight mass spectrometry. J. Chromatogr. A 2010, 1217, 4087–4099. [Google Scholar] [CrossRef] [PubMed]
- Moriwaki, H.; Tian, Y.S.; Kawashita, N.; Takagi, T. Mordred: A molecular descriptor calculator. J. Cheminform. 2018, 10, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- RDKit: Open-Source Cheminformatics. Available online: https://github.com/rdkit/rdkit (accessed on 15 May 2019).
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Model. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res. 2019, 47, D1102–D1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matyash, V.; Liebisch, G.; Kurzchalia, T.V.; Shevchenko, A.; Schwudke, D. Lipid extraction by methyl-tert-butyl ether for high-throughput lipidomics. J. Lipid Res. 2008, 49, 1137–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fahy, E.; Sud, M.; Cotter, D.; Subramaniam, S. LIPID MAPS online tools for lipid research. Nucleic Acids Res. 2007, 35, W606–W612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guijas, C.; Montenegro-Burke, J.R.; Domingo-Almenara, X.; Palermo, A.; Warth, B.; Hermann, G.; Koellensperger, G.; Huan, T.; Uritboonthai, W.; Aisporna, A.E.; et al. METLIN: A Technology Platform for Identifying Knowns and Unknowns. Anal. Chem. 2018, 90, 3156–3164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data-Set | -omics Type | Column | Published Model | Published # of Features | QSRR Automator Model | QSSR Automator # of Features |
|---|---|---|---|---|---|---|
| RP_Lipid [1] | Lipidomics | RP | SVR | 12 | SVR | 11–31 |
| RP_Met [7] | Metabolomics | RP | MLR | 3 | RF or SVR | 11–246 |
| HILIC_MLR1 [7] | Metabolomics | HILIC | MLR | 3 | SVR | 21–146 |
| HILIC_MLR2 [2] | Metabolomics | HILIC | MLR | 6 | SVR | 14–44 |
| HILIC_RF [6] | Metabolomics | HILIC | RF | 4 | RF or SVR | 14–294 |
| Data-Set | Published Best Fit Equation | Published r2 | QSRR Automater Best Fit Line | QSRR Automater r2 |
|---|---|---|---|---|
| RP_Lipid | n/a | 0.989 | y = 0.9778x + 0.1736 | 0.9942 |
| RP_Met | y = 0.8929x + 1.4018 | 0.7685 | y = 0.8466x + 1.824 | 0.7935 |
| HILIC_MLR1 | y = 0.5825x + 1.1667 | 0.65 | y = 0.5789x + 0.8424 | 0.5911 |
| HILIC_MLR2 | y = 0.8812x + 0.8575 | 0.8375 | y = 0.7909x + 2.0814 | 0.7385 |
| HILIC_RF | y = 0.9523x + 0.3217 | 0.8596 | y = 0.7729x + 2.8072 | 0.6667 |
| BEH Amide | cHILIC | HILIC-z | iHILIC | pHILIC | |
|---|---|---|---|---|---|
| Median Prediction Error | 0.009 | 0.044 | −0.022 | 0.029 | −0.025 |
| Median Prediction % Error | 0.18% | 0.49% | 0.59% | −0.28% | −0.29% |
| Average # of features per model | 72.4 | 58 | 19 | 59.73 | 58.73 |
| Elution time range (min) | 1.1–8.8 | 2.0–15 | 1.8–12 | 1.6–13 | 1.5–18 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naylor, B.C.; Catrow, J.L.; Maschek, J.A.; Cox, J.E. QSRR Automator: A Tool for Automating Retention Time Prediction in Lipidomics and Metabolomics. Metabolites 2020, 10, 237. https://doi.org/10.3390/metabo10060237
Naylor BC, Catrow JL, Maschek JA, Cox JE. QSRR Automator: A Tool for Automating Retention Time Prediction in Lipidomics and Metabolomics. Metabolites. 2020; 10(6):237. https://doi.org/10.3390/metabo10060237
Chicago/Turabian StyleNaylor, Bradley C., J. Leon Catrow, J. Alan Maschek, and James E. Cox. 2020. "QSRR Automator: A Tool for Automating Retention Time Prediction in Lipidomics and Metabolomics" Metabolites 10, no. 6: 237. https://doi.org/10.3390/metabo10060237
APA StyleNaylor, B. C., Catrow, J. L., Maschek, J. A., & Cox, J. E. (2020). QSRR Automator: A Tool for Automating Retention Time Prediction in Lipidomics and Metabolomics. Metabolites, 10(6), 237. https://doi.org/10.3390/metabo10060237