Comparison of Three Untargeted Data Processing Workflows for Evaluating LC-HRMS Metabolomics Data


 and
and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Study Design
2.2. Untargeted Metabolomics
2.2.1. Parameter Optimization for the Three Different Workflows of Untargeted Metabolomics
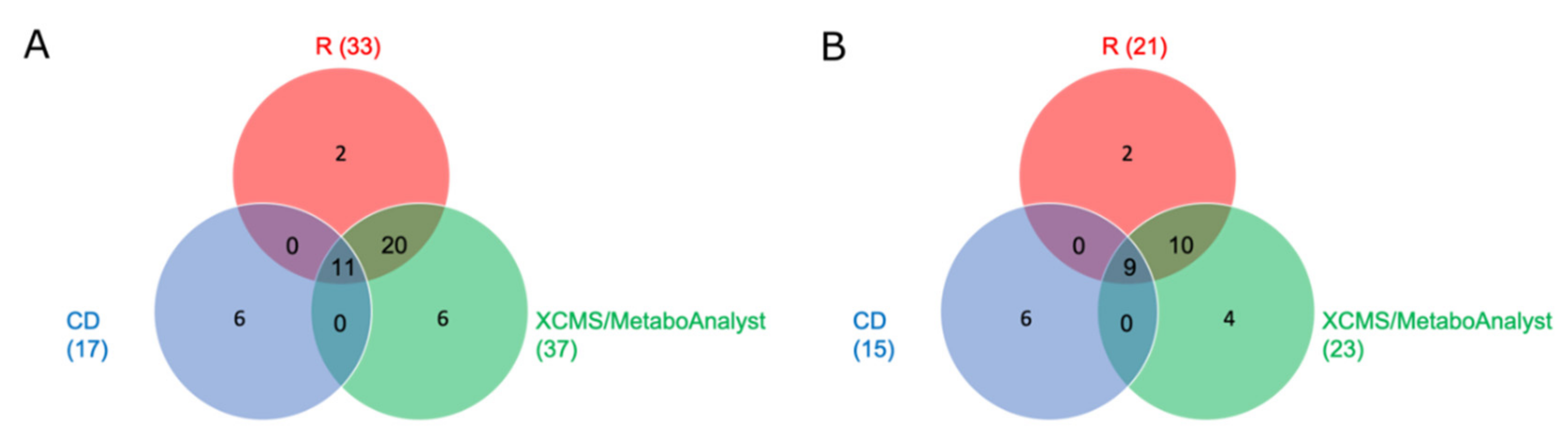
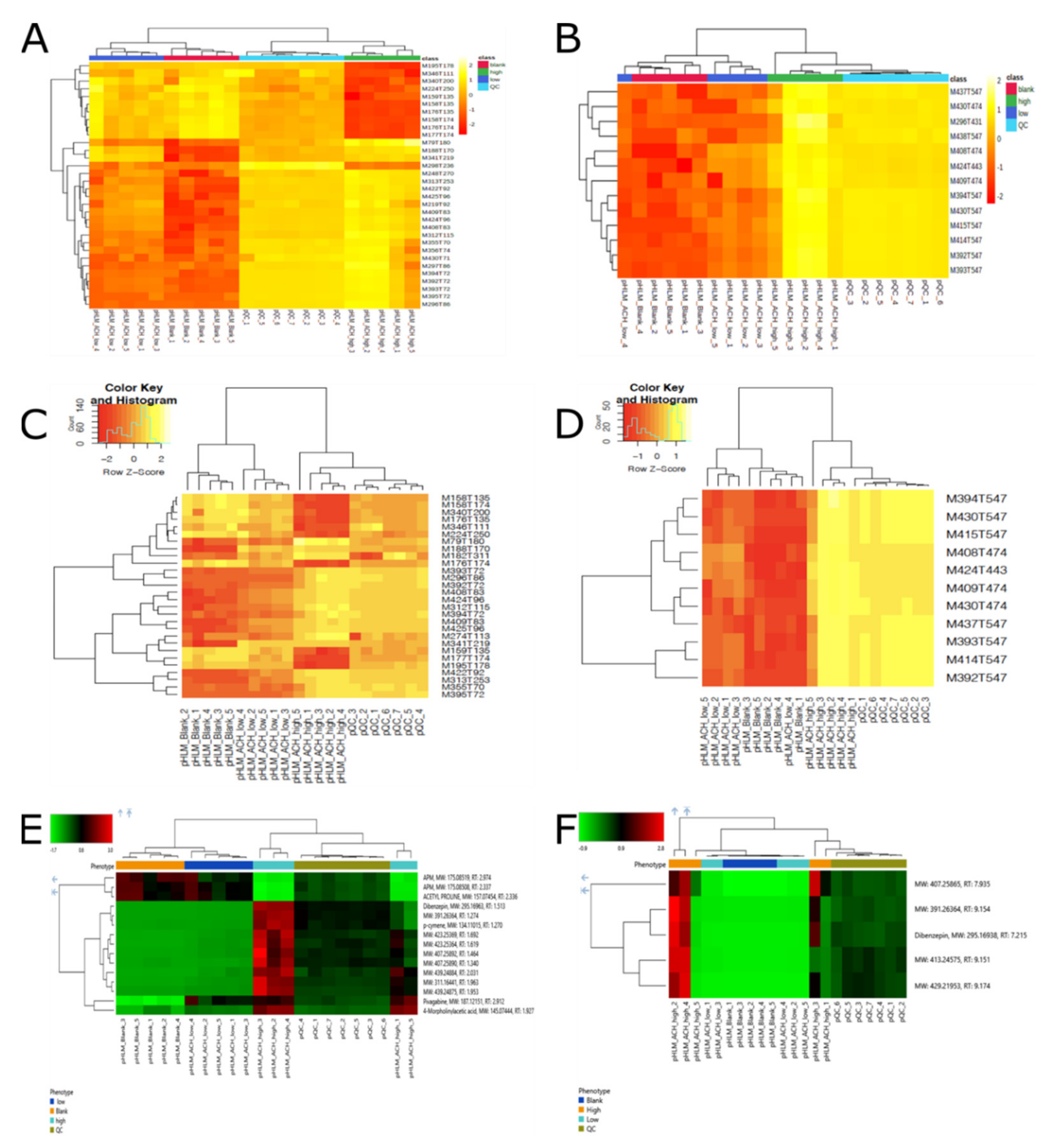
2.2.2. Comparison of Significant Features of the Three Different Workflows
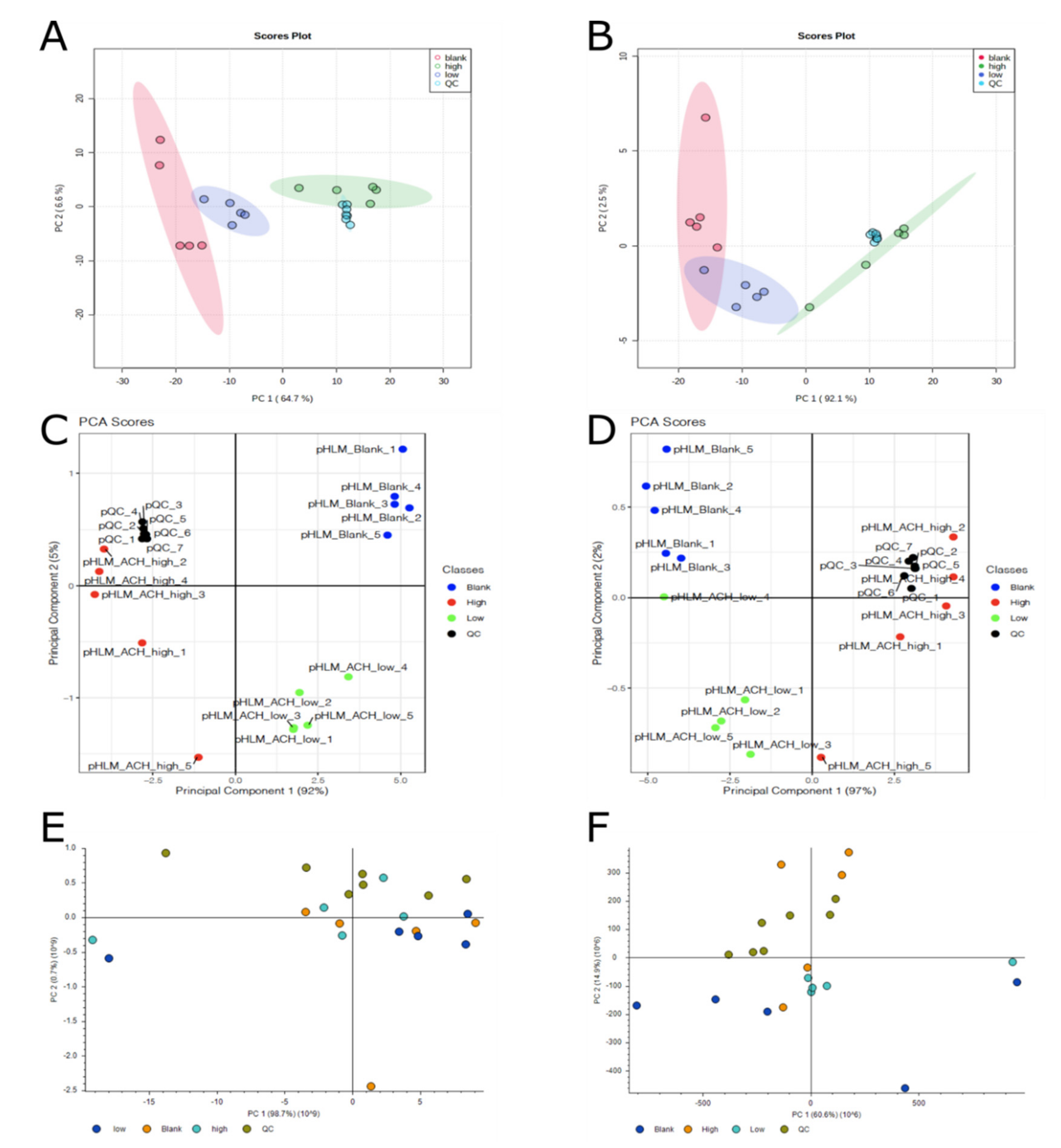
2.2.3. Comparison of Multivariate Statistics of the Three Different Software Workflows
2.3. Targeted Metabolomics
2.3.1. Identification of Significant Features
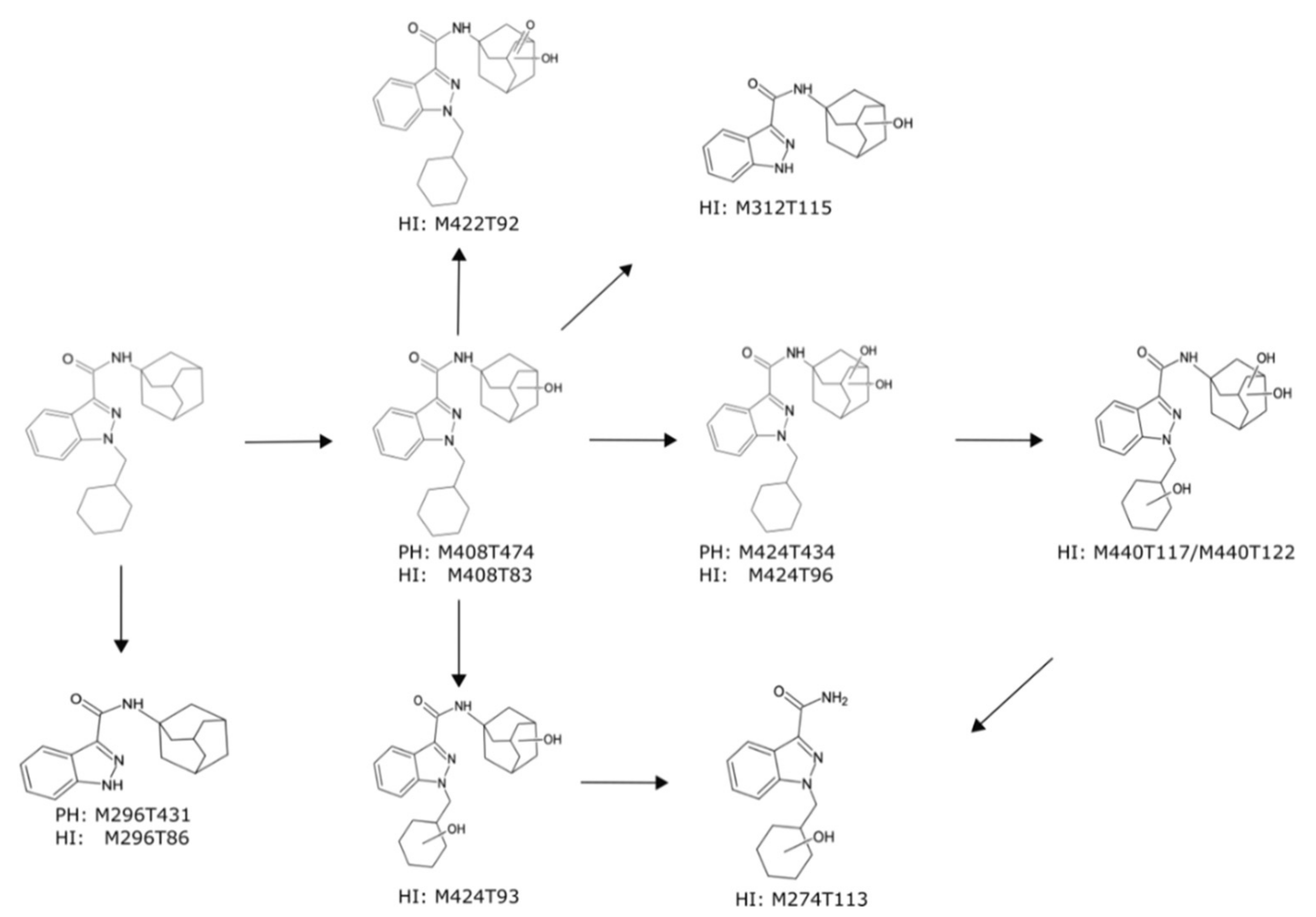
2.3.2. Metabolism of A-CHMINACA in pHLM
2.4. Comparison of the Three Software Workflows
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. pHLM Incubation
3.3. LC-HRMS/MS Apparatus
3.4. Dataset Processing with Different Software
3.5. Identification of Significant Features
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Barnes, S.; Benton, H.P.; Casazza, K.; Cooper, S.J.; Cui, X.; Du, X.; Engler, J.; Kabarowski, J.H.; Li, S.; Pathmasiri, W.; et al. Training in metabolomics research. I. Designing the experiment, collecting and extracting samples and generating metabolomics data. J. Mass Spectrom. 2016, 51, 461–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Locasale, J.W. Metabolomics: A Primer. Trends Biochem. Sci. 2017, 42, 274–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agin, A.; Heintz, D.; Ruhland, E.; Chao de la Barca, J.M.; Zumsteg, J.; Moal, V.; Gauchez, A.S.; Namer, I.J. Metabolomics—An overview. From basic principles to potential biomarkers (part 1). Méd. Nucl. 2016, 40, 4–10. [Google Scholar] [CrossRef]
- Naz, S.; Vallejo, M.; Garcia, A.; Barbas, C. Method validation strategies involved in non-targeted metabolomics. J. Chromatogr. A 2014, 1353, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Sheflin, A.M.; Broeckling, C.D.; Prenni, J.E. Data Processing for GC-MS- and LC-MS-Based Untargeted Metabolomics. Methods Mol. Biol. 2019, 1978, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Rurik, M.; Alka, O.; Aicheler, F.; Kohlbacher, O. Metabolomics Data Processing Using OpenMS. Methods Mol. Biol. 2020, 2104, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Ochoa, A.; Quirantes-Pine, R.; Borras-Linares, I.; Cadiz-Gurrea, M.L.; Precisesads Clinical, C.; Alarcon Riquelme, M.E.; Brunius, C.; Segura-Carretero, A. A Case Report of Switching from Specific Vendor-Based to R-Based Pipelines for Untargeted LC-MS Metabolomics. Metabolites 2020, 10. [Google Scholar] [CrossRef] [Green Version]
- Sugimoto, M.; Kawakami, M.; Robert, M.; Soga, T.; Tomita, M. Bioinformatics Tools for Mass Spectroscopy-Based Metabolomic Data Processing and Analysis. Curr. Bioinform. 2012, 7, 96–108. [Google Scholar] [CrossRef]
- Katajamaa, M.; Oresic, M. Data processing for mass spectrometry-based metabolomics. J. Chromatogr. A 2007, 1158, 318–328. [Google Scholar] [CrossRef] [PubMed]
- Castillo, S.; Gopalacharyulu, P.; Yetukuri, L.; Orešič, M. Algorithms and tools for the preprocessing of LC–MS metabolomics data. Chemom. Intell. Lab. Syst. 2011, 108, 23–32. [Google Scholar] [CrossRef]
- Spicer, R.; Salek, R.M.; Moreno, P.; Canueto, D.; Steinbeck, C. Navigating freely-available software tools for metabolomics analysis. Metabolomics 2017, 13, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cambiaghi, A.; Ferrario, M.; Masseroli, M. Analysis of metabolomic data: Tools, current strategies and future challenges for omics data integration. Brief. Bioinform. 2017, 18, 498–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Lu, Y.; Guo, Y.; Cao, H.; Wang, Q.; Shui, W. Comprehensive evaluation of untargeted metabolomics data processing software in feature detection, quantification and discriminating marker selection. Anal. Chim. Acta 2018, 1029, 50–57. [Google Scholar] [CrossRef]
- Smith, C.A.; Want, E.J.; O’Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: Processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Manier, S.K.; Keller, A.; Meyer, M.R. Automated optimization of XCMS parameters for improved peak picking of liquid chromatography-mass spectrometry data using the coefficient of variation and parameter sweeping for untargeted metabolomics. Drug Test Anal 2019, 11, 752–761. [Google Scholar] [CrossRef]
- Manier, S.K.; Keller, A.; Schaper, J.; Meyer, M.R. Untargeted metabolomics by high resolution mass spectrometry coupled to normal and reversed phase liquid chromatography as a tool to study the in vitro biotransformation of new psychoactive substances. Sci. Rep. 2019, 9, 2741. [Google Scholar] [CrossRef] [Green Version]
- Manier, S.K.; Wagmann, L.; Flockerzi, V.; Meyer, M.R. Toxicometabolomics of the new psychoactive substances alpha-PBP and alpha-PEP studied in HepaRG cell incubates by means of untargeted metabolomics revealed unexpected amino acid adducts. Arch. Toxicol. 2020, 94, 2047–2059. [Google Scholar] [CrossRef] [Green Version]
- Asha, S.; Vidyavathi, M. Role of human liver microsomes in in vitro metabolism of drugs-a review. Appl. Biochem. Biotechnol. 2010, 160, 1699–1722. [Google Scholar] [CrossRef]
- Myers, O.D.; Sumner, S.J.; Li, S.; Barnes, S.; Du, X. Detailed Investigation and Comparison of the XCMS and MZmine 2 Chromatogram Construction and Chromatographic Peak Detection Methods for Preprocessing Mass Spectrometry Metabolomics Data. Anal. Chem. 2017, 89, 8689–8695. [Google Scholar] [CrossRef]
- Lange, E.; Tautenhahn, R.; Neumann, S.; Gropl, C. Critical assessment of alignment procedures for LC-MS proteomics and metabolomics measurements. BMC Bioinform. 2008, 9, 375. [Google Scholar] [CrossRef] [Green Version]
- Garcia, C.J.; Yang, X.; Huang, D.; Tomas-Barberan, F.A. Can we trust biomarkers identified using different non-targeted metabolomics platforms? Multi-platform, inter-laboratory comparative metabolomics profiling of lettuce cultivars via UPLC-QTOF-MS. Metabolomics 2020, 16, 85. [Google Scholar] [CrossRef]
- Coble, J.B.; Fraga, C.G. Comparative evaluation of preprocessing freeware on chromatography/mass spectrometry data for signature discovery. J. Chromatogr. A 2014, 1358, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Broadhurst, D.I.; Kell, D.B. Statistical strategies for avoiding false discoveries in metabolomics and related experiments. Metabolomics 2006, 2, 171–196. [Google Scholar] [CrossRef] [Green Version]
- Barnes, S.; Benton, H.P.; Casazza, K.; Cooper, S.J.; Cui, X.; Du, X.; Engler, J.; Kabarowski, J.H.; Li, S.; Pathmasiri, W.; et al. Training in metabolomics research. II. Processing and statistical analysis of metabolomics data, metabolite identification, pathway analysis, applications of metabolomics and its future. J. Mass Spectrom. 2016, 51, 535–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, M.H.; Beger, R.; Daykin, C.A.; Fan, T.W.; Fiehn, O.; Goodacre, R.; Griffin, J.L.; et al. Proposed minimum reporting standards for chemical analysis Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics 2007, 3, 211–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wishart, D.S.; Tzur, D.; Knox, C.; Eisner, R.; Guo, A.C.; Young, N.; Cheng, D.; Jewell, K.; Arndt, D.; Sawhney, S.; et al. HMDB: The Human Metabolome Database. Nucleic Acids Res. 2007, 35, D521–D526. [Google Scholar] [CrossRef]
- Gandhi, A.S.; Zhu, M.; Pang, S.; Wohlfarth, A.; Scheidweiler, K.B.; Liu, H.F.; Huestis, M.A. First characterization of AKB-48 metabolism, a novel synthetic cannabinoid, using human hepatocytes and high-resolution mass spectrometry. AAPS J. 2013, 15, 1091–1098. [Google Scholar] [CrossRef] [PubMed]
- Kadomura, N.; Ito, T.; Kawashima, H.; Matsuhisa, T.; Kinoshita, T.; Soda, M.; Kohyama, E.; Iwaki, T.; Nagai, H.; Kitaichi, K. In vitro metabolic profiles of adamantyl positional isomers of synthetic cannabinoids. Forensic Toxicology 2020. [Google Scholar] [CrossRef]
- Erratico, C.; Negreira, N.; Norouzizadeh, H.; Covaci, A.; Neels, H.; Maudens, K.; van Nuijs, A.L. In vitro and in vivo human metabolism of the synthetic cannabinoid AB-CHMINACA. Drug Test Anal. 2015, 7, 866–876. [Google Scholar] [CrossRef] [PubMed]
- Welter, J.; Meyer, M.R.; Wolf, E.U.; Weinmann, W.; Kavanagh, P.; Maurer, H.H. 2-methiopropamine, a thiophene analogue of methamphetamine: Studies on its metabolism and detectability in the rat and human using GC-MS and LC-(HR)-MS techniques. Anal. Bioanal. Chem. 2013, 405, 3125–3135. [Google Scholar] [CrossRef] [PubMed]
- Richter, L.H.; Kaminski, Y.R.; Noor, F.; Meyer, M.R.; Maurer, H.H. Metabolic fate of desomorphine elucidated using rat urine, pooled human liver preparations, and human hepatocyte cultures as well as its detectability using standard urine screening approaches. Anal. Bioanal. Chem. 2016, 408, 6283–6294. [Google Scholar] [CrossRef] [PubMed]
- Maurer, H.H.; Pfleger, K.; Weber, A.A. Mass Spectral Data of Drugs, Poisons, Pesticides, Pollutants and Their Metabolites; Wiley-VCH: Weinheim, Germany, 2016. [Google Scholar]
- Maurer, H.H.; Meyer, M.R.; Helfer, A.G.; Weber, A.A. Maurer/Meyer/Helfer/Weber MMHW LC-HR-MS/MS Library of Drugs, Poisons, and Their Metabolites; Wiley-VCH: Weinheim, Germany, 2018. [Google Scholar]
- Wehrens, R.; Hageman, J.A.; van Eeuwijk, F.; Kooke, R.; Flood, P.J.; Wijnker, E.; Keurentjes, J.J.; Lommen, A.; van Eekelen, H.D.; Hall, R.D.; et al. Improved batch correction in untargeted MS-based metabolomics. Metabolomics 2016, 12, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adusumilli, R.; Mallick, P. Data Conversion with ProteoWizard msConvert. Methods Mol. Biol. 2017, 1550, 339–368. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Kuhl, C.; Tautenhahn, R.; Bottcher, C.; Larson, T.R.; Neumann, S. CAMERA: An integrated strategy for compound spectra extraction and annotation of liquid chromatography/mass spectrometry data sets. Anal. Chem. 2012, 84, 283–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature | Measured Mass, m/z | Retention Time, s | Found with | Identity |
|---|---|---|---|---|
| M296T431 | 296.1768 | 431 | XM, CD | A-CHMINACA-M (N-dealkyl-) |
| M424T443 | 424.2610 | 443 | R, XM | A-CHMINACA-M (di-HO-) |
| M408T474 | 408.2661 | 474 | R, XM, CD | A-CHMINACA-M (HO-) |
| M409T474 | 409.2693 | 474 | R, XM | A-CHMINACA-M (HO-) 13C isotope |
| M430T474 | 430.2481 | 474 | R, XM | A-CHMINACA-M (HO-) adduct [M + Na]+ |
| M392T547 | 392.2710 | 547 | R, XM, CD | A-CHMINACA |
| M393T547 | 393.2743 | 547 | R, XM | A-CHMINACA 13C isotope |
| M394T547 | 394.2775 | 547 | R, XM | A-CHMINACA 13C2 isotope |
| M414T547 | 414.2530 | 547 | R, XM, CD | A-CHMINACA adduct [M + Na]+ |
| M415T547 | 415.2562 | 547 | R, XM | A-CHMINACA adduct [M + Na]+ 13C isotope |
| M430T547 | 430.2270 | 547 | R, XM, CD | A-CHMINACA adduct [M + K]+ |
| M437T547 | 437.3290 | 547 | R, XM | A-CHMINACA adduct |
| M438T547 | 438.3320 | 547 | XM | A-CHMINACA adduct 13C isotope |
| Feature | Measured Mass, m/z | Retention Time, s | Found with | Identity |
|---|---|---|---|---|
| M355T70 | 355.2392 | 70 | R, XM | Unknown |
| M430T71 | 430.2270 | 71 | XM | A-CHMINACA adduct [M + K]+ |
| M392T72 | 392.2710 | 72 | R, XM, CD | A-CHMINACA |
| M393T72 | 393.2743 | 72 | R, XM | A-CHMINACA 13C isotope |
| M394T72 | 394.2775 | 72 | R, XM | A-CHMINACA 13C2 isotope |
| M395T72 | 395.2809 | 72 | R, XM | A-CHMINACA 13C3 isotope |
| M356T74 | 356.1802 | 74 | XM | Unknown |
| M135T76 | 135.1174 | 76 | CD | A-CHMINACA artifact (adamantyl-ring) |
| M408T83 | 408.2661 | 83 | R, XM, CD | A-CHMINACA-M (HO-) |
| M409T83 | 409.2693 | 83 | R, XM | A-CHMINACA-M (HO-) 13C isotope |
| M296T86 | 296.1768 | 86 | R, XM, CD | A-CHMINACA-M (N-dealkyl-) |
| M297T86 | 297.1800 | 86 | XM | A-CHMINACA-M (N-dealkyl-) 13C isotope |
| M408T88 | 408.2661 | 88 | CD | A-CHIMINACA-M (HO-) |
| M422T92 | 422.2453 | 92 | R, XM | A-CHIMINACA-M (HO, Oxo) |
| M424T93 | 424.2610 | 93 | CD | A-CHMINACA-M (di-HO-) |
| M424T96 | 424.2610 | 96 | R, XM, CD | A-CHMINACA-M (di-HO-) |
| M425T96 | 425.2644 | 96 | R, XM | A-CHMINACA-M (di-HO-) 13C isotope |
| M274T113 | 274.1559 | 113 | R, XM | A-CHMINACA-M (HO-) (N-dealkyl-) |
| M312T115 | 312.1715 | 115 | R, XM, CD | A-CHMINACA-M (HO-) (N-dealkyl-) |
| M146T116 | 146.0819 | 116 | CD | A-CHMINACA artifact (indazole-core) |
| M440T117 | 440.2561 | 117 | CD | A-CHMINACA-M (tri-HO-) |
| M440T122 | 440.2565 | 122 | CD | A-CHMINACA-M (tri-HO-) |
| M176T135 | 176.0924 | 135 | R, XM, CD | Unknown |
| M158T135 | 158.0818 | 135 | R, XM, CD | [M + H − H2O]+175.086 |
| M188T170 | 188.1288 | 170 | R, XM, CD | Unknown |
| M158T174 | 158.0818 | 174 | R, XM | [M + H − H2O]+175.086 |
| M176T174 | 176.0924 | 174 | R, XM, CD | Unknown |
| M341T219 | 341.2447 | 219 | R, XM | Unknown |
| M313T253 | 313.2649 | 253 | R, XM | Unknown |
| M248T270 | 248.2382 | 270 | XM | Unknown |
| Criteria | Compound Discoverer | XCMS Online/MetaboAnalyst 4.0 | Manually Programmed R Tool |
|---|---|---|---|
| Open source | - | + | + |
| Low false-positive rate | + | - | + |
| Flexibility | - | -/+ | + |
| Complex datasets | - | + | + |
| Using raw data | + | - | - |
| Required prior knowledge | - | - | + |
| Annotation of isotopes and adducts | - | + | + |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hemmer, S.; Manier, S.K.; Fischmann, S.; Westphal, F.; Wagmann, L.; Meyer, M.R. Comparison of Three Untargeted Data Processing Workflows for Evaluating LC-HRMS Metabolomics Data. Metabolites 2020, 10, 378. https://doi.org/10.3390/metabo10090378
Hemmer S, Manier SK, Fischmann S, Westphal F, Wagmann L, Meyer MR. Comparison of Three Untargeted Data Processing Workflows for Evaluating LC-HRMS Metabolomics Data. Metabolites. 2020; 10(9):378. https://doi.org/10.3390/metabo10090378
Chicago/Turabian StyleHemmer, Selina, Sascha K. Manier, Svenja Fischmann, Folker Westphal, Lea Wagmann, and Markus R. Meyer. 2020. "Comparison of Three Untargeted Data Processing Workflows for Evaluating LC-HRMS Metabolomics Data" Metabolites 10, no. 9: 378. https://doi.org/10.3390/metabo10090378
APA StyleHemmer, S., Manier, S. K., Fischmann, S., Westphal, F., Wagmann, L., & Meyer, M. R. (2020). Comparison of Three Untargeted Data Processing Workflows for Evaluating LC-HRMS Metabolomics Data. Metabolites, 10(9), 378. https://doi.org/10.3390/metabo10090378