Evaluation of Sample Preparation Methods for Inter-Laboratory Metabolomics Investigation of Streptomyces lividans TK24


 , and
, and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
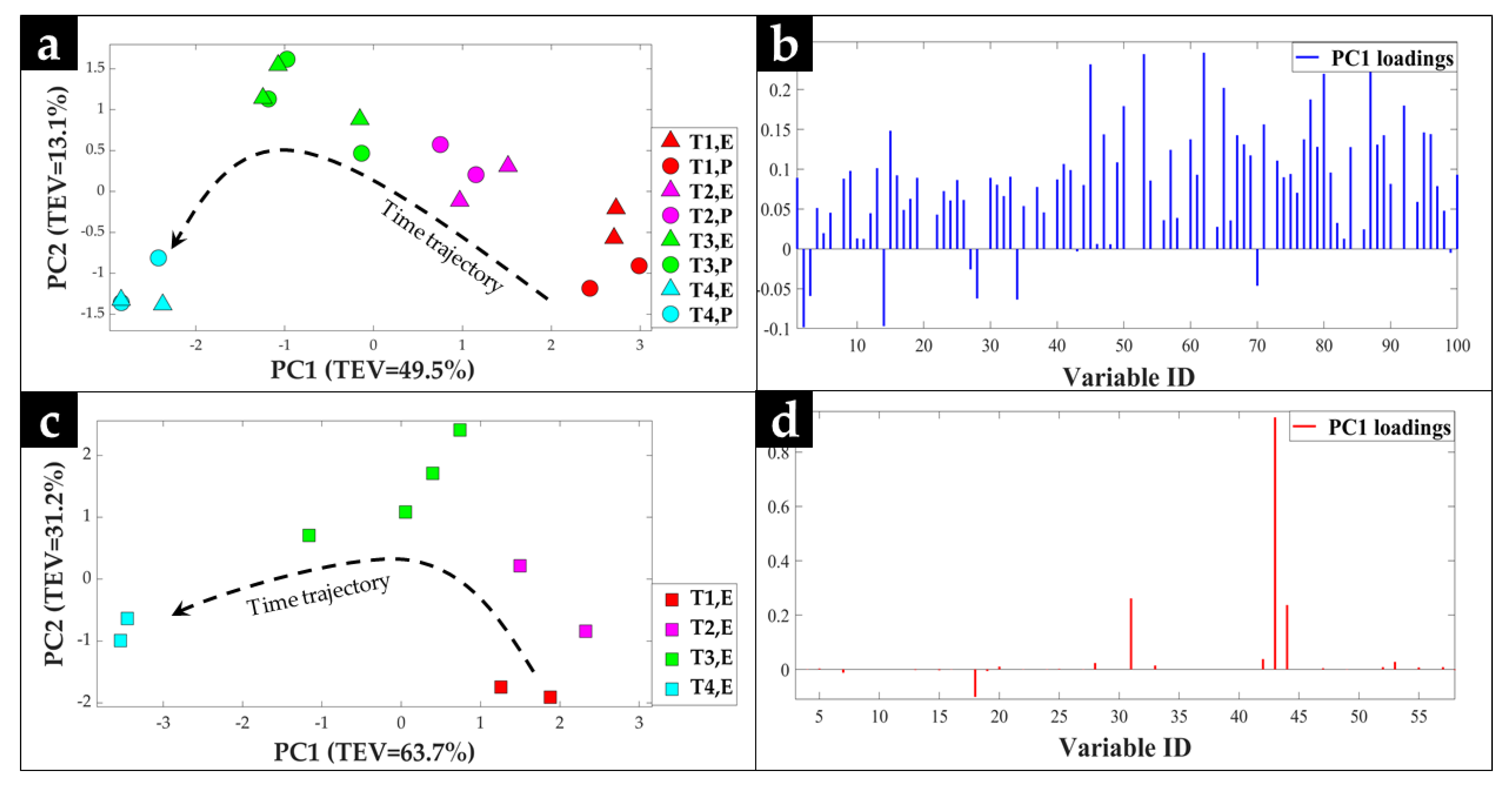
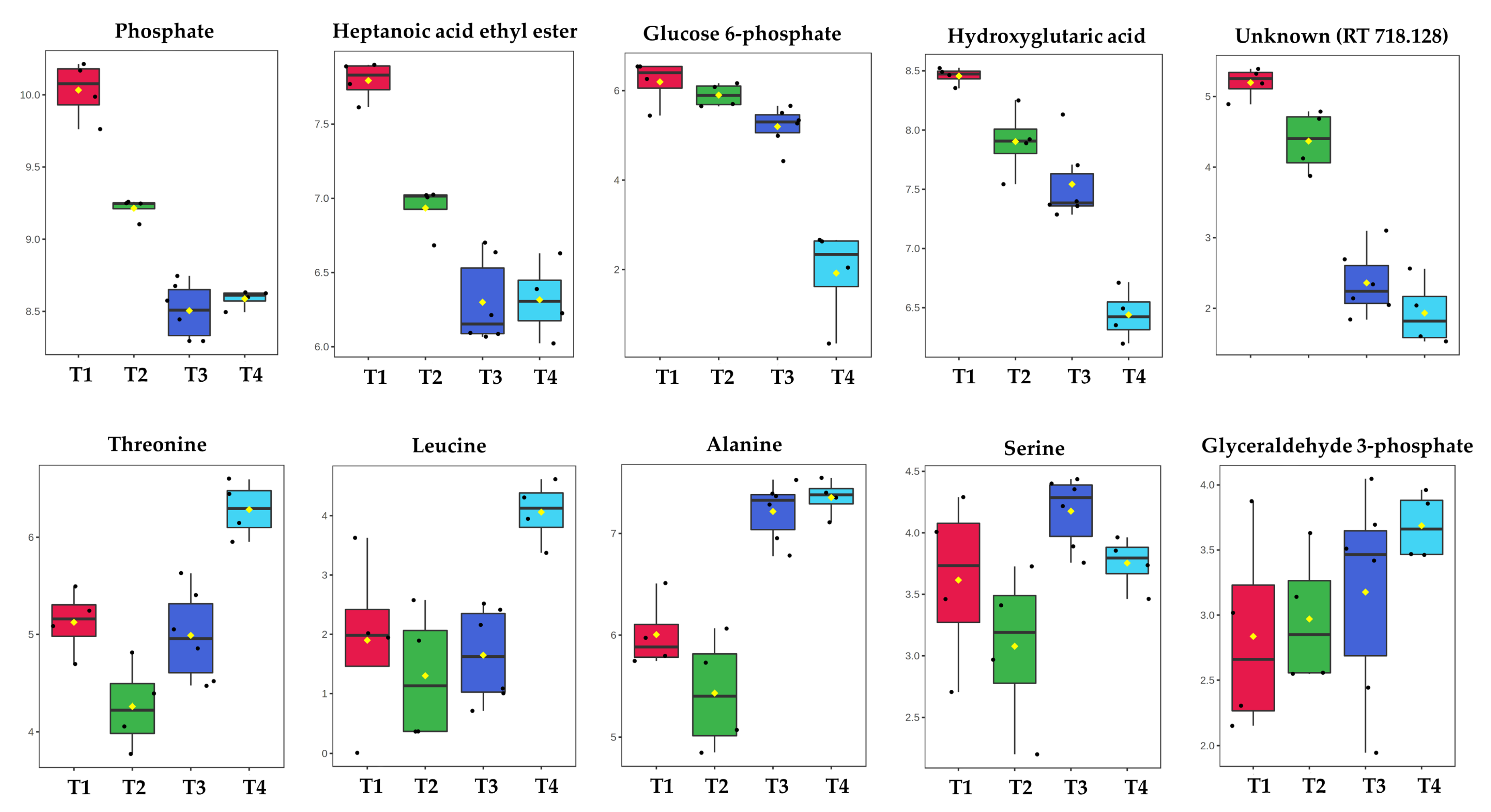
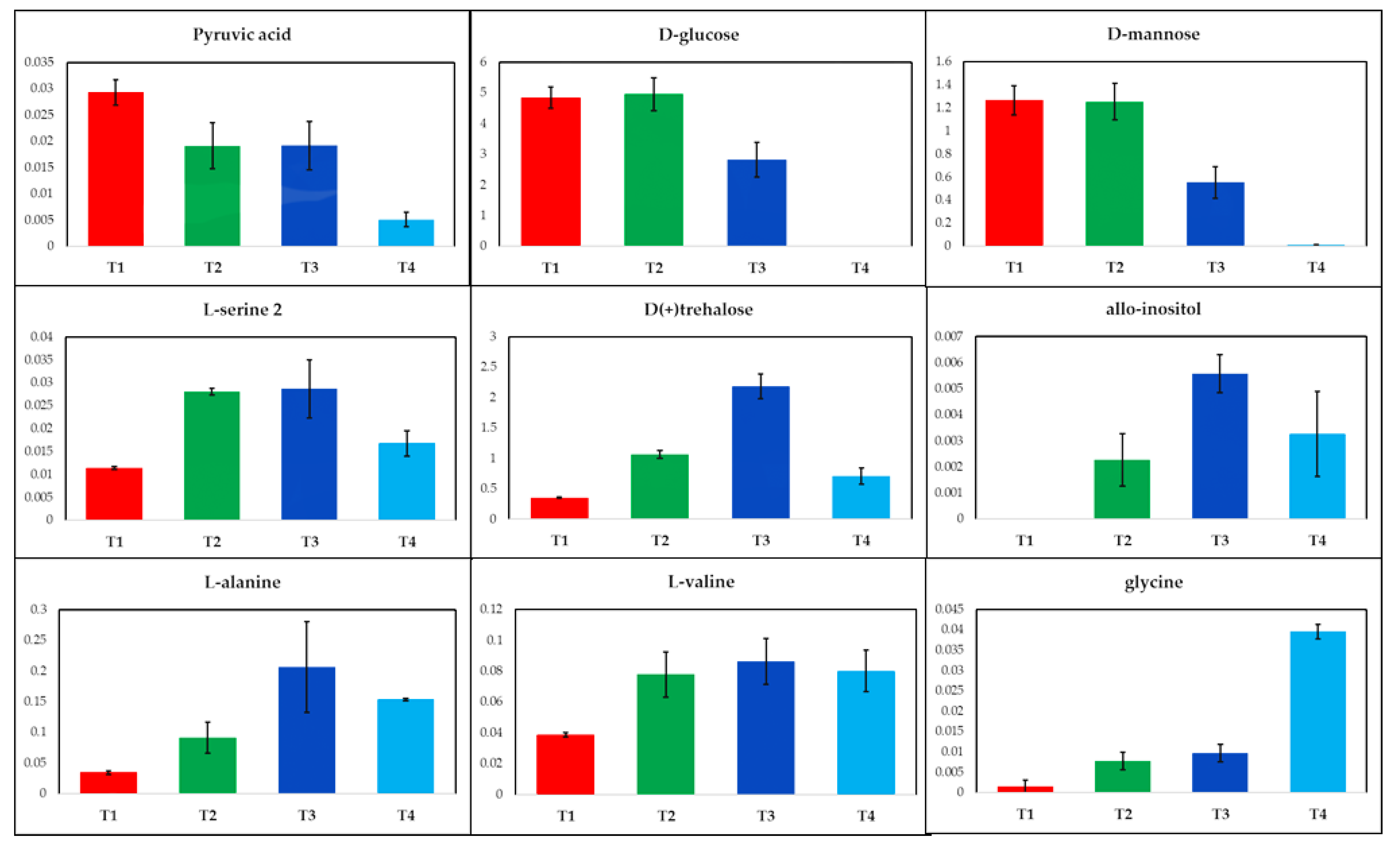
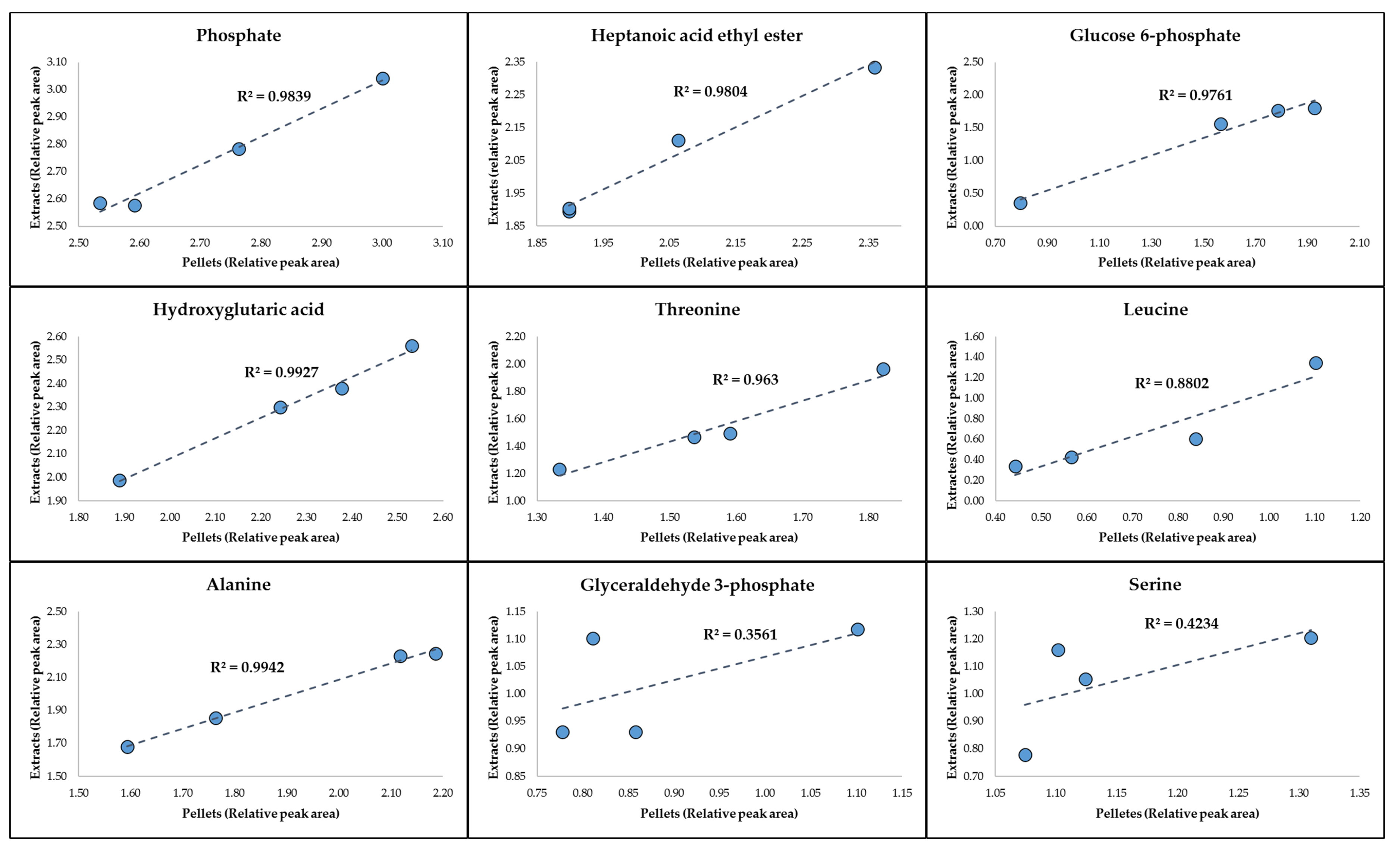
2.1. GC-MS Metabolic Profiles
- Derivatization: both labs used the same methoximation step, but the silylation reagent was MSTFA (Manchester) or BSTFA (Leuven).
- GC-MS: Agilent 6890N GC coupled to a Leco Pegasus III ToF-MS (Manchester) versus an Agilent 7890A GC with an Agilent 5975C VL MSD system (Leuven).
- Internal standard: this was either succinic-d4 acid (Manchester) or phenyl β-d-glucopyranoside (Leuven).
- Metabolite identification: Level 1 of metabolomics standards initiative (MSI) guidelines to an in-house library (Manchester) versus matching to the Golm library for MSI Level 2 (Leuven).
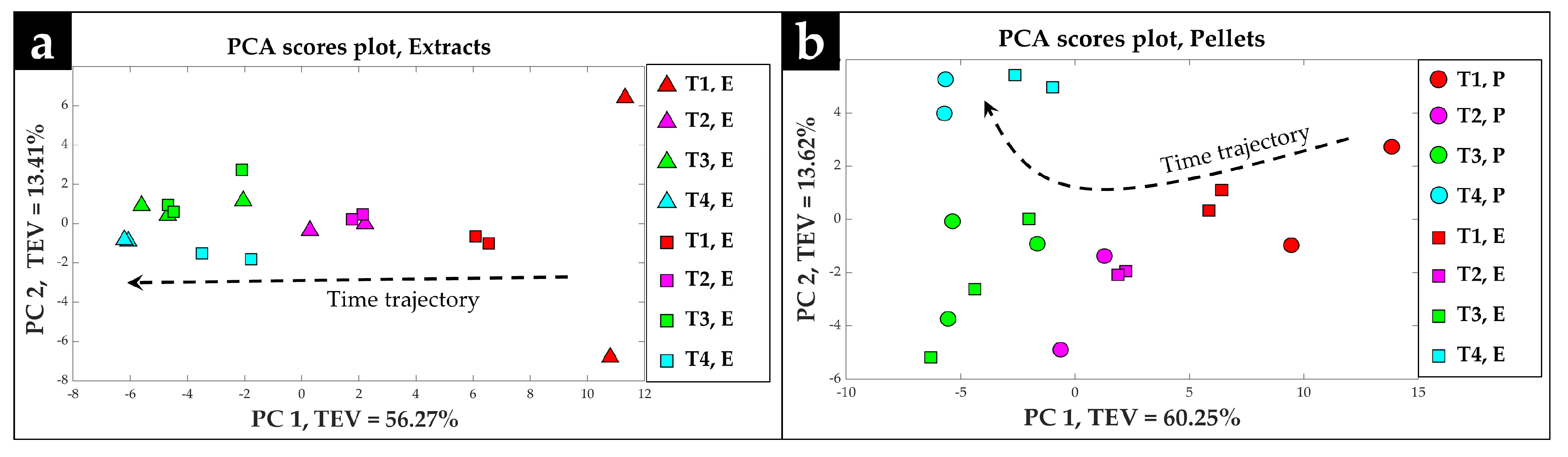
2.2. Procrustes Analysis of the GC-MS Data
3. Discussion
4. Materials and Methods
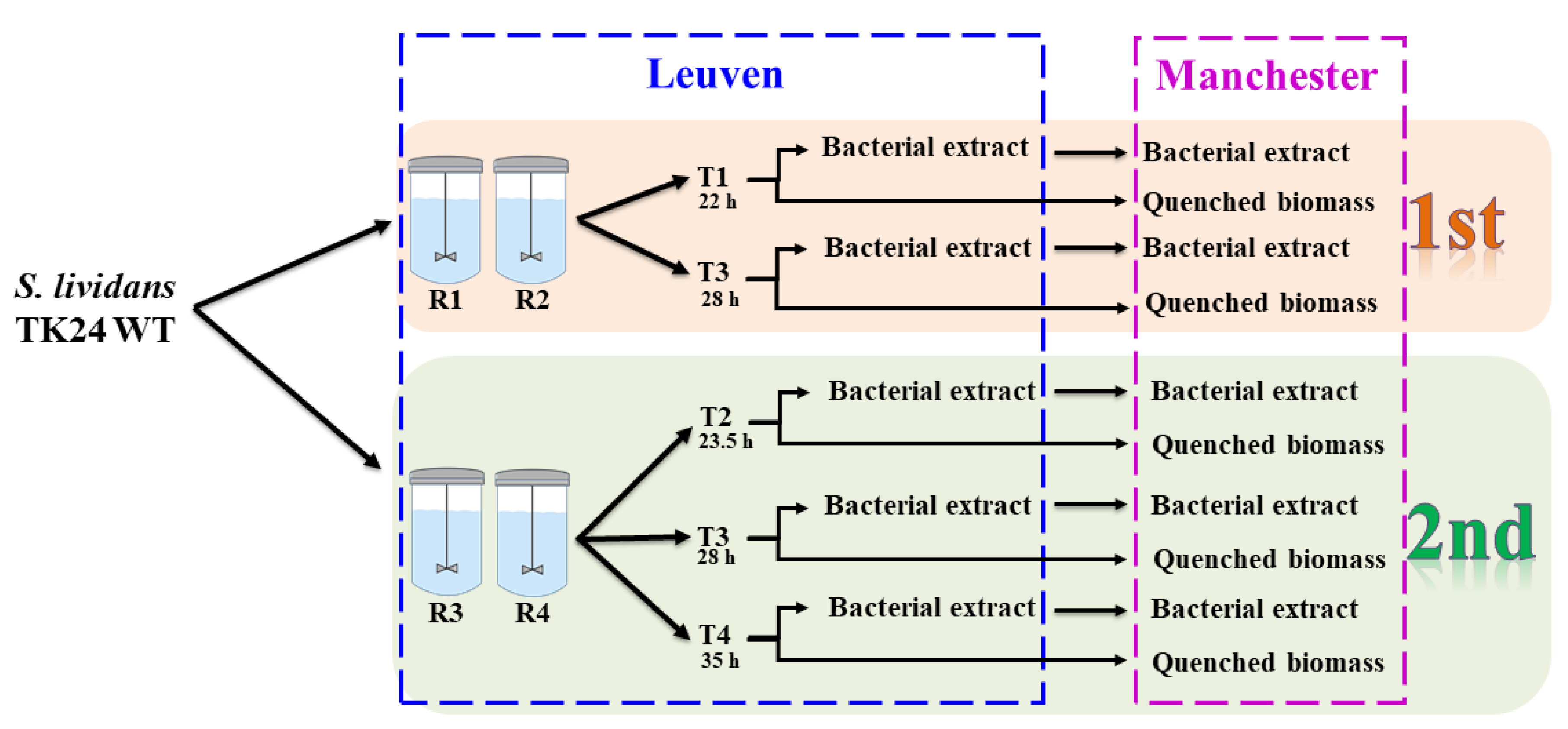
4.1. Bacterial Strain and Growth Condition
4.2. Quenching and Extraction
4.3. Derivatization
4.4. GC-MS Analysis
4.5. Data Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Liu, R.; Deng, Z.; Liu, T. Streptomyces species: Ideal chassis for natural product discovery and overproduction. Metab. Eng. 2018, 50, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Nakashima, T. Actinomycetes, an Inexhaustible Source of Naturally Occurring Antibiotics. Antibiotics 2018, 7, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalifa, S.A.M.; Elias, N.; Farag, M.A.; Chen, L.; Saeed, A.; Hegazy, M.F.; Moustafa, M.S.; Abd El-Wahed, A.; Al-Mousawi, S.M.; Musharraf, S.G.; et al. Marine Natural Products: A Source of Novel Anticancer Drugs. Mar. Drugs 2019, 17. [Google Scholar] [CrossRef] [Green Version]
- Bolourian, A.; Mojtahedi, Z. Immunosuppressants produced by Streptomyces: Evolution, hygiene hypothesis, tumour rapalog resistance and probiotics. Environ. Microbiol. Rep. 2018, 10, 123–126. [Google Scholar] [CrossRef]
- Chaudhary, A.K.; Dhakal, D.; Sohng, J.K. An Insight into the “-Omics” Based Engineering of Streptomycetes for Secondary Metabolite Overproduction. Biomed. Res. Int. 2013, 2013, 968518. [Google Scholar] [CrossRef] [Green Version]
- Doroghazi, J.R.; Albright, J.C.; Goering, A.W.; Ju, K.-S.; Haines, R.R.; Tchalukov, K.A.; Labeda, D.P.; Kelleher, N.L.; Metcalf, W.W. A roadmap for natural product discovery based on large-scale genomics and metabolomics. Nat. Chem. Biol. 2014, 10, 963–968. [Google Scholar] [CrossRef]
- Dhakal, D.; Pokhrel, A.R.; Shrestha, B.; Sohng, J.K. Marine Rare Actinobacteria: Isolation, Characterization, and Strategies for Harnessing Bioactive Compounds. Front. Microbiol 2017, 8, 1106. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Choi, Y.H.; Van Wezel, G.P. Metabolic profiling as a tool for prioritizing antimicrobial compounds. J. Ind. Microbiol. Biotechnol. 2016, 43, 299–312. [Google Scholar] [CrossRef] [Green Version]
- Milshteyn, A.; Schneider, J.S.; Brady, S.F. Mining the metabiome: Identifying novel natural products from microbial communities. Chem. Biol. 2014, 21, 1211–1223. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, C.T.; Dhakal, D.; Pham, V.T.T.; Nguyen, H.T.; Sohng, J.-K. Recent Advances in Strategies for Activation and Discovery/Characterization of Cryptic Biosynthetic Gene Clusters in Streptomyces. Microorganisms 2020, 8, 616. [Google Scholar] [CrossRef] [Green Version]
- Ellis, D.I.; Goodacre, R. Metabolomics-assisted synthetic biology. Curr. Opin. Biotechnol. 2012, 23, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Oliver, S.G.; Winson, M.K.; Kell, D.B.; Baganz, F. Systematic functional analysis of the yeast genome. Trends Biotechnol. 1998, 16, 373–378. [Google Scholar] [CrossRef]
- Goodacre, R.; Vaidyanathan, S.; Dunn, W.B.; Harrigan, G.G.; Kell, D.B. Metabolomics by numbers: Acquiring and understanding global metabolite data. Trends Biotechnol. 2004, 22, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Fiehn, O. Metabolomics–the link between genotypes and phenotypes. Plant. Mol. Biol. 2002, 48, 155–171. [Google Scholar] [CrossRef]
- Stefanovic, E.; Fitzgerald, G.; McAuliffe, O. Advances in the genomics and metabolomics of dairy lactobacilli: A review. Food Microbiol. 2017, 61, 33–49. [Google Scholar] [CrossRef] [PubMed]
- Muhamadali, H.; Xu, Y.; Morra, R.; Trivedi, D.K.; Rattray, N.J.W.; Dixon, N.; Goodacre, R. Metabolomic analysis of riboswitch containing E-coli recombinant expression system. Mol. Biosyst. 2016, 12, 350–361. [Google Scholar] [CrossRef] [Green Version]
- Santos, I.C.; Hildenbrand, Z.L.; Schug, K.A. Applications of MALDI-TOF MS in environmental microbiology. Analyst 2016, 141, 2827–2837. [Google Scholar] [CrossRef]
- Dunham, S.J.B.; Ellis, J.F.; Li, B.; Sweedler, J.V. Mass Spectrometry Imaging of Complex Microbial Communities. Acc. Chem. Res. 2017, 50, 96–104. [Google Scholar] [CrossRef]
- Melnik, A.V.; Da Silva, R.R.; Hyde, E.R.; Aksenov, A.A.; Vargas, F.; Bouslimani, A.; Protsyuk, I.; Jarmusch, A.K.; Tripathi, A.; Alexandrov, T.; et al. Coupling Targeted and Untargeted Mass Spectrometry for Metabolome-Microbiome-Wide Association Studies of Human Fecal Samples. Anal. Chem. 2017, 89, 7549–7559. [Google Scholar] [CrossRef]
- Chisanga, M.; Muhamadali, H.; Ellis, D.I.; Goodacre, R. Enhancing Disease Diagnosis: Biomedical Applications of Surface-Enhanced Raman Scattering. Appl. Sci. 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Dudzik, D.; Barbas-Bernardos, C.; García, A.; Barbas, C. Quality assurance procedures for mass spectrometry untargeted metabolomics. a review. J. Pharm. Biomed. Anal. 2018, 147, 149–173. [Google Scholar] [CrossRef] [PubMed]
- Engskog, M.K.R.; Haglof, J.; Arvidsson, T.; Pettersson, C. LC-MS based global metabolite profiling: The necessity of high data quality. Metabolomics 2016, 12. [Google Scholar] [CrossRef]
- Wang, X.Y.; Yang, F.X.; Zhang, Y.S.; Xu, G.W.; Liu, Y.; Tian, J.; Gao, P. Evaluation and optimization of sample preparation methods for metabolic profiling analysis of Escherichia coli. Electrophoresis 2015, 36, 2140–2147. [Google Scholar] [CrossRef]
- Yin, P.Y.; Lehmann, R.; Xu, G.W. Effects of pre-analytical processes on blood samples used in metabolomics studies. Anal. Bioanal. Chem. 2015, 407, 4879–4892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koning, W.D.; Dam, K.V. A method for the determination of changes of glycolytic metabolites in yeast on a subsecond time scale using extraction at neutral pH. Anal. Biochem. 1992, 204, 118–123. [Google Scholar] [CrossRef]
- Faijes, M.; Mars, A.E.; Smid, E.J. Comparison of quenching and extraction methodologies for metabolome analysis of Lactobacillus plantarum. Microb. Cell Factories 2007, 6, 27. [Google Scholar] [CrossRef] [Green Version]
- Goodacre, R. Metabolomics of a Superorganism. J. Nutr. 2007, 137, 259S–266S. [Google Scholar] [CrossRef] [Green Version]
- Muhamadali, H.; Xu, Y.; Ellis, D.I.; Trivedi, D.K.; Rattray, N.J.W.; Bernaerts, K.; Goodacre, R. Metabolomics investigation of recombinant mTNFα production in Streptomyces lividans. Microb. Cell Factories 2015, 14, 157. [Google Scholar] [CrossRef] [Green Version]
- Dryden, I.L.; Mardia, K.V. Procrustes analysis. In Statistical Shape Analysis, with Applications in R; Wiley: Hoboken, NJ, USA, 2016; pp. 125–173. [Google Scholar] [CrossRef]
- Kassama, Y.; Xu, Y.; Dunn, W.B.; Geukens, N.; Anné, J.; Goodacre, R. Assessment of adaptive focused acoustics versus manual vortex/freeze-thaw for intracellular metabolite extraction from Streptomyces lividans producing recombinant proteins using GC-MS and multi-block principal component analysis. Analyst 2010, 135, 934–942. [Google Scholar] [CrossRef]
- D'Huys, P.J.; Lule, I.; Van Hove, S.; Vercammen, D.; Wouters, C.; Bernaerts, K.; Anne, J.; Van Impe, J.F.M. Amino acid uptake profiling of wild type and recombinant Streptomyces lividans TK24 batch fermentations. J. Biotechnol. 2011, 152, 132–143. [Google Scholar] [CrossRef]
- Braβnta, A.F.; Eandez, C.M.; Dáiaaz, L.A.; Manzanal, M.B.; Hardisson, C. Glycogen and Trehalose Accumulation during Colony Development in Streptomyces antibioticus. Microbiology 1986, 132, 1319–1326. [Google Scholar] [CrossRef] [Green Version]
- McBride, M.J.; Ensign, J.C. Effects of intracellular trehalose content on Streptomyces griseus spores. J. Bacteriol. 1987, 169, 4995–5001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hincha, D.K.; Zuther, E.; Hellwege, E.M.; Heyer, A.G. Specific effects of fructo- and gluco-oligosaccharides in the preservation of liposomes during drying. Glycobiology 2002, 12, 103–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallagher, K.A.; Schumacher, M.A.; Bush, M.J.; Bibb, M.J.; Chandra, G.; Holmes, N.A.; Zeng, W.J.; Henderson, M.; Zhang, H.S.; Findlay, K.C.; et al. c-di-GMP Arms an Anti-sigma to Control Progression of Multicellular Differentiation in Streptomyces. Mol. Cell 2020, 77, 586. [Google Scholar] [CrossRef] [Green Version]
- Winder, C.L.; Dunn, W.B.; Schuler, S.; Broadhurst, D.; Jarvis, R.; Stephens, G.M.; Goodacre, R. Global Metabolic Profiling of Escherichia coli Cultures: An Evaluation of Methods for Quenching and Extraction of Intracellular Metabolites. Anal. Chem. 2008, 80, 2939–2948. [Google Scholar] [CrossRef]
- Fiehn, O.; Kopka, J.; Trethewey, R.N.; Willmitzer, L. Identification of Uncommon Plant Metabolites Based on Calculation of Elemental Compositions Using Gas Chromatography and Quadrupole Mass Spectrometry. Anal. Chem. 2000, 72, 3573–3580. [Google Scholar] [CrossRef]
- Wedge, D.C.; Allwood, J.W.; Dunn, W.; Vaughan, A.A.; Simpson, K.; Brown, M.; Priest, L.; Blackhall, F.H.; Whetton, A.D.; Dive, C.; et al. Is serum or plasma more appropriate for intersubject comparisons in metabolomic studies? An assessment in patients with small-cell lung cancer. Anal. Chem. 2011, 83, 6689–6697. [Google Scholar] [CrossRef]
- Begley, P.; Francis-McIntyre, S.; Dunn, W.B.; Broadhurst, D.I.; Halsall, A.; Tseng, A.; Knowles, J.; Goodacre, R.; Kell, D.B. Development and Performance of a Gas Chromatography−Time-of-Flight Mass Spectrometry Analysis for Large-Scale Nontargeted Metabolomic Studies of Human Serum. Anal. Chem. 2009, 81, 7038–7046. [Google Scholar] [CrossRef]
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, M.H.; Beger, R.; Daykin, C.A.; Fan, T.W.M.; Fiehn, O.; Goodacre, R.; Griffin, J.L.; et al. Proposed minimum reporting standards for chemical analysis Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics 2007, 3, 211–221. [Google Scholar] [CrossRef] [Green Version]
- Muhamadali, H.; Xu, Y.; Ellis, D.I.; Allwood, J.W.; Rattray, N.J.W.; Correa, E.; Alrabiah, H.; Lloyd, J.R.; Goodacre, R. Metabolic Profiling of Geobacter sulfurreducens during Industrial Bioprocess Scale-Up. Appl. Environ. Microbiol. 2015, 81, 3288–3298. [Google Scholar] [CrossRef] [Green Version]
- Hummel, J.; Selbig, J.; Walther, D.; Kopka, J. The Golm Metabolome Database: A database for GC-MS based metabolite profiling. In Metabolomics: A Powerful Tool in Systems Biology; Nielsen, J., Jewett, M.C., Eds.; Springer: Berlin/Heidelberg, Germany, 2007; pp. 75–95. [Google Scholar] [CrossRef]
- Wold, S.; Esbensen, K.; Geladi, P. Principal component analysis. Chemom. Intell. Lab. Syst. 1987, 2, 37–52. [Google Scholar] [CrossRef]
- Dryden, I.L.; Mardia, K.V. Statistical Shape Analysis; Wiley: Hoboken, NJ, USA, 1998. [Google Scholar]
- Jackson, D.A. PROTEST: A PROcrustean Randomization TEST of community environment concordance. Écoscience 1995, 2, 297–303. [Google Scholar] [CrossRef]
- Haug, K.; Cochrane, K.; Nainala, V.C.; Williams, M.; Chang, J.; Jayaseelan, K.V.; O’Donovan, C. MetaboLights: A resource evolving in response to the needs of its scientific community. Nucleic Acids Res. 2019, 48, D440–D444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muhamadali, H. Evaluation of sample preparation methods for inter-laboratory metabolomics investigation of Streptomyces lividans TK24. Available online: https://www.ebi.ac.uk/metabolights/MTBLS2013 (accessed on 22 September 2020).






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muhamadali, H.; Simoens, K.; Xu, Y.; Nicolai, B.; Bernaerts, K.; Goodacre, R. Evaluation of Sample Preparation Methods for Inter-Laboratory Metabolomics Investigation of Streptomyces lividans TK24. Metabolites 2020, 10, 379. https://doi.org/10.3390/metabo10090379
Muhamadali H, Simoens K, Xu Y, Nicolai B, Bernaerts K, Goodacre R. Evaluation of Sample Preparation Methods for Inter-Laboratory Metabolomics Investigation of Streptomyces lividans TK24. Metabolites. 2020; 10(9):379. https://doi.org/10.3390/metabo10090379
Chicago/Turabian StyleMuhamadali, Howbeer, Kenneth Simoens, Yun Xu, Bart Nicolai, Kristel Bernaerts, and Royston Goodacre. 2020. "Evaluation of Sample Preparation Methods for Inter-Laboratory Metabolomics Investigation of Streptomyces lividans TK24" Metabolites 10, no. 9: 379. https://doi.org/10.3390/metabo10090379
APA StyleMuhamadali, H., Simoens, K., Xu, Y., Nicolai, B., Bernaerts, K., & Goodacre, R. (2020). Evaluation of Sample Preparation Methods for Inter-Laboratory Metabolomics Investigation of Streptomyces lividans TK24. Metabolites, 10(9), 379. https://doi.org/10.3390/metabo10090379