Beyond Proteostasis: Lipid Metabolism as a New Player in ER Homeostasis


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
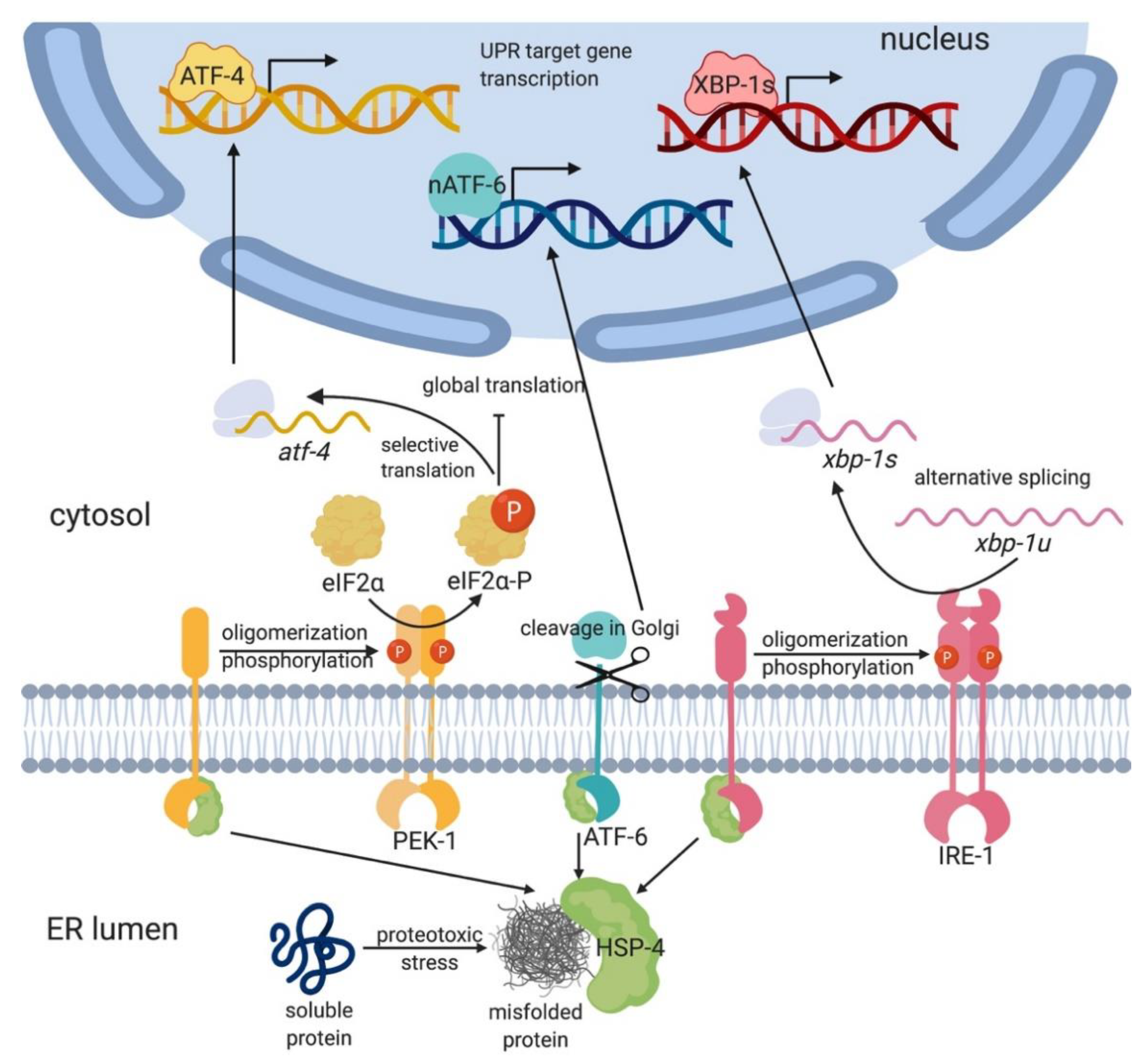
2. The Animal UPR-ER Is Composed of Three Branches
2.1. IRE1 Is the Most Highly Conserved and Ancient UPR-ER Transducer
2.2. The PERK/PEK-1 Branch of the UPR-ER Reprograms Translation
2.3. ATF6 Is a Parallel Sensor that Modulates UPR-ER Pathways
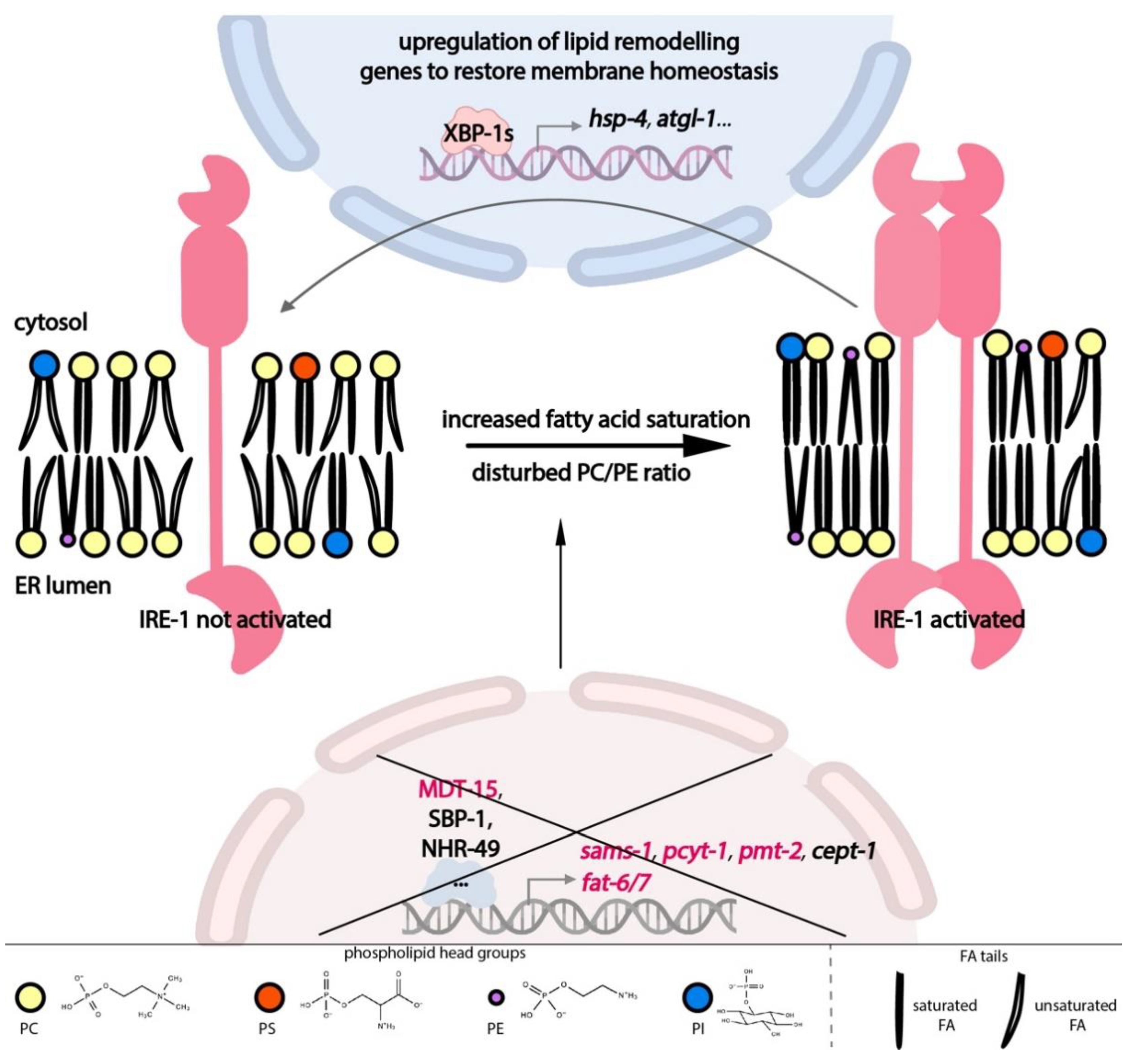
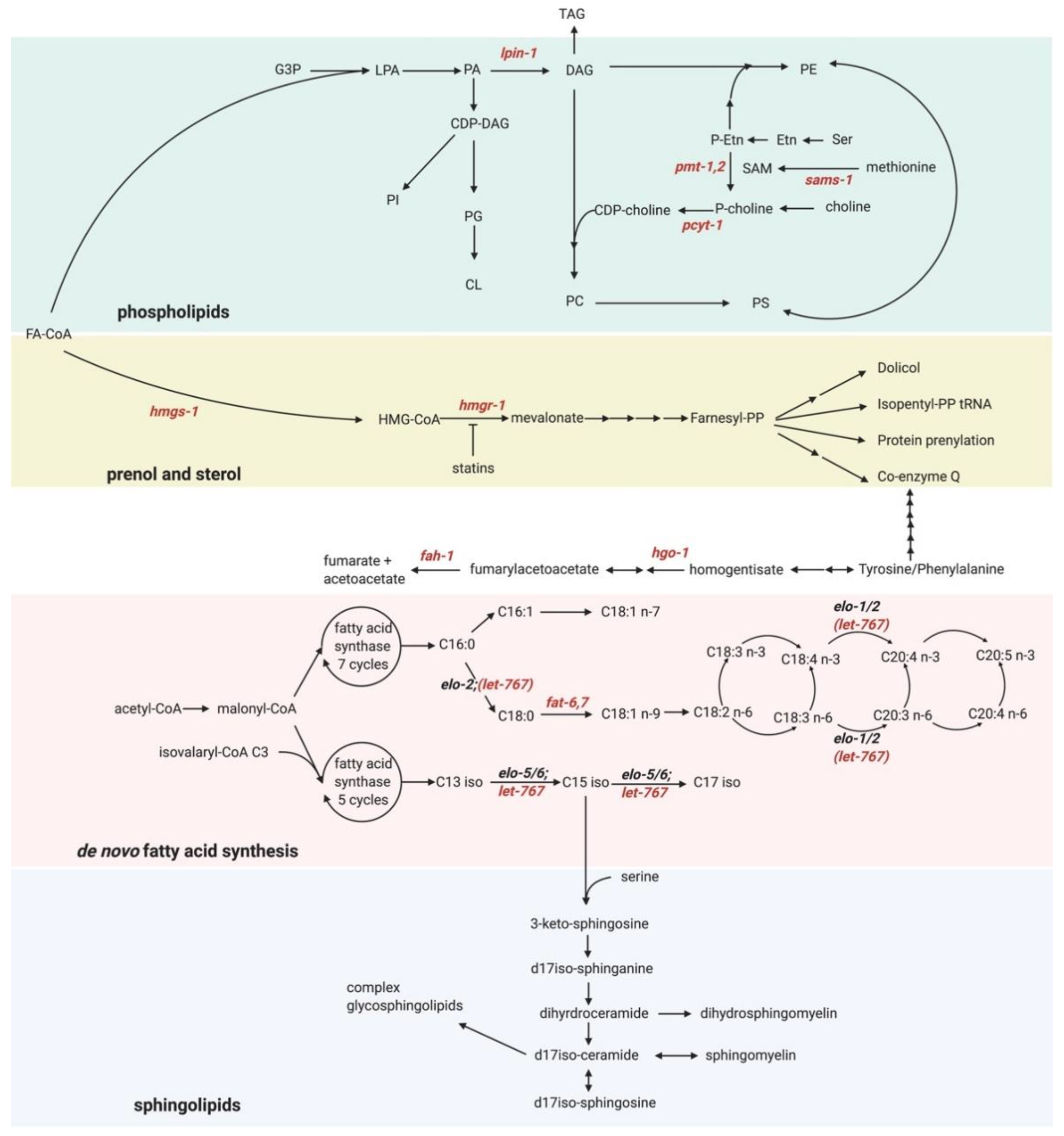
3. Membrane Lipids Are Critical for Normal ER Function
4. Bidirectional Interplay between Lipid Metabolism and the UPR-ER
5. The Role of Lipid Metabolism and ER Homeostasis in Human Diseases
6. Lipotoxicity Activates the UPR-ER through a Distinct Mechanism from Proteotoxicity
7. Crosstalk between Proteotoxicity- and Lipotoxicity-Induced UPR-ER
8. Functional Genomic Approaches Identify New UPR-ERLBS Components in C. elegans
9. Metabolomics, Lipidomics, and Label-Free Imaging Are Powerful Emerging Tools to Gain Insights into UPR-ERLBS Inputs and Outputs
10. Potential -omics Work Characterizing UPR-ER Inducing Metabolic Disturbances in C. elegans
11. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Senft, D.; Ronai, Z.A. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem. Sci. 2015, 40, 141–148. [Google Scholar] [CrossRef] [Green Version]
- Adams, C.J.; Kopp, M.C.; Larburu, N.; Nowak, P.R.; Ali, M.M.U. Structure and Molecular Mechanism of ER Stress Signaling by the Unfolded Protein Response Signal Activator IRE1. Front. Mol. Biosci. 2019, 6, 11. [Google Scholar] [CrossRef] [Green Version]
- Oakes, S.A.; Papa, F.R. The Role of Endoplasmic Reticulum Stress in Human Pathology. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 173–194. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C.; Papa, F.R. The Unfolded Protein Response and Cell Fate Control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef] [Green Version]
- Lindholm, D.; Korhonen, L.; Eriksson, O.; Kõks, S. Recent Insights into the Role of Unfolded Protein Response in ER Stress in Health and Disease. Front. Cell Dev. Biol. 2017, 5, 48. [Google Scholar] [CrossRef] [Green Version]
- McQuiston, A.; Diehl, J.A. Recent insights into PERK-dependent signaling from the stressed endoplasmic reticulum. F1000Research 2017, 6, 1897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hillary, R.F.; Fitzgerald, U. A lifetime of stress: ATF6 in development and homeostasis. J. Biomed. Sci. 2018, 25, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Walter, P.; Ron, D. The Unfolded Protein Response: From Stress Pathway to Homeostatic Regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [Green Version]
- Lai, C.-H.; Chou, C.-Y.; Ch’Ang, L.-Y.; Liu, C.-S.; Lin, W.-C. Identification of Novel Human Genes Evolutionarily Conserved in Caenorhabditis elegans by Comparative Proteomics. Genome Res. 2000, 10, 703–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaletta, T.; Hengartner, M.O. Finding function in novel targets: C. elegans as a model organism. Nat. Rev. Drug Discov. 2006, 5, 387–399. [Google Scholar] [CrossRef]
- Witting, M.; Schmitt-Kopplin, P. The Caenorhabditis elegans lipidome. Arch. Biochem. Biophys. 2016, 589, 27–37. [Google Scholar] [CrossRef]
- Zhang, Y.; Zou, X.; Ding, Y.; Wang, H.; Wu, X.; Liang, B. Comparative genomics and functional study of lipid metabolic genes in Caenorhabditis elegans. BMC Genom. 2013, 14, 164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, N.; Yap, W.S.; Xu, J.; Wu, H.; Koh, J.H.; Bin Goh, W.W.; George, B.; Chong, S.C.; Taubert, S.; Thibault, G. Stress sensor Ire1 deploys a divergent transcriptional program in response to lipid bilayer stress. J. Cell Biol. 2020, 219, 201909165. [Google Scholar] [CrossRef] [PubMed]
- Venz, R.; Korosteleva, A.; Jongsma, E.; Ewald, C.Y. Combining Auxin-Induced Degradation and RNAi Screening Identifies Novel Genes Involved in Lipid Bilayer Stress Sensing in Caenorhabditis elegans. G3 Genes Genomes Genet. 2020, 10, 3921–3928. [Google Scholar] [CrossRef]
- Singh, J.; Aballay, A. Endoplasmic Reticulum Stress Caused by Lipoprotein Accumulation Suppresses Immunity against Bacterial Pathogens and Contributes to Immunosenescence. mBio 2017, 8, e00778-17. [Google Scholar] [CrossRef] [Green Version]
- Marza, E.; Taouji, S.; Barroso, K.; Raymond, A.-A.; Guignard, L.; Bonneu, M.; Pallares-Lupon, N.; Dupuy, J.-W.; Fernandez-Zapico, M.E.; Rosenbaum, J.; et al. Genome-wide screen identifies a novel p97/CDC -48-dependent pathway regulating ER -stress-induced gene transcription. EMBO Rep. 2015, 16, 332–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, J.H.; Wang, L.; Beaudoin-Chabot, C.; Thibault, G. Lipid bilayer stress-activated IRE-1 modulates autophagy during endoplasmic reticulum stress. J. Cell Sci. 2018, 131, jcs217992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, X.; Ellis, R.E.; Sakaki, K.; Kaufman, R.J. Genetic Interactions Due to Constitutive and Inducible Gene Regulation Mediated by the Unfolded Protein Response in C. elegans. PLoS Genet. 2005, 1, e37. [Google Scholar] [CrossRef] [Green Version]
- Taylor, R.C.; Dillin, A. XBP-1 Is a Cell-Nonautonomous Regulator of Stress Resistance and Longevity. Cell 2013, 153, 1435–1447. [Google Scholar] [CrossRef] [Green Version]
- Imanikia, S.; Sheng, M.; Castro, C.; Griffin, J.L.; Taylor, R.C. XBP-1 Remodels Lipid Metabolism to Extend Longevity. Cell Rep. 2019, 28, 581–589.e4. [Google Scholar] [CrossRef] [Green Version]
- Özbey, N.P.; Imanikia, S.; Krueger, C.; Hardege, I.; Morud, J.; Sheng, M.; Schafer, W.R.; Casanueva, M.O.; Taylor, R.C. Tyramine Acts Downstream of Neuronal XBP-1s to Coordinate Inter-tissue UPRER Activation and Behavior in C. elegans. Dev. Cell 2020, 55, 754–770.e6. [Google Scholar] [CrossRef] [PubMed]
- Daniele, J.R.; Higuchi-Sanabria, R.; Durieux, J.; Monshietehadi, S.; Ramachandran, V.; Tronnes, S.U.; Kelet, N.; Sanchez, M.; Metcalf, M.G.; Garcia, G.; et al. UPRER promotes lipophagy independent of chaperones to extend life span. Sci. Adv. 2020, 6, eaaz1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikawa, J.-I.; Yamashita, S. IRE1 encodes a putative protein kinase containing a membrane-spanning domain and is required for inositol phototrophy in Saccharomyces cerevisiae. Mol. Microbiol. 1992, 6, 1441–1446. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.S.; Shamu, C.E.; Walter, P. Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell 1993, 73, 1197–1206. [Google Scholar] [CrossRef]
- Morl, K.; Ma, W.; Gething, M.-J.; Sambrook, J. A transmembrane protein with a cdc2+CDC28-related kinase activity is required for signaling from the ER to the nucleus. Cell 1993, 74, 743–756. [Google Scholar] [CrossRef]
- Carrara, M.; Prischi, F.; Nowak, P.R.; Ali, M.M.U. Crystal structures reveal transient PERK luminal domain tetramerization in endoplasmic reticulum stress signaling. EMBO J. 2015, 34, 1589–1600. [Google Scholar] [CrossRef]
- Lu, Y.; Liang, F.-X.; Wang, X. A Synthetic Biology Approach Identifies the Mammalian UPR RNA Ligase RtcB. Mol. Cell 2014, 55, 758–770. [Google Scholar] [CrossRef] [Green Version]
- Calfon, M.; Zeng, H.; Urano, F.; Till, J.H.; Hubbard, S.R.; Harding, H.P.; Clark, S.G.; Ron, D. IRE1 Couples Endoplasmic Reticulum Load to Secretory Capacity by Processing the XBP-1 MRNA. Nature 2002, 415, 92–96. [Google Scholar] [CrossRef]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA Is Induced by ATF6 and Spliced by IRE1 in Response to ER Stress to Produce a Highly Active Transcription Factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef] [Green Version]
- Travers, K.J.; Patil, C.K.; Wodicka, L.; Lockhart, D.J.; Weissman, J.S.; Walter, P. Functional and Genomic Analyses Reveal an Essential Coordination between the Unfolded Protein Response and ER-Associated Degradation. Cell 2000, 101, 249–258. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C.; Glimcher, L.H. Fine-Tuning of the Unfolded Protein Response: Assembling the IRE1α Interactome. Mol. Cell 2009, 35, 551–561. [Google Scholar] [CrossRef] [Green Version]
- Hollien, J.; Weissman, J.S. Decay of Endoplasmic Reticulum-Localized mRNAs During the Unfolded Protein Response. Science 2006, 313, 104–107. [Google Scholar] [CrossRef] [Green Version]
- Cross, B.C.S.; Bond, P.J.; Sadowski, P.G.; Jha, B.K.; Zak, J.; Goodman, J.M.; Silverman, R.H.; Neubert, T.A.; Baxendale, I.R.; Ron, D.; et al. The molecular basis for selective inhibition of unconventional mRNA splicing by an IRE1-binding small molecule. Proc. Natl. Acad. Sci. USA 2012, 109, E869–E878. [Google Scholar] [CrossRef] [Green Version]
- Kimmig, P.; Diaz, M.; Zheng, J.; Williams, C.C.; Lang, A.; Aragón, T.; Li, H.; Walter, P. The unfolded protein response in fission yeast modulates stability of select mRNAs to maintain protein homeostasis. eLife 2012, 1, e00048. [Google Scholar] [CrossRef]
- Ho, N.; Xu, C.; Thibault, G. From the unfolded protein response to metabolic diseases—Lipids under the spotlight. J. Cell Sci. 2018, 131, jcs199307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, M.H.; Ploegh, H.L.; Weissman, J.S. Road to Ruin: Targeting Proteins for Degradation in the Endoplasmic Reticulum. Science 2011, 334, 1086–1090. [Google Scholar] [CrossRef] [Green Version]
- Struwe, W.B.; Hughes, B.L.; Osborn, D.W.; Boudreau, E.D.; Shaw, K.M.D.; Warren, C.E. Modeling a congenital disorder of glycosylation type I in C. elegans: A genome-wide RNAi screen for N-glycosylation-dependent loci. Glycobiology 2009, 19, 1554–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, C.E.; Kooistra, T.G.; Kim, D.H. An essential role for XBP-1 in host protection against immune activation in C. elegans. Nat. Cell Biol. 2010, 463, 1092–1095. [Google Scholar] [CrossRef] [Green Version]
- Salzberg, Y.; Coleman, A.J.; Celestrin, K.; Cohen-Berkman, M.; Biederer, T.; Henis-Korenblit, S.; Bülow, H.E. Reduced Insulin/Insulin-Like Growth Factor Receptor Signaling Mitigates Defective Dendrite Morphogenesis in Mutants of the ER Stress Sensor IRE-1. PLoS Genet. 2017, 13, e1006579. [Google Scholar] [CrossRef] [Green Version]
- Ryoo, H.D.; Domingos, P.M.; Kang, M.-J.; Steller, H. Unfolded protein response in a Drosophila model for retinal degeneration. EMBO J. 2006, 26, 242–252. [Google Scholar] [CrossRef] [Green Version]
- Reimold, A.M.; Etkin, A.; Clauss, I.; Perkins, A.; Friend, D.S.; Zhang, J.; Horton, H.F.; Scott, A.; Orkin, S.H.; Byrne, M.C.; et al. An essential role in liver development for transcription factor XBP-1. Genome Res. 2000, 14, 152–157. [Google Scholar]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef]
- Novoa, I.; Zeng, H.; Harding, H.P.; Ron, D. Feedback Inhibition of the Unfolded Protein Response by GADD34-Mediated Dephosphorylation of eIF2α. J. Cell Biol. 2001, 153, 1011–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rousakis, A.; Vlassis, A.; Vlanti, A.; Patera, S.; Thireos, G.; Syntichaki, P. The general control nonderepressible-2 kinase mediates stress response and longevity induced by target of rapamycin inactivation inCaenorhabditis elegans. Aging Cell 2013, 12, 742–751. [Google Scholar] [CrossRef] [Green Version]
- Statzer, C.; Venz, R.; Bland, M.; Robida-Stubbs, S.; Meng, J.; Patel, K.; Emsley, R.; Petrovic, D.; Liu, P.; Morantte, I.; et al. ATF-4 and Hydrogen Sulfide Signalling Mediate Longevity from Inhibition of Translation or MTORC1. bioRxiv 2020. [Google Scholar] [CrossRef]
- Shen, X.; Ellis, R.E.; Lee, K.; Liu, C.-Y.; Yang, K.; Solomon, A.; Yoshida, H.; Morimoto, R.; Kurnit, D.M.; Mori, K.; et al. Complementary Signaling Pathways Regulate the Unfolded Protein Response and Are Required for C. elegans Development. Cell 2001, 107, 893–903. [Google Scholar] [CrossRef] [Green Version]
- Harding, H.P.; Zhang, Y.; Bertolotti, A.; Zeng, H.; Ron, D. Perk Is Essential for Translational Regulation and Cell Survival during the Unfolded Protein Response. Mol. Cell 2000, 5, 897–904. [Google Scholar] [CrossRef]
- Harding, H.P.; Zeng, H.; Zhang, Y.; Jungries, R.; Chung, P.; Plesken, H.; Sabatini, D.D.; Ron, D. Diabetes Mellitus and Exocrine Pancreatic Dysfunction in Perk −/− Mice Reveals a Role for Translational Control in Secretory Cell Survival. Mol. Cell 2001, 7, 1153–1163. [Google Scholar] [CrossRef]
- Delépine, M.; Nicolino, M.; Barrett, T.G.; Golamaully, M.; Lathrop, G.M.; Julier, C. EIF2AK3, encoding translation initiation factor 2-α kinase 3, is mutated in patients with Wolcott-Rallison syndrome. Nat. Genet. 2000, 25, 406–409. [Google Scholar] [CrossRef]
- Lee, K.; Tirasophon, W.; Shen, X.; Michalak, M.; Prywes, R.; Okada, T.; Yoshida, H.; Mori, K.; Kaufman, R.J. IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev. 2002, 16, 452–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, K.; Sato, T.; Matsui, T.; Sato, M.; Okada, T.; Yoshida, H.; Harada, A.; Mori, K. Transcriptional Induction of Mammalian ER Quality Control Proteins Is Mediated by Single or Combined Action of ATF6α and XBP1. Dev. Cell 2007, 13, 365–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoulders, M.D.; Ryno, L.M.; Genereux, J.C.; Moresco, J.J.; Tu, P.G.; Wu, C.; Yates, J.R.; Su, A.I.; Kelly, J.W.; Wiseman, R.L. Stress-Independent Activation of XBP1s and/or ATF6 Reveals Three Functionally Diverse ER Proteostasis Environments. Cell Rep. 2013, 3, 1279–1292. [Google Scholar] [CrossRef] [Green Version]
- Waldherr, S.M.; Strovas, T.J.; Vadset, T.A.; Liachko, N.F.; Kraemer, B.C. Constitutive XBP-1s-mediated activation of the endoplasmic reticulum unfolded protein response protects against pathological tau. Nat. Commun. 2019, 10, 4443. [Google Scholar] [CrossRef]
- Wu, J.; Rutkowski, D.T.; Dubois, M.; Swathirajan, J.; Saunders, T.; Wang, J.; Song, B.; Yau, G.D.-Y.; Kaufman, R.J. ATF6α Optimizes Long-Term Endoplasmic Reticulum Function to Protect Cells from Chronic Stress. Dev. Cell 2007, 13, 351–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Hu, B.; Ding, Z.; Dang, Y.; Wu, J.; Li, D.; Liu, X.; Xiao, B.; Zhang, W.; Ren, R.; et al. ATF6 safeguards organelle homeostasis and cellular aging in human mesenchymal stem cells. Cell Discov. 2018, 4, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohl, S.; Zobor, D.; Chiang, W.-C.; Weisschuh, N.; Staller, J.; Menendez, I.G.; Chang, S.; Beck, S.C.; Garrido, M.G.; Sothilingam, V.; et al. Mutations in the unfolded protein response regulator ATF6 cause the cone dysfunction disorder achromatopsia. Nat. Genet. 2015, 47, 757–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Havel, R.J.; Eder, H.A.; Bragdon, J.H. The Distribution and Chemical Composition of Ultracentrifugally Separated Lipoproteins in Human Serum. J. Clin. Investig. 1955, 34, 1345–1353. [Google Scholar] [CrossRef] [Green Version]
- Fahy, E.; Subramaniam, S.; Brown, H.A.; Glass, C.K.; Merrill, A.H.; Murphy, R.C.; Raetz, C.R.H.; Russell, D.W.; Seyama, Y.; Shaw, W.; et al. A comprehensive classification system for lipids. J. Lipid Res. 2005, 46, 839–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haucke, V.; Di Paolo, G. Lipids and lipid modifications in the regulation of membrane traffic. Curr. Opin. Cell Biol. 2007, 19, 426–435. [Google Scholar] [CrossRef] [Green Version]
- Parks, L.W.; Casey, W.M. Physiological Implications of Sterol Biosynthesis in Yeast. Annu. Rev. Microbiol. 1995, 49, 95–116. [Google Scholar] [CrossRef]
- Van Meer, G.; De Kroon, A.I.P.M. Lipid map of the mammalian cell. J. Cell Sci. 2010, 124, 5–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vance, J.E. Phospholipid Synthesis and Transport in Mammalian Cells. Traffic 2014, 16, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Potting, C.; Tatsuta, T.; König, T.; Haag, M.; Wai, T.; Aaltonen, M.J.; Langer, T. TRIAP1/PRELI Complexes Prevent Apoptosis by Mediating Intramitochondrial Transport of Phosphatidic Acid. Cell Metab. 2013, 18, 287–295. [Google Scholar] [CrossRef] [Green Version]
- Mesmin, B. Mitochondrial lipid transport and biosynthesis: A complex balance. J. Cell Biol. 2016, 214, 9–11. [Google Scholar] [CrossRef]
- Wang, R.; Li, B.; Lam, S.-M.; Shui, G. Integration of lipidomics and metabolomics for in-depth understanding of cellular mechanism and disease progression. J. Genet. Genom. 2020, 47, 69–83. [Google Scholar] [CrossRef] [PubMed]
- Ariyama, H.; Kono, N.; Matsuda, S.; Inoue, T.; Arai, H. Decrease in Membrane Phospholipid Unsaturation Induces Unfolded Protein Response. J. Biol. Chem. 2010, 285, 22027–22035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pineau, L.; Ferreira, T. Lipid-induced ER stress in yeast and β cells: Parallel trails to a common fate. FEMS Yeast Res. 2010, 10, 1035–1045. [Google Scholar] [CrossRef] [PubMed]
- Pineau, L.; Colas, J.; Dupont, S.; Beney, L.; Fleurat-Lessard, P.; Berjeaud, J.-M.; Bergès, T.; Ferreira, T. Lipid-Induced ER Stress: Synergistic Effects of Sterols and Saturated Fatty Acids. Traffic 2009, 10, 673–690. [Google Scholar] [CrossRef]
- Volmer, R.; Van Der Ploeg, K.; Ron, D. Membrane lipid saturation activates endoplasmic reticulum unfolded protein response transducers through their transmembrane domains. Proc. Natl. Acad. Sci. USA 2013, 110, 4628–4633. [Google Scholar] [CrossRef] [Green Version]
- Sommerweiss, D.; Gorski, T.; Richter, S.; Garten, A.; Kiess, W. Oleate rescues INS-1E β-cells from palmitate-induced apoptosis by preventing activation of the unfolded protein response. Biochem. Biophys. Res. Commun. 2013, 441, 770–776. [Google Scholar] [CrossRef] [Green Version]
- Hou, N.S.; Gutschmidt, A.; Choi, D.Y.; Pather, K.; Shi, X.; Watts, J.L.; Hoppe, T.; Taubert, S. Activation of the endoplasmic reticulum unfolded protein response by lipid disequilibrium without disturbed proteostasis in vivo. Proc. Natl. Acad. Sci. USA 2014, 111, E2271–E2280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thibault, G.; Shui, G.; Kim, W.; McAlister, G.C.; Ismail, N.; Gygi, S.P.; Wenk, M.R.; Ng, D.T. The Membrane Stress Response Buffers Lethal Effects of Lipid Disequilibrium by Reprogramming the Protein Homeostasis Network. Mol. Cell 2012, 48, 16–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, S.; Yang, L.; Li, P.; Hofmann, O.; Dicker, L.; Hide, W.; Lin, X.; Watkins, S.M.; Ivanov, A.R.; Hotamisligil, G.S. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nat. Cell Biol. 2011, 473, 528–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, H.; Shim, J.; Lee, J.H.; Lee, J.; Kim, J.B. IRE-1 and HSP-4 Contribute to Energy Homeostasis via Fasting-Induced Lipases in C. elegans. Cell Metab. 2009, 9, 440–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, J.H.; Wang, L.; Beaudoin-Chabot, C.; Thibault, G. Lipid Perturbation-Activated IRE-1 Modulates Autophagy and Lipolysis during Endoplasmic Reticulum Stress. bioRxiv 2018. [Google Scholar] [CrossRef]
- Kyriakakis, E.; Charmpilas, N.; Tavernarakis, N. Differential adiponectin signalling couples ER stress with lipid metabolism to modulate ageing in C. elegans. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.-W.; Zeng, X.; Rhim, T.; Ron, D.; Ryoo, H.D. The requirement of IRE1 and XBP1 in resolving physiological stress during Drosophila development. J. Cell Sci. 2017, 130, 3040–3049. [Google Scholar] [CrossRef] [Green Version]
- Harayama, T.; Riezman, H. Understanding the diversity of membrane lipid composition. Nat. Rev. Mol. Cell Biol. 2018, 19, 281–296. [Google Scholar] [CrossRef]
- Wortmann, S.B.; Espeel, M.; Almeida, L.; Reimer, A.; Bosboom, D.; Roels, F.; De Brouwer, A.P.; Wevers, R.A. Inborn errors of metabolism in the biosynthesis and remodelling of phospholipids. J. Inherit. Metab. Dis. 2014, 38, 99–110. [Google Scholar] [CrossRef]
- Liu, X.; Strable, M.S.; Ntambi, J.M. Stearoyl CoA Desaturase 1: Role in Cellular Inflammation and Stress. Adv. Nutr. 2011, 2, 15–22. [Google Scholar] [CrossRef] [Green Version]
- Dobrzyń, A. The Role of Stearoyl-CoA Desaturase in Body Weight Regulation. Trends Cardiovasc. Med. 2004, 14, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Matsui, H.; Yokoyama, T.; Sekiguchi, K.; Iijima, D.; Sunaga, H.; Maniwa, M.; Ueno, M.; Iso, T.; Arai, M.; Kurabayashi, M. Stearoyl-CoA Desaturase-1 (SCD1) Augments Saturated Fatty Acid-Induced Lipid Accumulation and Inhibits Apoptosis in Cardiac Myocytes. PLoS ONE 2012, 7, e33283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauvoisin, D.; Mounier, C. Hormonal and nutritional regulation of SCD1 gene expression. Biochimie 2011, 93, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Shao, W.; Ip, W.; Song, Z.; Badakhshi, Y.; Jin, T. The developmental Wnt signaling pathway effector β-catenin/TCF mediates hepatic functions of the sex hormone estradiol in regulating lipid metabolism. PLoS Biol. 2019, 17, e3000444. [Google Scholar] [CrossRef] [PubMed]
- Samuel, W.; Nagineni, C.N.; Kutty, R.K.; Parks, W.T.; Gordon, J.S.; Prouty, S.M.; Hooks, J.J.; Wiggert, B. Transforming Growth Factor-β Regulates Stearoyl Coenzyme A Desaturase Expression through a Smad Signaling Pathway. J. Biol. Chem. 2002, 277, 59–66. [Google Scholar] [CrossRef] [Green Version]
- Samuel, W.; Kutty, R.K.; Nagineni, S.; Gordon, J.S.; Prouty, S.M.; Chandraratna, R.A.S.; Wiggert, B. Regulation of Stearoyl Coenzyme A Desaturase Expression in Human Retinal Pigment Epithelial Cells by Retinoic Acid. J. Biol. Chem. 2001, 276, 28744–28750. [Google Scholar] [CrossRef] [Green Version]
- Mori, H.; Dugan, C.E.; Nishii, A.; Benchamana, A.; Li, Z.; Cadenhead, T.S.; Das, A.K.; Evans, C.R.; Overmyer, K.A.; Romanelli, S.M.; et al. The Molecular and Metabolic Program for Adaptation of White Adipocytes to Cool Physiologic Temperatures. bioRxiv 2020. [Google Scholar] [CrossRef]
- Chen, M.; Huang, J. The expanded role of fatty acid metabolism in cancer: New aspects and targets. Precis. Clin. Med. 2019, 2, 183–191. [Google Scholar] [CrossRef] [Green Version]
- Cheng, M.; Bhujwalla, Z.M.; Glunde, K. Targeting Phospholipid Metabolism in Cancer. Front. Oncol. 2016, 6, 266. [Google Scholar] [CrossRef] [Green Version]
- Sharma, B.; Kanwar, S.S. Phosphatidylserine: A cancer cell targeting biomarker. Semin. Cancer Biol. 2018, 52, 17–25. [Google Scholar] [CrossRef]
- Huang, J.; Fan, X.-X.; He, J.; Pan, H.; Liyan, H.; Huang, L.; Jiang, Z.; Yao, X.-J.; Liu, L.; Leung, E.L.-H.; et al. SCD1 is associated with tumor promotion, late stage and poor survival in lung adenocarcinoma. Oncotarget 2016, 7, 39970–39979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ackerman, D.; Simon, M.C. Hypoxia, lipids, and cancer: Surviving the harsh tumor microenvironment. Trends Cell Biol. 2014, 24, 472–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munir, R.; Lisec, J.; Swinnen, J.V.; Zaidi, N. Lipid metabolism in cancer cells under metabolic stress. Br. J. Cancer 2019, 120, 1090–1098. [Google Scholar] [CrossRef]
- Oakes, S.A. Endoplasmic Reticulum Stress Signaling in Cancer Cells. Am. J. Pathol. 2020, 190, 934–946. [Google Scholar] [CrossRef] [Green Version]
- Mahameed, M.; Boukeileh, S.; Obiedat, A.; Darawshi, O.; Dipta, P.; Rimon, A.; McLennan, G.; Fassler, R.; Reichmann, D.; Karni, R.; et al. Pharmacological induction of selective endoplasmic reticulum retention as a strategy for cancer therapy. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Raymundo, D.P.; Doultsinos, D.; Guillory, X.; Carlesso, A.; Eriksson, L.A.; Chevet, E. Pharmacological Targeting of IRE1 in Cancer. Trends Cancer 2020, 6, 1018–1030. [Google Scholar] [CrossRef]
- Pagliassotti, M.J.; Kim, P.Y.; Estrada, A.L.; Stewart, C.M.; Gentile, C.L. Endoplasmic reticulum stress in obesity and obesity-related disorders: An expanded view. Metabolism 2016, 65, 1238–1246. [Google Scholar] [CrossRef] [Green Version]
- Basseri, S.; Austin, R.C. Endoplasmic Reticulum Stress and Lipid Metabolism: Mechanisms and Therapeutic Potential. Biochem. Res. Int. 2011, 2012, 1–13. [Google Scholar] [CrossRef]
- Gregor, M.F.; Yang, L.; Fabbrini, E.; Mohammed, B.S.; Eagon, J.C.; Hotamisligil, G.S.; Klein, S. Endoplasmic Reticulum Stress Is Reduced in Tissues of Obese Subjects After Weight Loss. Diabetes 2008, 58, 693–700. [Google Scholar] [CrossRef] [Green Version]
- Sohn, M.; Ivanova, P.; Brown, H.A.; Toth, D.J.; Varnai, P.; Kim, Y.J.; Balla, T. Lenz-Majewski mutations in PTDSS1 affect phosphatidylinositol 4-phosphate metabolism at ER-PM and ER-Golgi junctions. Proc. Natl. Acad. Sci. USA 2016, 113, 4314–4319. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Carvalho, P.; Voeltz, G.K. Here, there, and everywhere: The importance of ER membrane contact sites. Science 2018, 361, eaan5835. [Google Scholar] [CrossRef] [Green Version]
- Funato, K.; Riezman, H.; Muñiz, M. Vesicular and non-vesicular lipid export from the ER to the secretory pathway. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158453. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, U.; Yilmaz, E.; Özcan, L.; Furuhashi, M.; Vaillancourt, E.; Smith, R.O.; Görgün, C.Z.; Hotamisligil, G.S. Chemical Chaperones Reduce ER Stress and Restore Glucose Homeostasis in a Mouse Model of Type 2 Diabetes. Science 2006, 313, 1137–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mai, C.T.; Le, Q.G.; Ishiwata-Kimata, Y.; Takagi, H.; Kohno, K.; Kimata, Y. 4-Phenylbutyrate suppresses the unfolded protein response without restoring protein folding in Saccharomyces cerevisiae. FEMS Yeast Res. 2018, 18, 16. [Google Scholar] [CrossRef] [Green Version]
- Nissar, A.U.; Sharma, L.; Mudasir, M.A.; Nazir, L.A.; Umar, S.A.; Sharma, P.R.; Vishwakarma, R.A.; Tasduq, S.A. Chemical chaperone 4-phenyl butyric acid (4-PBA) reduces hepatocellular lipid accumulation and lipotoxicity through induction of autophagy. J. Lipid Res. 2017, 58, 1855–1868. [Google Scholar] [CrossRef] [Green Version]
- Arai, Y.; Choi, B.; Kim, B.J.; Rim, W.; Park, S.; Park, H.; Ahn, J.; Park, H. Tauroursodeoxycholic acid (TUDCA) counters osteoarthritis by regulating intracellular cholesterol levels and membrane fluidity of degenerated chondrocytes. Biomater. Sci. 2019, 7, 3178–3189. [Google Scholar] [CrossRef]
- Lee, H.; Lee, G.; Kim, H.; Lee, Y.; Chae, H. Phosphatidylinositol 3-kinase-δ controls endoplasmic reticulum membrane fluidity and permeability in fungus-induced allergic inflammation in mice. Br. J. Pharmacol. 2020, 177, 1556–1567. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Wang, D.; Gentile, C.L.; Pagliassotti, M.J. Reduced endoplasmic reticulum luminal calcium links saturated fatty acid-mediated endoplasmic reticulum stress and cell death in liver cells. Mol. Cell. Biochem. 2009, 331, 31–40. [Google Scholar] [CrossRef] [Green Version]
- Bi, J.; Wang, W.; Liu, Z.; Huang, X.; Jiang, Q.; Liu, G.; Wang, Y.; Huang, X. Seipin Promotes Adipose Tissue Fat Storage through the ER Ca2+-ATPase SERCA. Cell Metab. 2014, 19, 861–871. [Google Scholar] [CrossRef] [Green Version]
- Kitai, Y.; Ariyama, H.; Kono, N.; Oikawa, D.; Iwawaki, T.; Arai, H. Membrane lipid saturation activates IRE1α without inducing clustering. Genes Cells 2013, 18, 798–809. [Google Scholar] [CrossRef]
- Promlek, T.; Ishiwata-Kimata, Y.; Shido, M.; Sakuramoto, M.; Kohno, K.; Kimata, Y. Membrane aberrancy and unfolded proteins activate the endoplasmic reticulum stress sensor Ire1 in different ways. Mol. Biol. Cell 2011, 22, 3520–3532. [Google Scholar] [CrossRef] [PubMed]
- Deguil, J.; Pineau, L.; Snyder, E.C.R.; Dupont, S.; Beney, L.; Gil, A.; Frapper, G.; Ferreira, T. Modulation of Lipid-Induced ER Stress by Fatty Acid Shape. Traffic 2011, 12, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Shyu, P.; Ng, B.S.H.; Ho, N.; Chaw, R.; Seah, Y.L.; Marvalim, C.; Thibault, G. Membrane phospholipid alteration causes chronic ER stress through early degradation of homeostatic ER-resident proteins. Sci. Rep. 2019, 9, 8637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boslem, E.; Weir, J.M.; MacIntosh, G.; Sue, N.; Cantley, J.; Meikle, P.J.; Biden, T.J. Alteration of Endoplasmic Reticulum Lipid Rafts Contributes to Lipotoxicity in Pancreatic β-Cells. J. Biol. Chem. 2013, 288, 26569–26582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharpe, H.J.; Stevens, T.J.; Munro, S. A Comprehensive Comparison of Transmembrane Domains Reveals Organelle-Specific Properties. Cell 2010, 142, 158–169. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Trikha, S.; Bhowmick, D.C.; Sarkar, A.A.; Jeremic, A.M. Role of Cholesterol and Phospholipids in Amylin Misfolding, Aggregation and Etiology of Islet Amyloidosis. Cannabinoids Neuropsychiatr. Disord. 2015, 855, 95–116. [Google Scholar] [CrossRef] [Green Version]
- Bogdanov, M.; Dowhan, W. Phospholipid-assisted protein folding: Phosphatidylethanolamine is required at a late step of the conformational maturation of the polytopic membrane protein lactose permease. EMBO J. 1998, 17, 5255–5264. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-S.; Mendez, R.; Heng, H.H.; Yang, Z.-Q.; Zhang, K. Pharmacological ER stress promotes hepatic lipogenesis and lipid droplet formation. Am. J. Transl. Res. 2012, 4, 102–113. [Google Scholar]
- Kim, S.H.; Kwon, D.-Y.; Kwak, J.-H.; Lee, S.; Lee, Y.-H.; Yun, J.; Son, T.G.; Jung, Y.-S. Tunicamycin-Induced ER Stress is Accompanied with Oxidative Stress via Abrogation of Sulfur Amino Acids Metabolism in the Liver. Int. J. Mol. Sci. 2018, 19, 4114. [Google Scholar] [CrossRef] [Green Version]
- Qin, X.; Su, T.; Yu, W.; Kojima, S. Lipid desaturation-associated endoplasmic reticulum stress regulates MYCN gene expression in hepatocellular carcinoma cells. Cell Death Dis. 2020, 11, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Bik, E.; Mielniczek, N.; Jarosz, M.; Denbigh, J.; Budzynska, R.; Baranska, M.; Majzner, K. Tunicamycin induced endoplasmic reticulum changes in endothelial cells investigated in vitro by confocal Raman imaging. Analyst 2019, 144, 6561–6569. [Google Scholar] [CrossRef] [PubMed]
- Reinhard, J.; Mattes, C.; Väth, K.; Radanović, T.; Surma, M.A.; Klose, C.; Ernst, R. A Quantitative Analysis of Cellular Lipid Compositions During Acute Proteotoxic ER Stress Reveals Specificity in the Production of Asymmetric Lipids. Front. Cell Dev. Biol. 2020, 8, 756. [Google Scholar] [CrossRef] [PubMed]
- Witting, M.; Hastings, J.; Rodriguez, N.; Joshi, C.J.; Hattwell, J.P.N.; Ebert, P.R.; Van Weeghel, M.; Gao, A.W.; Wakelam, M.J.O.; Houtkooper, R.H.; et al. Modeling Meets Metabolomics—The WormJam Consensus Model as Basis for Metabolic Studies in the Model Organism Caenorhabditis elegans. Front. Mol. Biosci. 2018, 5, 96. [Google Scholar] [CrossRef] [PubMed]
- Artyukhin, A.B.; Zhang, Y.K.; Akagi, A.E.; Panda, O.; Sternberg, P.W.; Schroeder, F.C. Metabolomic “Dark Matter” Dependent on Peroxisomal β-Oxidation in Caenorhabditis elegans. J. Am. Chem. Soc. 2018, 140, 2841–2852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, R.; Ramanathan, A. The Aging Metabolome—Biomarkers to Hub Metabolites. Proteomics 2020, 20, e1800407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.T.M.; An, Y.J.; Cha, J.W.; Ko, Y.-J.; Lee, H.; Chung, C.H.; Jeon, S.-M.; Lee, J.; Park, S. Real-Time In-Organism NMR Metabolomics Reveals Different Roles of AMP-Activated Protein Kinase Catalytic Subunits. Anal. Chem. 2020, 92, 7382–7387. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-W.; Lemieux, G.A.; Camp, C.H.; Chang, T.-C.; Ashrafi, K.; Cicerone, M.T. Spectroscopic coherent Raman imaging of Caenorhabditis elegans reveals lipid particle diversity. Nat. Chem. Biol. 2020, 16, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Gebauer, J.; Gentsch, C.; Mansfeld, J.; Schmeißer, K.; Waschina, S.; Brandes, S.; Klimmasch, L.; Zamboni, N.; Zarse, K.; Schuster, S.; et al. A Genome-Scale Database and Reconstruction of Caenorhabditis elegans Metabolism. Cell Syst. 2016, 2, 312–322. [Google Scholar] [CrossRef] [Green Version]
- Yilmaz, L.S.; Walhout, A.J.M. A Caenorhabditis elegans Genome-Scale Metabolic Network Model. Cell Syst. 2016, 2, 297–311. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Chan, A.H.C.; Hattwell, J.; Ebert, P.; Schirra, H.J. Systems Biology Analysis Using a Genome-Scale Metabolic Model Shows That Phosphine Triggers Global Metabolic Suppression in a Resistant Strain of C. Elegans. bioRxiv 2017. [Google Scholar] [CrossRef] [Green Version]
- Hastings, J.; Mains, A.; Artal-Sanz, M.; Bergmann, S.; Braeckman, B.P.; Bundy, J.G.; Cabreiro, F.; Dobson, P.; Ebert, P.; Hattwell, J.; et al. WormJam: A consensus C. elegans Metabolic Reconstruction and Metabolomics Community and Workshop Series. Worm 2017, 6, e1373939. [Google Scholar] [CrossRef] [Green Version]
- Orth, J.D.; Thiele, I.; Palsson, B.O. What is flux balance analysis? Nat. Biotechnol. 2010, 28, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Hastings, J.; Mains, A.; Virk, B.; Rodriguez, N.; Murdoch, S.; Pearce, J.; Bergmann, S.; Le Novère, N.; Casanueva, O. Multi-Omics and Genome-Scale Modeling Reveal a Metabolic Shift During C. elegans Aging. Front. Mol. Biosci. 2019, 6, 2. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Zhao, Q.; Luo, J.; Ma, W.; Jin, Y.; Li, C.; Hou, Y.; Feng, M.; Wang, Y.; Chen, J.; et al. Integration of proteomics, lipidomics, and metabolomics reveals novel metabolic mechanisms underlying N, N-dimethylformamide induced hepatotoxicity. Ecotoxicol. Environ. Saf. 2020, 205, 111166. [Google Scholar] [CrossRef] [PubMed]
- Yuan, P.; Dong, M.; Lei, H.; Xu, G.; Chen, G.; Song, Y.; Ma, J.; Cheng, L.; Zhang, L. Targeted metabolomics reveals that 2,3,7,8-tetrachlorodibenzofuran exposure induces hepatic steatosis in male mice. Environ. Pollut. 2020, 259, 113820. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, V.B.; Dennis, E.A.; Wakelam, M.J.O.; Subramaniam, S. LIPID MAPS: Serving the next generation of lipid researchers with tools, resources, data, and training. Sci. Signal. 2019, 12, eaaw2964. [Google Scholar] [CrossRef] [Green Version]
- Walker, A.K.; Jacobs, R.L.; Watts, J.L.; Rottiers, V.; Jiang, K.; Finnegan, D.M.; Shioda, T.; Hansen, M.; Yang, F.; Niebergall, L.J.; et al. A Conserved SREBP-1/Phosphatidylcholine Feedback Circuit Regulates Lipogenesis in Metazoans. Cell 2011, 147, 840–852. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Zhang, F.; Tay, L.W.R.; Boroda, S.; Nian, W.; Levental, K.R.; Levental, I.; Harris, T.E.; Chang, J.T.; Du, G. Lipin-1 regulation of phospholipid synthesis maintains endoplasmic reticulum homeostasis and is critical for triple-negative breast cancer cell survival. FASEB J. 2017, 31, 2893–2904. [Google Scholar] [CrossRef] [Green Version]
- Golden, A.; Liu, J.; Cohen-Fix, O. Inactivation of the C. elegans lipin homolog leads to ER disorganization and to defects in the breakdown and reassembly of the nuclear envelope. J. Cell Sci. 2009, 122, 1970–1978. [Google Scholar] [CrossRef] [Green Version]
- Jung, Y.; Kwon, S.; Ham, S.; Lee, D.; Park, H.H.; Yamaoka, Y.; Jeong, D.; Artan, M.; Altintas, O.; Park, S.; et al. Caenorhabditis elegans Lipin 1 moderates the lifespan-shortening effects of dietary glucose by maintaining ω-6 polyunsaturated fatty acids. Aging Cell 2020, 19, 13150. [Google Scholar] [CrossRef]
- Sapir, A.; Tsur, A.; Koorman, T.; Ching, K.; Mishra, P.; Bardenheier, A.; Podolsky, L.; Bening-Abu-Shach, U.; Boxem, M.; Chou, T.-F.; et al. Controlled sumoylation of the mevalonate pathway enzyme HMGS-1 regulates metabolism during aging. Proc. Natl. Acad. Sci. USA 2014, 111, E3880–E3889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.-C.; Wu, M.-L.; Huang, K.-C.; Lin, W.-W. HMG-CoA reductase inhibitors activate the unfolded protein response and induce cytoprotective GRP78 expression. Cardiovasc. Res. 2008, 80, 138–150. [Google Scholar] [CrossRef] [Green Version]
- Watts, J.L.; Ristow, M. Lipid and Carbohydrate Metabolism in Caenorhabditis elegans. Genetics 2017, 207, 413–446. [Google Scholar] [PubMed]
- Thinon, E.; Serwa, R.A.; Broncel, M.; Brannigan, J.A.; Brassat, U.; Wright, M.H.; Heal, W.P.; Wilkinson, A.J.; Mann, D.J.; Tate, E.W. Global profiling of co- and post-translationally N-myristoylated proteomes in human cells. Nat. Commun. 2014, 5, 4919. [Google Scholar] [CrossRef] [Green Version]
- Thinon, E.; Morales-Sanfrutos, J.; Mann, D.J.; Tate, E.W. N-Myristoyltransferase Inhibition Induces ER-Stress, Cell Cycle Arrest, and Apoptosis in Cancer Cells. ACS Chem. Biol. 2016, 11, 2165–2176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiavone, M.L.; Millucci, L.; Bernardini, G.; Giustarini, D.; Rossi, R.; Marzocchi, B.; Santucci, A. Homogentisic acid affects human osteoblastic functionality by oxidative stress and alteration of the Wnt/β-catenin signaling pathway. J. Cell. Physiol. 2020, 235, 6808–6816. [Google Scholar] [CrossRef] [PubMed]
- Fisher, A.L.; Page, K.E.; Lithgow, G.J.; Nash, L. The Caenorhabditis elegans K10C2.4 Gene Encodes a Member of the Fumarylacetoacetate Hydrolase Family. J. Biol. Chem. 2008, 283, 9127–9135. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Chen, H.; Hao, G.; Yang, B.; Feng, Y.; Wang, Y.; Feng, L.; Zhao, J.; Song, Y.; Zhang, H.; et al. Role of the Phenylalanine-Hydroxylating System in Aromatic Substance Degradation and Lipid Metabolism in the Oleaginous Fungus Mortierella alpina. Appl. Environ. Microbiol. 2013, 79, 3225–3233. [Google Scholar] [CrossRef] [Green Version]
- Moseley, K.D.; Koch, R.; Moser, A. Lipid Status and Long-Chain Polyunsaturated Fatty Acid Concentrations in Adults and Adolescents with Phenylketonuria on Phenylalanine-Restricted Diet. J. Inherit. Metab. Dis. 2002, 25, 56–64. [Google Scholar] [CrossRef]
- Perkins, R.J.; Vaida, V. Phenylalanine Increases Membrane Permeability. J. Am. Chem. Soc. 2017, 139, 14388–14391. [Google Scholar] [CrossRef] [Green Version]
- Entchev, E.V.; Schwudke, D.; Zagoriy, V.; Matyash, V.; Bogdanova, A.; Habermann, B.; Zhu, L.; Shevchenko, A.; Kurzchalia, T.V. LET-767 Is Required for the Production of Branched Chain and Long Chain Fatty Acids inCaenorhabditis elegans. J. Biol. Chem. 2008, 283, 17550–17560. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Abraham, N.; Khan, L.A.; Hall, D.H.; Fleming, J.T.; Göbel, V. Apicobasal domain identities of expanding tubular membranes depend on glycosphingolipid biosynthesis. Nat. Cell Biol. 2011, 13, 1189–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuervers, L.M.; Jones, C.L.; O’Neil, N.J.; Baillie, D.L. The sterol modifying enzyme LET-767 is essential for growth, reproduction and development in Caenorhabditis elegans. Mol. Genet. Genom. 2003, 270, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Galles, C.; Prez, G.M.; Penkov, S.; Boland, S.; Porta, E.O.J.; Altabe, S.G.; Labadie, G.R.; Schmidt, U.; Knölker, H.-J.; Kurzchalia, T.V.; et al. Endocannabinoids in Caenorhabditis elegans are essential for the mobilization of cholesterol from internal reserves. Sci. Rep. 2018, 8, 6398. [Google Scholar] [CrossRef] [PubMed]
- Garcia, G. Lipid Homeostasis Is Essential for Endoplasmic Reticulum Protein Quality Control. Ph.D. Thesis, University of California, Berkeley, CA, USA, 2019. [Google Scholar]
- Kniazeva, M.; Crawford, Q.T.; Seiber, M.; Wang, C.-Y.; Han, M. Monomethyl Branched-Chain Fatty Acids Play an Essential Role in Caenorhabditis elegans Development. PLoS Biol. 2004, 2, e257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tam, A.B.; Roberts, L.S.; Chandra, V.; Rivera, I.G.; Nomura, D.; Forbes, U.J.; Niwa, M. The UPR Activator ATF6 Responds to Proteotoxic and Lipotoxic Stress by Distinct Mechanisms. Dev. Cell 2018, 46, 327–343.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Micoogullari, Y.; Basu, S.S.; Ang, J.; Weisshaar, N.; Schmitt, N.D.; Abdelmoula, W.M.; Lopez, B.; Agar, J.N.; Agar, N.; Hanna, J. Dysregulation of very-long-chain fatty acid metabolism causes membrane saturation and induction of the unfolded protein response. Mol. Biol. Cell 2020, 31, 7–17. [Google Scholar] [CrossRef]
- Williamson, C.D.; Wong, D.S.; Bozidis, P.; Zhang, A.; Colberg-Poley, A.M. Isolation of Endoplasmic Reticulum, Mitochondria, and Mitochondria-Associated Membrane and Detergent Resistant Membrane Fractions from Transfected Cells and from Human Cytomegalovirus-Infected Primary Fibroblasts. Curr. Protoc. Cell Biol. 2015, 68, 3.27.1–3.27.33. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, J.; Taubert, S. Beyond Proteostasis: Lipid Metabolism as a New Player in ER Homeostasis. Metabolites 2021, 11, 52. https://doi.org/10.3390/metabo11010052
Xu J, Taubert S. Beyond Proteostasis: Lipid Metabolism as a New Player in ER Homeostasis. Metabolites. 2021; 11(1):52. https://doi.org/10.3390/metabo11010052
Chicago/Turabian StyleXu, Jiaming, and Stefan Taubert. 2021. "Beyond Proteostasis: Lipid Metabolism as a New Player in ER Homeostasis" Metabolites 11, no. 1: 52. https://doi.org/10.3390/metabo11010052
APA StyleXu, J., & Taubert, S. (2021). Beyond Proteostasis: Lipid Metabolism as a New Player in ER Homeostasis. Metabolites, 11(1), 52. https://doi.org/10.3390/metabo11010052