
Approaches to Integrating Metabolomics and Multi-Omics Data: A Primer


Abstract
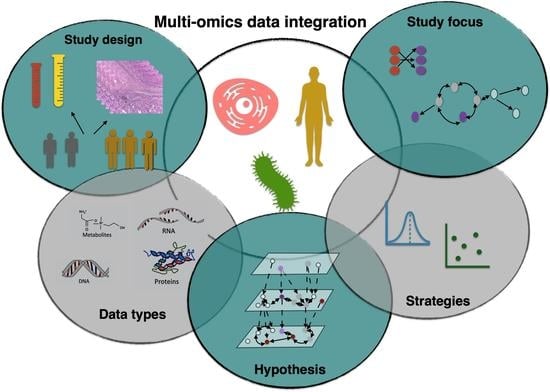
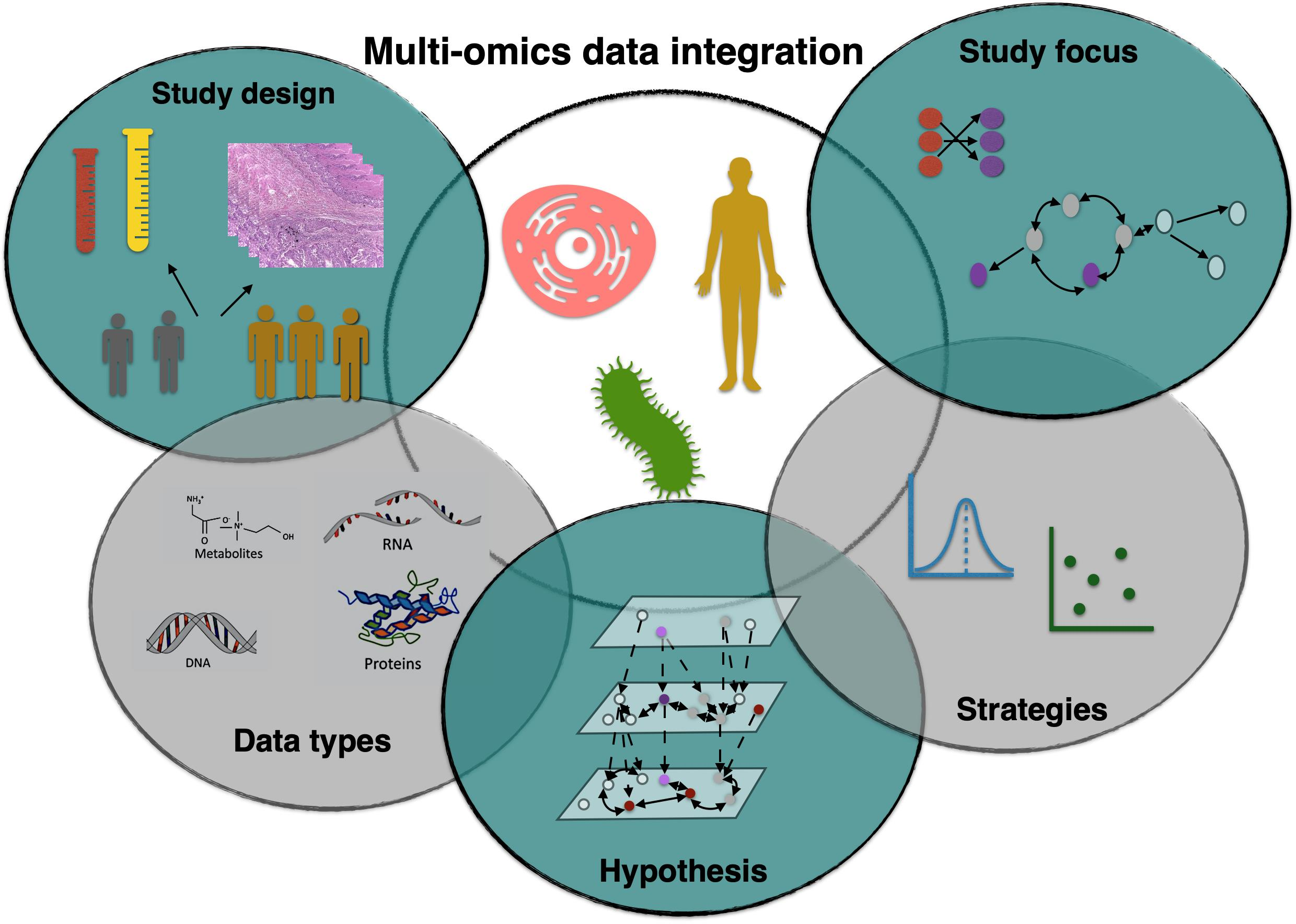
:
1. Introduction
2. Challenges in Metabolomics and Multi-Omics Data Integration
3. Study Design
4. Data Types
5. Hypothesis
6. Data Integration Strategies
7. Study Focus
8. Discussion
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bylesjö, M.; Eriksson, D.; Kusano, M.; Moritz, T.; Trygg, J. Data integration in plant biology: The O2PLS method for combined modeling of transcript and metabolite data. Plant J. 2007, 52, 1181–1191. [Google Scholar] [CrossRef] [PubMed]
- Griffin, J.L.; Blenkiron, C.; Valonen, P.K.; Caldas, C.; Kauppinen, R.A. High-resolution magic angle spinning 1H NMR spectroscopy and reverse transcription-PCR analysis of apoptosis in a rat glioma. Anal. Chem. 2006, 78, 1546–1552. [Google Scholar] [CrossRef] [PubMed]
- Lindon, J.C.; Nicholson, J.K.; Holmes, E.; Antti, H.; Bollard, M.E.; Keun, H.; Beckonert, O.; Ebbels, T.M.; Reily, M.D.; Robertson, D.; et al. Contemporary issues in toxicology the role of metabonomics in toxicology and its evaluation by the COMET project. Toxicol. Appl. Pharmacol. 2003, 187, 137–146. [Google Scholar] [CrossRef]
- Li, S.; Todor, A.; Luo, R. Blood transcriptomics and metabolomics for personalized medicine. Comput. Struct. Biotechnol. J. 2016, 14, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Robinson, O.; Chadeau, H.M.; Karaman, I.; Climaco, P.R.; Ala-Korpela, M.; Handakas, E.; Fiorito, G.; Gao, H.; Heard, A.; Jarvelin, M.R.; et al. Determinants of accelerated metabolomic and epigenetic ageing in a UK cohort. Aging Cell 2020, 19, 1–13. [Google Scholar] [CrossRef]
- Karaman, I.; Ferreira, D.; Boulange, C.; Kaluarachchi, M.; Herrington, D.; Dona, A.; Castagné, R.; Moayyeri, A.; Lehne, B.; Loh, M.; et al. A workflow for integrated processing of multi-cohort untargeted 1H NMR metabolomics data in large scale metabolic epidemiology. J. Proteome Res. 2016, 15, 4188–4194. [Google Scholar] [CrossRef]
- Valcárcel, B.; Ebbels, T.M.; Kangas, A.J.; Soininen, P.; Elliot, P.; Ala-Korpela, M.; Järvelin, M.R.; de Iorio, M. Genome metabolome integrated network analysis to uncover connections between genetic variants and complex traits: An application to obesity. J. R. Soc. Interface 2014, 11, 20130908. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, J.K.; Wilson, I.D. Understanding ’global’ systems biology: Metabonomics and the continuum of metabolism. Nat. Rev. Drug Discov. 2003, 2, 668. [Google Scholar] [CrossRef] [PubMed]
- Haukaas, T.H.; Euceda, L.R.; Giskeødegård, G.F.; Bathen, T.F. Metabolic portraits of breast cancer by HR MAS MR spectroscopy of intact tissue samples. Metabolites 2017, 7, 18. [Google Scholar] [CrossRef] [Green Version]
- Pazoki, R.; Evangelou, E.; Mosen-Ansorena, D.; Pinto, R.; Karaman, I.; Blakeley, P.; Gill, D.; Zuber, V.; Elliott, P.; Tzoulaki, I.; et al. Pathways underlying urinary sodium and potassium excretion and the link to blood pressure and cardiovascular disease. J. Hypertens. 2019, 37, e74. [Google Scholar] [CrossRef]
- Rantalainen, M.; Cloarec, O.; Beckonert, O.; Wilson, I.; Jackson, D.; Tonge, R.; Rowlinson, R.; Rayner, S.; Nickson, J.; Wilkinson, R.W.; et al. Statistically integrated metabonomic- proteomic studies on a human prostate cancer xenograft model in mice. J. Proteome Res. 2006, 5, 2642–2655. [Google Scholar] [CrossRef]
- Jendoubi, T.; Ebbels, T.M. Integrative analysis of time course metabolic data and biomarker discovery. BMC Bioinform. 2020, 21, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Ruepp, S.U.; Tonge, R.P.; Shaw, J.; Wallis, N.; Pognan, F. Genomics and proteomics analysis of acetaminophen toxicity in mouse liver. Toxicol. Sci. 2002, 65, 135–150. [Google Scholar] [CrossRef] [Green Version]
- Dumas, M.E.; Wilder, S.P.; Bihoreau, M.T.; Barton, R.H.; Fearnside, J.F.; Argoud, K.; D’Amato, L.; Wallis, R.H.; Blancher, C.; Keun, H.C.; et al. Direct quantitative trait locus mapping of mammalian metabolic phenotypes in diabetic and normoglycemic rat models. Nat. Genet. 2007, 39, 666–672. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, G.; Rantalainen, M.; Li, J.V.; Maher, A.D.; Malmodin, D.; Ahmadi, K.R.; Faber, J.H.; Barrett, A.; Min, J.L.; Rayner, N.W.; et al. A genome-wide metabolic QTL analysis in Europeans implicates two loci shaped by recent positive selection. PLoS Genet. 2011, 7, e1002270. [Google Scholar] [CrossRef] [Green Version]
- Clayton, T.A.; Lindon, J.C.; Cloarec, O.; Antti, H.; Charuel, C.; Hanton, G.; Provost, J.P.; Le Net, J.L.; Baker, D.; Walley, R.J.; et al. Pharmaco-metabonomic phenotyping and personalized drug treatment. Nature 2006, 440, 1073–1077. [Google Scholar] [CrossRef] [PubMed]
- Teitsma, X.M.; Yang, W.; Jacobs, J.W.; Pethö-Schramm, A.; Borm, M.E.; Harms, A.C.; Hankemeier, T.; van Laar, J.M.; Bijlsma, J.W.; Lafeber, F.P. Baseline metabolic profiles of early rheumatoid arthritis patients achieving sustained drug-free remission after initiating treat-to-target tocilizumab, methotrexate, or the combination: Insights from systems biology. Arthritis Res. Ther. 2018, 20, 230. [Google Scholar] [CrossRef] [Green Version]
- Griffin, J.L.; Bonney, S.A.; Mann, C.; Hebbachi, A.M.; Gibbons, G.F.; Nicholson, J.K.; Shoulders, C.C.; Scott, J. An integrated reverse functional genomic and metabolic approach to understanding orotic acid-induced fatty liver. Physiol. Genom. 2004, 17, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Raamsdonk, L.M.; Teusink, B.; Broadhurst, D.; Zhang, N.; Hayes, A.; Walsh, M.C.; Berden, J.A.; Brindle, K.M.; Kell, D.B.; Rowland, J.J.; et al. A functional genomics strategy that uses metabolome data to reveal the phenotype of silent mutations. Nat. Biotechnol. 2001, 19, 45–50. [Google Scholar] [CrossRef]
- Lindon, J.C.; Nicholson, J.K. Spectroscopic and statistical techniques for information recovery in metabonomics and metabolomics. Annu. Rev. Anal. Chem. 2008, 1, 45–69. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, J.K.; Holmes, E.; Lindon, J.C.; Wilson, I.D. The challenges of modeling mammalian biocomplexity. Nat. Biotechnol. 2004, 22, 1268–1274. [Google Scholar] [CrossRef] [PubMed]
- Gieger, C.; Geistlinger, L.; Altmaier, E.; De Angelis, M.H.; Kronenberg, F.; Meitinger, T.; Mewes, H.W.; Wichmann, H.E.; Weinberger, K.M.; Adamski, J.; et al. Genetics meets metabolomics: A genome-wide association study of metabolite profiles in human serum. PLoS Genet. 2008, 4, e1000282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kathiresan, S.; Manning, A.K.; Demissie, S.; D’agostino, R.B.; Surti, A.; Guiducci, C.; Gianniny, L.; Burtt, N.P.; Melander, O.; Orho-Melander, M.; et al. A genome-wide association study for blood lipid phenotypes in the Framingham Heart Study. BMC Med Genet. 2007, 8, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teslovich, T.M.; Musunuru, K.; Smith, A.V.; Edmondson, A.C.; Stylianou, I.M.; Koseki, M.; Pirruccello, J.P.; Ripatti, S.; Chasman, D.I.; Willer, C.J.; et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature 2010, 466, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Vineis, P.; van Veldhoven, K.; Chadeau-Hyam, M.; Athersuch, T.J. Advancing the application of omics-based biomarkers in environmental epidemiology. Environ. Mol. Mutagen. 2013, 54, 461–467. [Google Scholar] [CrossRef]
- Suhre, K.; Wallaschofski, H.; Raffler, J.; Friedrich, N.; Haring, R.; Michael, K.; Wasner, C.; Krebs, A.; Kronenberg, F.; Chang, D.; et al. A genome-wide association study of metabolic traits in human urine. Nat. Genet. 2011, 43, 565. [Google Scholar] [CrossRef]
- Rattray, N.J.; Deziel, N.C.; Wallach, J.D.; Khan, S.A.; Vasiliou, V.; Ioannidis, J.P.; Johnson, C.H. Beyond genomics: Understanding exposotypes through metabolomics. Hum. Genom. 2018, 12, 4. [Google Scholar] [CrossRef] [Green Version]
- Mishra, P.; Roger, J.M.; Jouan-Rimbaud-Bouveresse, D.; Biancolillo, A.; Marini, F.; Nordon, A.; Rutledge, D.N. Recent trends in multi-block data analysis in chemometrics for multi-source data integration. TrAC Trends Anal. Chem. 2021, 137, 116206. [Google Scholar] [CrossRef]
- Kirk, P.; Griffin, J.E.; Savage, R.S.; Ghahramani, Z.; Wild, D.L. Bayesian correlated clustering to integrate multiple datasets. Bioinformatics 2012, 28, 3290–3297. [Google Scholar] [CrossRef]
- Liang, M.; Li, Z.; Chen, T.; Zeng, J. Integrative data analysis of multi-platform cancer data with a multimodal deep learning approach. IEEE/ACM Trans. Comput. Biol. Bioinform. 2014, 12, 928–937. [Google Scholar] [CrossRef]
- Le, V.; Quinn, T.P.; Tran, T.; Venkatesh, S. Deep in the Bowel: Highly interpretable neural encoder-decoder networks predict gut metabolites from gut microbiome. BMC Genom. 2020, 21, 1–15. [Google Scholar] [CrossRef]
- Richardson, S.; Tseng, G.C.; Sun, W. Statistical methods in integrative genomics. Annu. Rev. Stat. Its Appl. 2016, 3, 181–209. [Google Scholar] [CrossRef] [Green Version]
- Hamid, J.S.; Hu, P.; Roslin, N.M.; Ling, V.; Greenwood, C.M.; Beyene, J. Data integration in genetics and genomics: Methods and challenges. Hum. Genom. Proteom. HGP 2009, 2009, 869093. [Google Scholar] [CrossRef]
- Tseng, G.; Ghosh, D.; Zhou, X.J. Integrating Omics Data; Cambridge University Press: Cambridge, UK, 2015. [Google Scholar]
- O’Shea, K.; Misra, B.B. Software tools, databases and resources in metabolomics: Updates from 2018 to 2019. Metabolomics 2020, 16, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Chu, S.H.; Huang, M.; Kelly, R.S.; Benedetti, E.; Siddiqui, J.K.; Zeleznik, O.A.; Pereira, A.; Herrington, D.; Wheelock, C.E.; Krumsiek, J.; et al. Integration of metabolomic and other omics data in population-based study designs: An epidemiological perspective. Metabolites 2019, 9, 117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wörheide, M.A.; Krumsiek, J.; Kastenmüller, G.; Arnold, M. Multi-omics integration in biomedical research–A metabolomics-centric review. Anal. Chim. Acta 2020, 1141, 144–162. [Google Scholar] [CrossRef] [PubMed]
- Cavill, R.; Jennen, D.; Kleinjans, J.; Briedé, J.J. Transcriptomic and metabolomic data integration. Briefings Bioinform. 2015, 17, 891–901. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Y.; Savage, R.S.; Markowetz, F. Patient-specific data fusion defines prognostic cancer subtypes. PLoS Comput. Biol. 2011, 7, e1002227. [Google Scholar] [CrossRef] [Green Version]
- Evangelou, E.; Ioannidis, J.P. Meta-analysis methods for genome-wide association studies and beyond. Nat. Rev. Genet. 2013, 14, 379. [Google Scholar] [CrossRef]
- Krumsiek, J.; Suhre, K.; Illig, T.; Adamski, J.; Theis, F.J. Gaussian graphical modeling reconstructs pathway reactions from high-throughput metabolomics data. BMC Syst. Biol. 2011, 5, 21. [Google Scholar] [CrossRef] [Green Version]
- Smolinska, A.; Blanchet, L.; Coulier, L.; Ampt, K.A.M.; Luider, T.; Hintzen, R.Q.; Wijmenga, S.S.; Buydens, L.M.C. Interpretation and visualization of non-linear data fusion in kernel space: Study on metabolomic characterization of progression of multiple sclerosis. PLoS ONE 2012, 7, e38163. [Google Scholar] [CrossRef] [Green Version]
- Witten, D.M.; Tibshirani, R.J. Extensions of sparse canonical correlation analysis with applications to genomic data. Stat. Appl. Genet. Mol. Biol. 2009, 8, 1–27. [Google Scholar] [CrossRef]
- Daemen, A.; Gevaert, O.; Ojeda, F.; Debucquoy, A.; Suykens, J.A.; Sempoux, C.; Machiels, J.P.; Haustermans, K.; De Moor, B. A kernel-based integration of genome-wide data for clinical decision support. Genome Med. 2009, 1, 39. [Google Scholar] [CrossRef] [Green Version]
- Fridley, B.L.; Lund, S.; Jenkins, G.D.; Wang, L. A Bayesian Integrative Genomic Model for Pathway Analysis of Complex Traits. Genet. Epidemiol. 2012, 36, 352–359. [Google Scholar] [CrossRef] [Green Version]
- Le Van, T.; van Leeuwen, M.; Carolina Fierro, A.; De Maeyer, D.; Van den Eynden, J.; Verbeke, L.; De Raedt, L.; Marchal, K.; Nijssen, S. Simultaneous discovery of cancer subtypes and subtype features by molecular data integration. Bioinformatics 2016, 32, i445–i454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanckriet, G.R.; De Bie, T.; Cristianini, N.; Jordan, M.I.; Noble, W.S. A statistical framework for genomic data fusion. Bioinformatics 2004, 20, 2626–2635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Gao, L.; Wei, C.; Yang, X.; Zhao, Y.; Dong, A. A computational method based on the integration of heterogeneous networks for predicting disease-gene associations. PLoS ONE 2011, 6, e24171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acharjee, A.; Ament, Z.; West, J.A.; Stanley, E.; Griffin, J.L. Integration of metabolomics, lipidomics and clinical data using a machine learning method. BMC Bioinform. 2016, 17, 440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, R.; Mo, Q.; Schultz, N.; Seshan, V.E.; Olshen, A.B.; Huse, J.; Ladanyi, M.; Sander, C. Integrative subtype discovery in glioblastoma using iCluster. PLoS ONE 2012, 7, e35236. [Google Scholar] [CrossRef]
- Hirai, M.Y.; Klein, M.; Fujikawa, Y.; Yano, M.; Goodenowe, D.B.; Yamazaki, Y.; Kanaya, S.; Nakamura, Y.; Kitayama, M.; Suzuki, H.; et al. Elucidation of gene-to-gene and metabolite-to-gene networks in Arabidopsis by integration of metabolomics and transcriptomics. J. Biol. Chem. 2005, 280, 25590–25595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavill, R.; Kamburov, A.; Ellis, J.K.; Athersuch, T.J.; Blagrove, M.S.; Herwig, R.; Ebbels, T.M.; Keun, H.C. Consensus-phenotype integration of transcriptomic and metabolomic data implies a role for metabolism in the chemosensitivity of tumour cells. PLoS Comput. Biol. 2011, 7, e1001113. [Google Scholar] [CrossRef]
- Safo, S.E.; Li, S.; Long, Q. Integrative analysis of transcriptomic and metabolomic data via sparse canonical correlation analysis with incorporation of biological information. Biometrics 2018, 74, 300–312. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.; Chen, X.; Jin, L.; Xiong, M. Canonical correlation analysis for RNA-seq co-expression networks. Nucleic Acids Res. 2013, 41, e95. [Google Scholar] [CrossRef]
- Devlin, T.M. Textbook of Biochemistry; John Wiley & Sons: Hoboken, NJ, USA, 2011. [Google Scholar]
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, M.H.; Beger, R.; Daykin, C.A.; Fan, T.W.M.; Fiehn, O.; Goodacre, R.; Griffin, J.L.; et al. Proposed minimum reporting standards for chemical analysis. Metabolomics 2007, 3, 211–221. [Google Scholar] [CrossRef] [Green Version]
- Holliday, R. DNA methylation and epigenetic inheritance. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1990, 326, 329–338. [Google Scholar] [CrossRef]
- Richelle, A.; Joshi, C.; Lewis, N.E. Assessing key decisions for transcriptomic data integration in biochemical networks. PLoS Comput. Biol. 2019, 15, e1007185. [Google Scholar] [CrossRef] [Green Version]
- Jendoubi, T.; Strimmer, K. A whitening approach to probabilistic canonical correlation analysis for omics data integration. BMC Bioinform. 2019, 20, 1–13. [Google Scholar] [CrossRef]
- Riekeberg, E.; Powers, R. New frontiers in metabolomics: From measurement to insight. F1000Research 2017, 6. [Google Scholar] [CrossRef] [Green Version]
- Keun, H.C.; Ebbels, T.M.; Antti, H.; Bollard, M.E.; Beckonert, O.; Schlotterbeck, G.; Senn, H.; Niederhauser, U.; Holmes, E.; Lindon, J.C.; et al. Analytical reproducibility in 1H NMR-based metabonomic urinalysis. Chem. Res. Toxicol. 2002, 15, 1380–1386. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.R.; Eckel-Mahan, K.; Sassone-Corsi, P.; Baldi, P. CircadiOmics: Integrating circadian genomics, transcriptomics, proteomics and metabolomics. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Canzler, S.; Schor, J.; Busch, W.; Schubert, K.; Rolle-Kampczyk, U.E.; Seitz, H.; Kamp, H.; von Bergen, M.; Buesen, R.; Hackermüller, J. Prospects and challenges of multi-omics data integration in toxicology. Arch. Toxicol. 2020, 94, 371–388. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, J.K.; Lindon, J.C.; Holmes, E. ’Metabonomics’: Understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological NMR spectroscopic data. Xenobiotica 1999, 29, 1181–1189. [Google Scholar] [CrossRef]
- Reo, N.V. NMR-based metabolomics. DRug Chem. Toxicol. 2002, 25, 375–382. [Google Scholar] [CrossRef]
- Dettmer, K.; Aronov, P.A.; Hammock, B.D. Mass spectrometry-based metabolomics. Mass Spectrom. Rev. 2007, 26, 51–78. [Google Scholar] [CrossRef]
- Suhre, K.; Gieger, C. Genetic variation in metabolic phenotypes: Study designs and applications. Nat. Rev. Genet. 2012, 13, 759–769. [Google Scholar] [CrossRef]
- Marshall, D.D.; Powers, R. Beyond the paradigm: Combining mass spectrometry and nuclear magnetic resonance for metabolomics. Prog. Nucl. Magn. Reson. Spectrosc. 2017, 100, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Raffler, J.; Römisch-Margl, W.; Petersen, A.K.; Pagel, P.; Blöchl, F.; Hengstenberg, C.; Illig, T.; Meisinger, C.; Stark, K.; Wichmann, H.E.; et al. Identification and MS-assisted interpretation of genetically influenced NMR signals in human plasma. Genome Med. 2013, 5, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Bhinderwala, F.; Wase, N.; DiRusso, C.; Powers, R. Combining Mass Spectrometry and NMR Improves Metabolite Detection and Annotation. J. Proteome Res. 2018, 17, 4017–4022. [Google Scholar] [CrossRef]
- Pan, Z.; Raftery, D. Comparing and combining NMR spectroscopy and mass spectrometry in metabolomics. Anal. Bioanal. Chem. 2007, 387, 525–527. [Google Scholar] [CrossRef]
- Marshall, D.D.; Lei, S.; Worley, B.; Huang, Y.; Garcia-Garcia, A.; Franco, R.; Dodds, E.D.; Powers, R. Combining DI-ESI–MS and NMR datasets for metabolic profiling. Metabolomics 2015, 11, 391–402. [Google Scholar] [CrossRef] [Green Version]
- Yusufi, F.N.K.; Lakshmanan, M.; Ho, Y.S.; Loo, B.L.W.; Ariyaratne, P.; Yang, Y.; Ng, S.K.; Tan, T.R.M.; Yeo, H.C.; Lim, H.L.; et al. Mammalian systems biotechnology reveals global cellular adaptations in a recombinant CHO cell line. Cell Syst. 2017, 4, 530–542. [Google Scholar] [CrossRef] [Green Version]
- Gulston, M.K.; Rubtsov, D.V.; Atherton, H.J.; Clarke, K.; Davies, K.E.; Lilley, K.S.; Griffin, J.L. A combined metabolomic and proteomic investigation of the effects of a failure to express dystrophin in the mouse heart. J. Proteome Res. 2008, 7, 2069–2077. [Google Scholar] [CrossRef]
- Kaluarachchi, M.R.; Boulangé, C.L.; Garcia-Perez, I.; Lindon, J.C.; Minet, E.F. Multiplatform serum metabolic phenotyping combined with pathway mapping to identify biochemical differences in smokers. Bioanalysis 2016, 8, 2023–2043. [Google Scholar] [CrossRef] [Green Version]
- Toro-Domínguez, D.; Villatoro-García, J.A.; Martorell-Marugán, J.; Román-Montoya, Y.; Alarcón-Riquelme, M.E.; Carmona-Sáez, P. A survey of gene expression meta-analysis: Methods and applications. Briefings Bioinform. 2020, bbaa019. [Google Scholar] [CrossRef]
- Ritchie, M.D.; Holzinger, E.R.; Li, R.; Pendergrass, S.A.; Kim, D. Methods of integrating data to uncover genotype–phenotype interactions. Nat. Rev. Genet. 2015, 16, 85. [Google Scholar] [CrossRef] [PubMed]
- Ebbels, T.M.; Cavill, R. Bioinformatic methods in NMR-based metabolic profiling. Prog. Nucl. Magn. Reson. Spectrosc. 2009, 4, 361–374. [Google Scholar] [CrossRef]
- Riedl, A.; Gieger, C.; Hauner, H.; Daniel, H.; Linseisen, J. Metabotyping and its application in targeted nutrition: An overview. Br. J. Nutr. 2017, 117, 1631–1644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanckriet, G.; Deng, M.; Cristianini, N.; Jordan, M.; Noble, W. Kernel-based data fusion and its application to protein function prediction in yeast. In Biocomputing 2004, Proceedings of the Pacific Symposium, Waimea, HI, USA, 6–10 January 2004; World Scientific: Singapore, 2004; pp. 300–311. [Google Scholar]
- Davis, D.A.; Chawla, N.V. Exploring and exploiting disease interactions from multi-relational gene and phenotype networks. PLoS ONE 2011, 6, e22670. [Google Scholar] [CrossRef]
- Forshed, J.; Idborg, H.; Jacobsson, S.P. Evaluation of different techniques for data fusion of LC/MS and 1H-NMR. Chemom. Intell. Lab. Syst. 2007, 85, 102–109. [Google Scholar] [CrossRef]
- Žitnik, M.; Zupan, B. Data fusion by matrix factorization. IEEE Trans. Pattern Anal. Mach. Intell. 2015, 37, 41–53. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Baladandayuthapani, V.; Morris, J.S.; Broom, B.M.; Manyam, G.; Do, K.A. iBAG: Integrative Bayesian analysis of high-dimensional multiplatform genomics data. Bioinformatics 2012, 29, 149–159. [Google Scholar] [CrossRef]
- Kleemann, R.; Verschuren, L.; van Erk, M.J.; Nikolsky, Y.; Cnubben, N.H.; Verheij, E.R.; Smilde, A.K.; Hendriks, H.F.; Zadelaar, S.; Smith, G.J.; et al. Atherosclerosis and liver inflammation induced by increased dietary cholesterol intake: A combined transcriptomics and metabolomics analysis. Genome Biol. 2007, 8, R200. [Google Scholar] [CrossRef] [Green Version]
- Santos, E.M.; Ball, J.S.; Williams, T.D.; Wu, H.; Ortega, F.; Van Aerle, R.; Katsiadaki, I.; Falciani, F.; Viant, M.R.; Chipman, J.K.; et al. Identifying health impacts of exposure to copper using transcriptomics and metabolomics in a fish model. Environ. Sci. Technol. 2009, 44, 820–826. [Google Scholar] [CrossRef]
- Verhoeckx, K.C.; Bijlsma, S.; Jespersen, S.; Ramaker, R.; Verheij, E.R.; Witkamp, R.F.; van der Greef, J.; Rodenburg, R.J. Characterization of anti-inflammatory compounds using transcriptomics, proteomics, and metabolomics in combination with multivariate data analysis. Int. Immunopharmacol. 2004, 4, 1499–1514. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Stewart, D.A.; Sandhu, R.; Kirk, E.L.; Pathmasiri, W.W.; McRitchie, S.L.; Clark, R.F.; Troester, M.A.; Sumner, S.J. Correlated metabolomic, genomic, and histologic phenotypes in histologically normal breast tissue. PLoS ONE 2018, 13, e0193792. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Lin, C.C.; Spasojevic, I.; Iversen, E.S.; Chi, J.T.; Marks, J.R. A joint analysis of metabolomics and genetics of breast cancer. Breast Cancer Res. 2014, 16, 415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manikandan, P.; Ramyachitra, D.; Banupriya, D. Detection of overlapping protein complexes in gene expression, phenotype and pathways of Saccharomyces cerevisiae using Prorank based Fuzzy algorithm. Gene 2016, 580, 144–158. [Google Scholar] [CrossRef]
- Becker, E.; Robisson, B.; Chapple, C.E.; Guénoche, A.; Brun, C. Multifunctional proteins revealed by overlapping clustering in protein interaction network. Bioinformatics 2012, 28, 84–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, G.; Sze, S.H.; Thon, M.R. Identifying clusters of functionally related genes in genomes. Bioinformatics 2007, 23, 1053–1060. [Google Scholar] [CrossRef] [Green Version]
- Savage, R.S.; Ghahramani, Z.; Griffin, J.E.; De La Cruz, B.J.; Wild, D.L. Discovering transcriptional modules by Bayesian data integration. Bioinformatics 2010, 26, i158–i167. [Google Scholar] [CrossRef]
- Gromski, P.S.; Muhamadali, H.; Ellis, D.I.; Xu, Y.; Correa, E.; Turner, M.L.; Goodacre, R. A tutorial review: Metabolomics and partial least squares-discriminant analysis–a marriage of convenience or a shotgun wedding. Anal. Chim. Acta 2015, 879, 10–23. [Google Scholar] [CrossRef] [PubMed]
- Mendez, K.M.; Broadhurst, D.I.; Reinke, S.N. Migrating from partial least squares discriminant analysis to artificial neural networks: A comparison of functionally equivalent visualisation and feature contribution tools using jupyter notebooks. Metabolomics 2020, 16, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinu, F.R.; Beale, D.J.; Paten, A.M.; Kouremenos, K.; Swarup, S.; Schirra, H.J.; Wishart, D. Systems biology and multi-omics integration: Viewpoints from the metabolomics research community. Metabolites 2019, 9, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McIntyre, L.M.; Huertas, F.; Moskalenko, O.; Llansola, M.; Felipo, V.; Morse, A.M.; Conesa, A. GAIT-GM: Galaxy tools for modeling metabolite changes as a function of gene expression. bioRxiv 2020. [Google Scholar] [CrossRef]
- Koh, H.W.; Fermin, D.; Vogel, C.; Choi, K.P.; Ewing, R.M.; Choi, H. iOmicsPASS: Network-based integration of multiomics data for predictive subnetwork discovery. NPJ Syst. Biol. Appl. 2019, 5, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Zuo, Y.; Cui, Y.; Di Poto, C.; Varghese, R.S.; Yu, G.; Li, R.; Ressom, H.W. INDEED: Integrated differential expression and differential network analysis of omic data for biomarker discovery. Methods 2016, 111, 12–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harbig, T.A.; Fratte, J.; Krone, M.; Nieselt, K.K. OmicsTIDE: Interactive Exploration of Trends in Multi-Omics Data. bioRxiv 2021. [Google Scholar] [CrossRef]
- Baum, A.; Vermue, L. Multiblock PLS: Block dependent prediction modeling for Python. J. Open Source Softw. 2019, 4, 1190. [Google Scholar] [CrossRef]
- Xia, J.; Psychogios, N.; Young, N.; Wishart, D.S. MetaboAnalyst: A web server for metabolomic data analysis and interpretation. Nucleic Acids Res. 2009, 37, W652–W660. [Google Scholar] [CrossRef] [Green Version]
- Hinshaw, S.J.; HY Lee, A.; Gill, E.E.; Hancock, R.E.W. MetaBridge: Enabling network-based integrative analysis via direct protein interactors of metabolites. Bioinformatics 2018, 34, 3225–3227. [Google Scholar] [CrossRef]
- Cottret, L.; Wildridge, D.; Vinson, F.; Barrett, M.P.; Charles, H.; Sagot, M.F.; Jourdan, F. MetExplore: A web server to link metabolomic experiments and genome-scale metabolic networks. Nucleic Acids Res. 2010, 38, W132–W137. [Google Scholar] [CrossRef]
- Rohart, F.; Gautier, B.; Singh, A.; Lê Cao, K.A. mixOmics: An R package for ‘omics feature selection and multiple data integration. PLoS Comput. Biol. 2017, 13, e1005752. [Google Scholar] [CrossRef] [Green Version]
- Tal, O.; Selvaraj, G.; Medina, S.; Ofaim, S.; Freilich, S. NetMet: A Network-Based Tool for Predicting Metabolic Capacities of Microbial Species and their Interactions. Microorganisms 2020, 8, 840. [Google Scholar] [CrossRef] [PubMed]
- García-Alcalde, F.; García-López, F.; Dopazo, J.; Conesa, A. Paintomics: A web based tool for the joint visualization of transcriptomics and metabolomics data. Bioinformatics 2011, 27, 137–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández-de Diego, R.; Tarazona, S.; Martínez-Mira, C.; Balzano-Nogueira, L.; Furió-Tarí, P.; Pappas, G.J., Jr.; Conesa, A. PaintOmics 3: A web resource for the pathway analysis and visualization of multi-omics data. Nucleic Acids Res. 2018, 46, W503–W509. [Google Scholar] [CrossRef] [Green Version]
- Liland, K.H.; Næs, T.; Indahl, U.G. ROSA—A fast extension of partial least squares regression for multiblock data analysis. J. Chemom. 2016, 30, 651–662. [Google Scholar] [CrossRef]
- Joyce, A.R.; Palsson, B.∅. The model organism as a system: Integrating’omics’ data sets. Nat. Rev. Mol. Cell Biol. 2006, 7, 198–210. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Integrative Analysis | Description | Examples |
|---|---|---|
| study design | ||
| Repeated study | In a repeated study the experiment is repeated in another time or place to generate a second type of data. | Cavill et al. [38] |
| Replicate matched study | In a replicate matched study, biological replicates are used to generate additional types of data. | Cavill et al. [38] |
| Split sample study | In a split sample study, the same biological sample is split for profiling with different omics technologies. | Cavill et al. [38] |
| Source matched study | In a source matched study, different samples from the same biological organism are extracted and used to generate different types of data. | Cavill et al. [38] |
| data types | ||
| Horizontal or homogeneous data integration (meta-analysis) | Horizontal integration involves combining measurements of the same omics entities across various cohorts, labs or studies. | Richardson et al. [32], Yuan et al. [39] |
| Vertical or heterogeneous data integration | Vertical integration involves combining entities from different omics levels, often measured using different platforms. | Richardson et al. [32], Evangelou and Ioannidis [40] |
| hypothesis | ||
| Multi-staged | In multi-staged integration, inter-omics variation (variation between omics) is assumed to be unidirectional from the genome to the metabolome | Nicholson et al. [15], Gieger et al. [22], Krumsiek et al. [41] |
| Meta-dimensional | In meta-dimensional integration, inter-omics variation is assumed to be multi-directional or simultaneous. | Smolinska et al. [42], Witten and Tibshirani [43], Daemen et al. [44] |
| strategy | ||
| Early integration | Early integration combines two datasets by simply concatenating them into one data. | Fridley et al. [45], Le Van et al. [46] |
| Intermediate integration | Intermediate integration involves a data transformation step to be performed prior to modeling. | Le et al. [31], Smolinska et al. [42], Lanckriet et al. [47], Guo et al. [48] |
| Late integration | Late integration consists of combining single data models into a high level model. | Acharjee et al. [49] |
| study focus | Depending on the study focus, integrative analysis seeks to answer the following: | |
| Sequential analysis | Does the additional data type enhance understanding of the first data type? | Yuan et al. [39], Le Van et al. [46], Shen et al. [50] |
| Biological analysis | What are the underlying processes leading to phenotypical changes? Which mechanisms explain the prevalence of a phenotype? | Hirai et al. [51], Cavill et al. [52], Safo et al. [53], Hong et al. [54] |
| Model-based analysis | Which variables are phenotypically relevant? significantly associated? Can predictive ability be improved? | Smolinska et al. [42], Witten and Tibshirani [43], Daemen et al. [44] |
| Workflow | Considerations | Choices and Comments |
|---|---|---|
| Example from Le et al. [31] | ||
| Study focus | Research questions | Is it possible to predict metabolite abundance from bacteria abundance in inflammatory bowel disease (IBD)? Can we learn the synergistic relationship between the gut microbiome and their surrounding metabolites? These questions suggest an interest in complex associations between the metabolome and the microbiome which will be investigated through model-based analysis. The choice of a model-based analysis highly affects the integrative strategy while requiring it to comply with the hypothesis. |
| Hypothesis | As suggested by the research question, the authors assume that there exists intermediate factors that act in the middle of the process that transforms microbes to metabolites and that the processes in which microbes affect metabolites are highly interdependent following a multi-staged integrative approach. | |
| Study design, sample collection and data acquisition | Study type | Paired data from a cohort of inflammatory bowel disease patients. |
| Omics layers | Microbiome and metabolome | |
| Biological samples | Fecal samples | |
| Platforms | Next-Generation Sequencing (NGS) and LC-MS | |
| Preprocessing | In addition to the standard pre-processing workflow applied to each platform, the authors used compositional methods e.g., centered log-ratio transformation, to ensure that their workflow will generalize to any pair of omics data. | |
| Data types | Vertical data integration on paired data with heterogeneous features: microbe abundance and metabolite abundance. | |
| Data analysis | Strategies | Intermediate integration via neural encoder-decoder networks. Non negative weights are imposed on the networks to enforce a unidirectional variation from the microbiome to the metabolome. |
| Data interpretation | Hypothesis | Microbe abundance is able to reliably predict abundance of a range of metabolites while empowering clinically relevant relationships. The findings also suggest that the “microbe-metabolite axis itself, not just the microbes and metabolites alone, is an IBD-specific biomarker signature.” |
| Example from Nicholson et al. [15] | ||
| Study focus | Research question | Are there 1H NMR-detectable metabolites in urine or plasma that are strongly influenced by common single-locus genetic variation? This question involves, but not restricted to, a model-based integrative analysis and will guide the study design, data analysis and data interpretation. |
| Hypothesis | Variation is unidirectional downstream from genes to metabolites. | |
| Study design, sample collection and data acquisition | Study type | Cohort study |
| Omics layers | Genome and metabolome | |
| Biological samples | Whole-blood, plasma and urine | |
| Platforms | Untargeted 1H NMR and targeted flow-injection tandem MS: The sets of metabolites observed from the two platforms were minimally overlapping and therefore complementary. The genotyping assay used Illumina arrays. | |
| Longitudinal profiling | Measurements of heterogeneous omics entities were recorded at the same time point. The longitudinal design allowed detailed variance-components analysis of the sources of population variation in metabolite levels. | |
| Preprocessing | Preprocessing including metabolite annotation was performed using standard pipelines for each platform. | |
| Data types | The authors considered two cohorts from the MolPAGE study with the aim of using one cohort to replicate findings of the other one (Sequential integration). Vertical data integration has been performed on Genome-wide SNP genotypes and metabolic features. | |
| Data analysis | Strategies | Early integration through Genome-Wide Metabolic QTL Analysis to identify associations. |
| Data interpretation | Hypothesis | The mQTLs explained a significant biological population variation in the corresponding metabolites’ concentrations which is well aligned with the hypothesis of a multi-staged integrative analysis. This is also coherent with the research question (study focus) and strategy adopted. |
| Resource | Core Integrative Analysis Tasks | Interface | Study Focus | Reference |
|---|---|---|---|---|
| GAIT-GM | Annotation, network modeling and pathway analysis | Python | Sequential analysis & Biological-based integration | McIntyre et al. [97] |
| iOmicsPASS | Network-based analysis and predictive feature selection | C++ | Model-based integration & Biological-based integration | Koh et al. [98] |
| INDEED | Network analysis | R | Model-based integration | Zuo et al. [99] |
| OmicsTIDE | Clustering and visualisation | online | Model-based integration & Sequential integration | Harbig et al. [100] |
| mbpls | Dimension reduction (Multi-block PLS) | Python | Model-based analysis & Sequential integration | Baum and Vermue [101] |
| MetaboAnalyst | Enrichment analysis | online, R | Biological-based integration | Xia et al. [102] |
| MetaBridge | Pathway mapping | online | Biological-based integration | Hinshaw et al. [103] |
| MetExplore | Pathway mapping and graph-based analysis | online | Biological-based integration | Cottret et al. [104] |
| mixOmics | Dimension reduction and feature selection | R | Model-based integration | Rohart et al. [105] |
| multiGSEA | Enrichment analysis | R | Biological-based integration | Canzler et al. [63] |
| NetMet | Network modeling | online | Biological-based integration | Tal et al. [106] |
| paintOmics 3 | Pathway visualisation | online | Biological-based integration | García-Alcalde et al. [107], Hernández-de Diego et al. [108] |
| ROSA | Dimension reduction (Multi-block PLS) | R | Model-based analysis & Sequential integration | Liland et al. [109] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jendoubi, T. Approaches to Integrating Metabolomics and Multi-Omics Data: A Primer. Metabolites 2021, 11, 184. https://doi.org/10.3390/metabo11030184
Jendoubi T. Approaches to Integrating Metabolomics and Multi-Omics Data: A Primer. Metabolites. 2021; 11(3):184. https://doi.org/10.3390/metabo11030184
Chicago/Turabian StyleJendoubi, Takoua. 2021. "Approaches to Integrating Metabolomics and Multi-Omics Data: A Primer" Metabolites 11, no. 3: 184. https://doi.org/10.3390/metabo11030184
APA StyleJendoubi, T. (2021). Approaches to Integrating Metabolomics and Multi-Omics Data: A Primer. Metabolites, 11(3), 184. https://doi.org/10.3390/metabo11030184