
NMR Methods for Determining Lipid Turnover via Stable Isotope Resolved Metabolomics

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Results
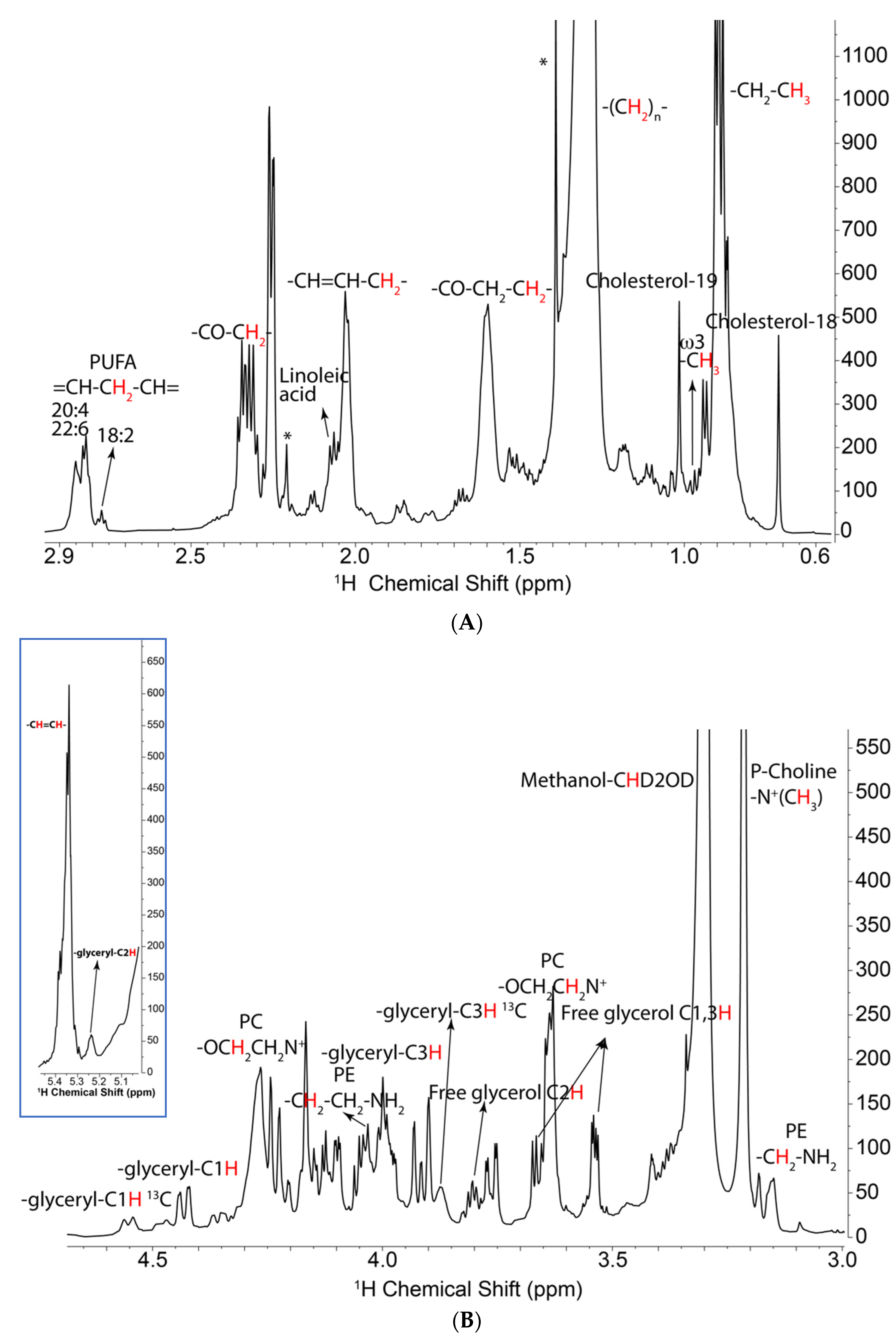
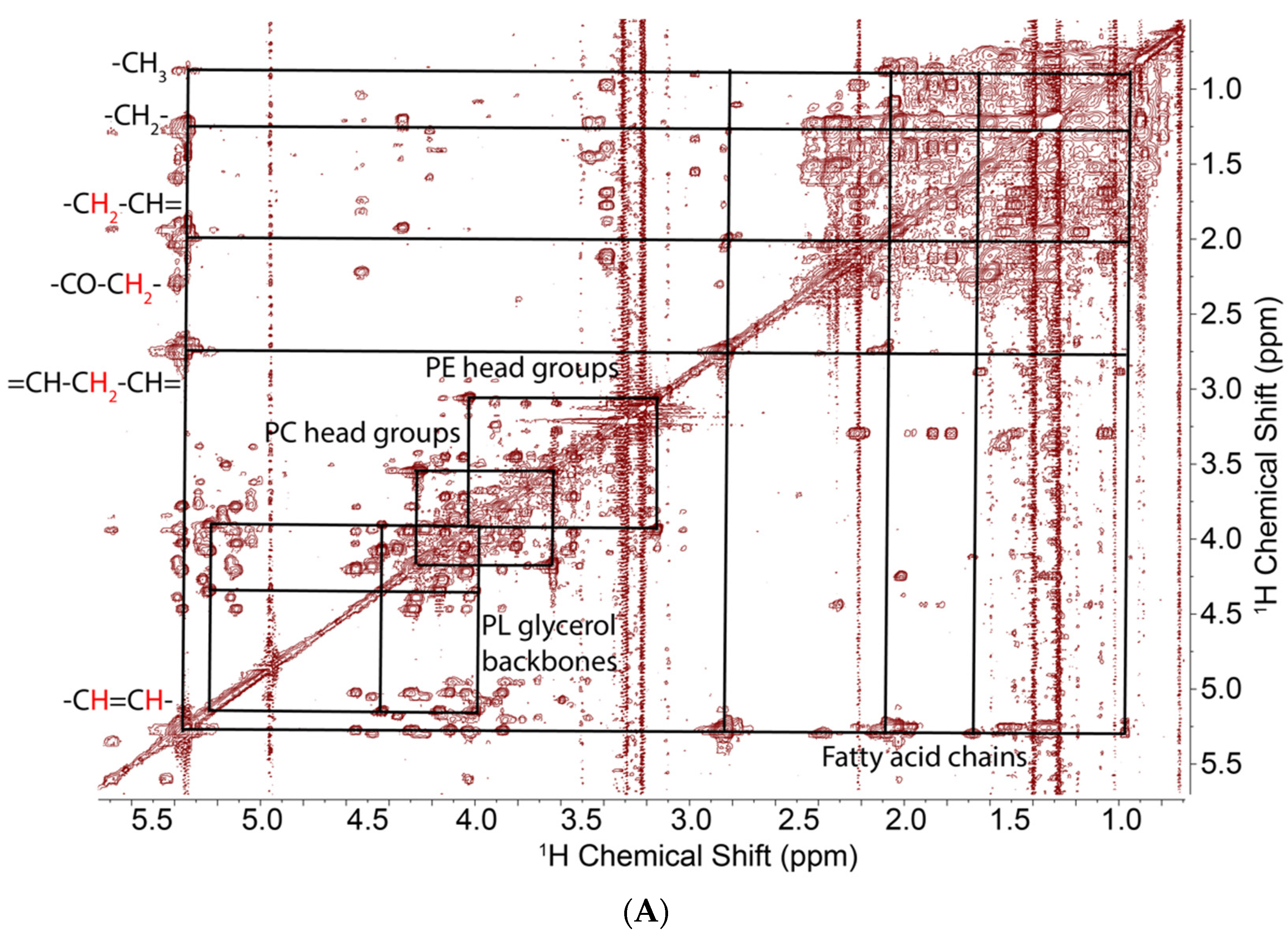
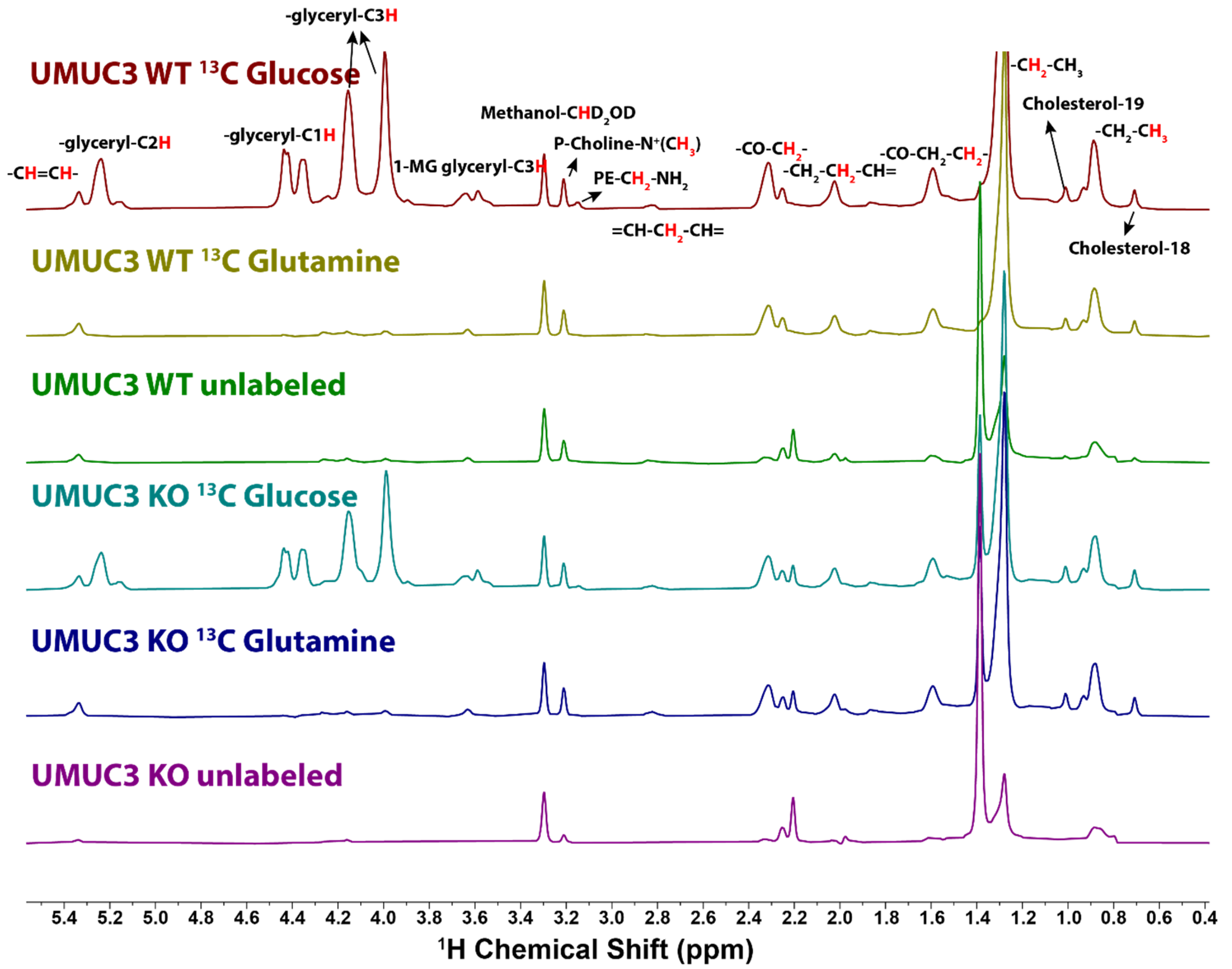
2.1. NMR Analysis
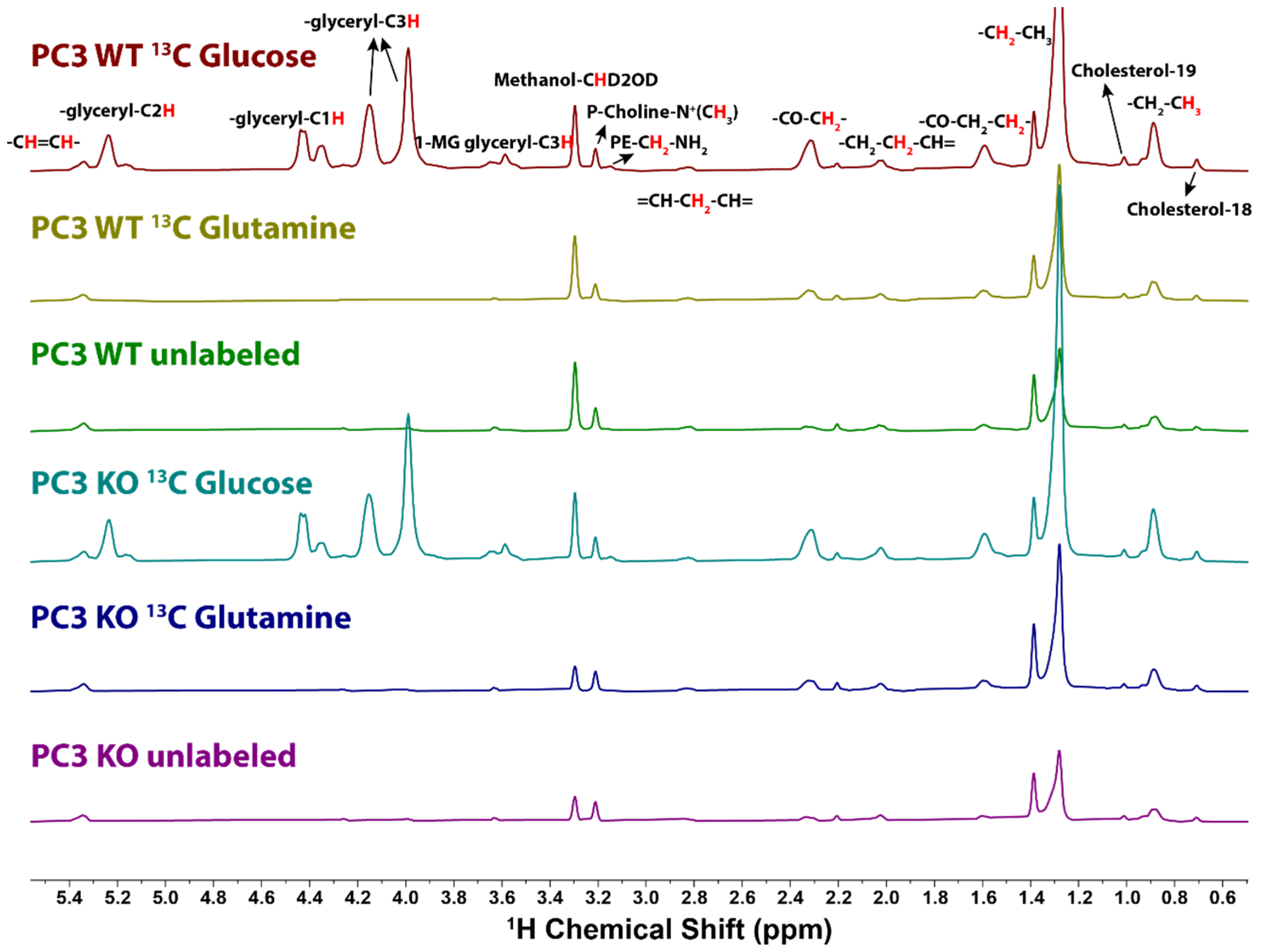
2.2. 13C Incorporation from Glucose and Glutamine by 1H NMR
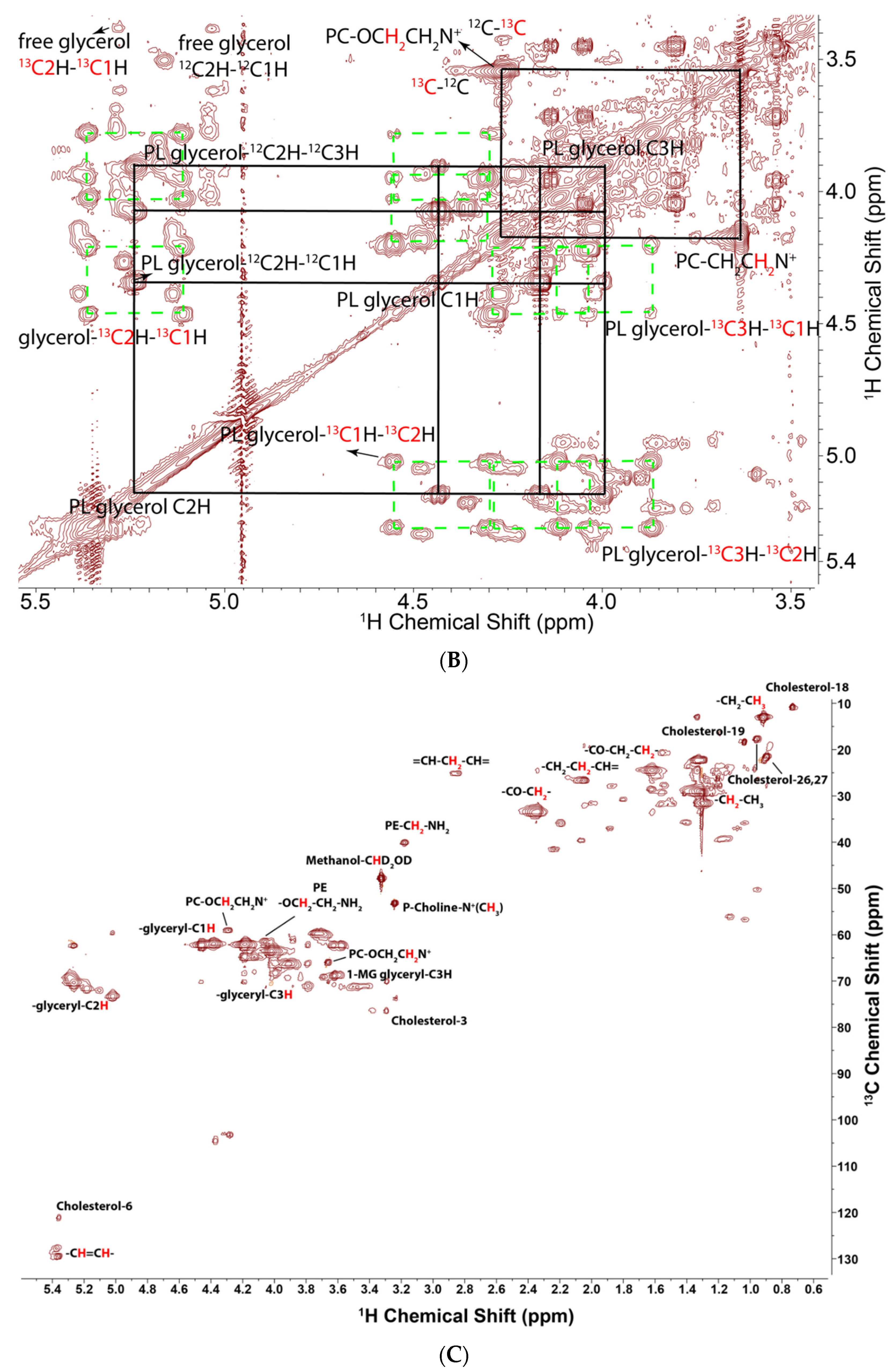
2.3. Determination of 13C Incorporation from Glucose and Glutamine by 1H{13C}-HSQC
2.4. Estimation of Lipid Distributions from NMR
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fahy, E.; Subramaniam, S.; Brown, H.A.; Glass, C.K.; Merrill, A.H., Jr.; Murphy, R.C.; Raetz, C.R.H.; Russell, D.W.; Seyama, Y.; Shaw, W.; et al. A comprehensive classification system for lipids. J. Lipid Res. 2005, 46, 839–861. [Google Scholar] [CrossRef] [Green Version]
- Fahy, E.; Subramaniam, S.; Murphy, R.C.; Nishijima, M.; Raetz, C.R.H.; Shimizu, T.; Spener, F.; van Meer, G.; Wakelam, M.J.O.; Dennis, E.A. Update of the LIPID MAPS comprehensive classification system for lipids. J. Lipid Res. 2009, 50, S9–S14. [Google Scholar] [CrossRef] [Green Version]
- Fan, T.W.M.; Zhang, X.; Wang, C.; Yang, Y.; Kang, W.-Y.; Arnold, S.; Higashi, R.M.; Liu, J.; Lane, A.N. Exosomal lipids for classifying early and late stage non-small cell lung cancer. Anal. Chim. Acta 2018, 1037, 256–264. [Google Scholar] [CrossRef]
- Sud, M.; Fahy, E.; Cotter, D.; Brown, A.; Dennis, E.A.; Glass, C.K.; Merrill, A.H., Jr.; Murphy, R.C.; Raetz, C.R.H.; Russell, D.W.; et al. LMSD: LIPID MAPS structure database. Nucleic Acids Res. 2007, 35, D527–D532. [Google Scholar] [CrossRef] [Green Version]
- German, J.B. Dietary lipids from an evolutionary perspective:sources, structures and functions. Matern. Child Nutr. 2011, 7, 2–16. [Google Scholar] [CrossRef]
- Han, X.L.; Gross, R.W. Shotgun lipidomics: Multidimensional MS analysis of cellular lipidomes. Expert Rev. Proteom. 2005, 2, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Quehenberger, O.; Dennis, E.A. The Human Plasma Lipidome. N. Engl. J. Med. 2011, 365, 1812–1823. [Google Scholar] [CrossRef]
- Torkhovskaya, T.I.; Zakharova, T.S.; Korotkevich, E.I.; Ipatova, O.M.; Markin, S.S. Human Blood Plasma Lipidome: Opportunities and Prospects of Its Analysis in Medical Chemistry. Russ. J. Bioorg. Chem. 2019, 45, 335–346. [Google Scholar] [CrossRef]
- Wang, C.; Timári, I.; Zhang, B.; Li, D.-W.; Leggett, A.; Amer, A.O.; Bruschweiler-Li, L.; Kopec, R.E.; Brüschweiler, R. COLMAR Lipids Web Server and Ultrahigh-Resolution Methods for Two-Dimensional Nuclear Magnetic Resonance- and Mass Spectrometry-Based Lipidomics. J. Proteome Res. 2020, 19, 1674–1683. [Google Scholar] [CrossRef]
- Alexandri, E.; Ahmed, R.; Siddiqui, H.; Choudhary, M.I.; Tsiafoulis, C.G.; Gerothanassis, I.P. High Resolution NMR Spectroscopy as a Structural and Analytical Tool for Unsaturated Lipids in Solution. Molecules 2017, 22, 1663. [Google Scholar] [CrossRef] [PubMed]
- Neumann, E.K.; Comi, T.J.; Rubakhin, S.S.; Sweedler, J.V. Lipid Heterogeneity between Astrocytes and Neurons Revealed by Single-Cell MALDI-MS Combined with Immunocytochemical Classification. Angew. Chem. Int. Ed. 2019, 58, 5910–5914. [Google Scholar] [CrossRef]
- Liebisch, G.; Vizcaíno, J.A.; Köfeler, H.; Trötzmüller, M.; Griffiths, W.J.; Schmitz, G.; Spener, F.; Wakelam, M.J.O. Shorthand notation for lipid structures derived from mass spectrometry. J. Lipid Res. 2013, 54, 1523–1530. [Google Scholar] [CrossRef] [Green Version]
- Triebl, A.; Wenk, M.R. Analytical Considerations of Stable Isotope Labelling in Lipidomics. Biomolecules 2018, 8, 151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ecker, J.; Liebisch, G. Application of stable isotopes to investigate the metabolism of fatty acids, glycerophospholipid and sphingolipid species. Prog. Lipid Res. 2014, 54, 14–31. [Google Scholar] [CrossRef] [PubMed]
- Stuani, L.; Riols, F.; Millard, P.; Sabatier, M.; Batut, A.; Saland, E.; Viars, F.; Tonini, L.; Zaghdoudi, S.; Linares, L.K.; et al. Stable Isotope Labeling Highlights Enhanced Fatty Acid and Lipid Metabolism in Human Acute Myeloid Leukemia. Int. J. Mol. Sci. 2018, 19, 3325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umpleby, A.M. HORMONE MEASUREMENT GUIDELINES Tracing lipid metabolism: The value of stable isotopes. J. Endocrinol. 2015, 226, G1–G10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parks, E.J.; Hellerstein, M.K. Recent advances in liver triacylglycerol and fatty acid metabolism using stable isotope labeling techniques. J. Lipid Res. 2006, 47, 1651–1660. [Google Scholar] [CrossRef] [Green Version]
- Hellerstein, M.K. New stable isotope-mass spectrometric techniques for measuring fluxes through intact metabolic pathways in mammalian systems: Introduction of moving pictures into functional genomics and biochemical phenotyping. Metab. Eng. 2004, 6, 85–100. [Google Scholar] [CrossRef]
- Turner, S.M.; Murphy, E.J.; Neese, R.A.; Antelo, F.; Thomas, T.; Agarwal, A.; Go, C.; Hellerstein, M.K. Measurement of TG synthesis and turnover in vivo by (2HO)-O-2 incorporation into the glycerol moiety and application of MIDA. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E790–E803. [Google Scholar] [CrossRef] [Green Version]
- Lane, A.N.; Fan, T.W.M.; Xie, Z.; Moseley, H.N.B.; Higashi, R.M. Isotopomer analysis of lipid biosynthesis by high resolution mass spectrometry and NMR. Anal. Chim. Acta 2009, 651, 201–208. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Boroughs, L.K.; DeBerardinis, R.J. Metabolic pathways promoting cancer cell survival and growth. Nat. Cell Biol. 2015, 17, 351–359. [Google Scholar] [CrossRef] [Green Version]
- Lane, A.N.; Higashi, R.M.; Fan, T.W.M. Metabolic reprogramming in tumors: Contributions of the tumor microenvironment. Genes Dis. 2020, 7, 185–198. [Google Scholar] [CrossRef]
- Dang, C.V.; Hamaker, M.; Sun, P.; Le, A.; Gao, P. Therapeutic targeting of cancer cell metabolism. J. Mol. Med. JMM 2011, 89, 205–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Cañaveras, J.C.; Chen, L.; Rabinowitz, J.D. The Tumor Metabolic Microenvironment: Lessons from Lactate. Cancer Res. 2019, 79, 3155–3162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuneva, M.O.; Fan, T.W.-M.; Allen, T.D.; Higashi, R.M.; Ferraris, D.V.; Tsukamoto, T.; Matés, J.M.; Alonso, F.J.; Wang, C.; Seo, Y.; et al. The Metabolic Profile of Tumors Depends on Both the Responsible Genetic Lesion and Tissue Type. Cell Metab. 2012, 15, 157–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, C.; Thompson, C.B. Metabolic Regulation of Epigenetics. Cell Metab. 2012, 16, 9–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, T.W.M.; Lorkiewicz, P.K.; Sellers, K.; Moseley, H.N.B.; Higashi, R.M.; Lane, A.N. Stable isotope-resolved metabolomics and applications for drug development. Pharmacol. Ther. 2012, 133, 366–391. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Lane, A.N.; Ricketts, C.J.; Carole Sourbier, C.; Wei, M.-H.; Shuch, B.; Pike, L.; Wu, M.; Rouault, T.A.; Boros, L.G.; et al. Metabolic Reprogramming for Producing Energy and Reducing Power in Fumarate Hydratase Null Cells from Hereditary Leiomyomatosis Renal Cell Carcinoma. PLoS ONE 2013, 8, e72179. [Google Scholar] [CrossRef] [PubMed]
- Deng, P.; Higashi, R.M.; Lane, A.N.; Bruntz, R.C.; Sun, R.C.; Ramakrishnam Raju, M.V.; Nantz, M.H.; Qi, Z.; Fan, T.W. Quantitative profiling of carbonyl metabolites directly in crude biological extracts using chemoselective tagging and nanoESI-FTMS. Analyst 2017, 143, 311–322. [Google Scholar] [CrossRef]
- Schlame, M.; Xu, Y.; Erdjument-Bromage, H.; Neubert, T.A.; Ren, M. Lipidome-wide 13 C flux analysis: A novel tool to estimate the turnover of lipids in organisms and cultures. J. Lipid Res. 2020, 61, 95–104. [Google Scholar] [CrossRef]
- Kang, W.Y.; Thompson, P.T.; El-Amouri, S.S.; Fan, T.W.M.; Lane, A.N.; Higashi, R.M. Improved segmented-scan spectral stitching for stable isotope resolved metabolomics (SIRM) by ultra-high-resolution Fourier transform mass spectrometry. Anal. Chim Acta 2019, 1080, 104–115. [Google Scholar] [CrossRef]
- Di Buono, M.; Jones, P.J.; Beaumier, L.; Wykes, L.J. Comparison of deuterium incorporation and mass isotopomer distribution analysis for measurement of human cholesterol biosynthesis. J. Lipid Res. 2000, 41, 1516–1523. [Google Scholar] [CrossRef]
- Crooks, D.R.; Maio, N.; Lane, A.N.; Jarnik, M.; Higashi, R.M.; Haller, R.G.; Yang, Y.; Fan, T.W.; Linehan, W.M.; Rouault, T.A. Acute loss of iron-sulfur clusters results in metabolic reprogramming and generation of lipid droplets in mammalian cells. J. Biol. Chem. 2018, 293, 8297–8311. [Google Scholar] [CrossRef] [Green Version]
- Lane, A.N.; Tan, J.; Wang, Y.; Yan, J.; Higashi, R.M.; Fan, T.W.-M. Probing the metabolic phenotype of breast cancer cells by multiple tracer Stable Isotope Resolved Metabolomics. Metab. Eng. 2017, 43, 125–136. [Google Scholar] [CrossRef] [Green Version]
- Freriksen, A.; Seykens, D.; Scharloo, W.; Heinstra, P.W. Alcohol dehydrogenase controls the flux from ethanol into lipids in Drosophila larvae. A 13C NMR study. J. Biol. Chem. 1991, 266, 21399–21403. [Google Scholar] [PubMed]
- Guo, W.; Choi, J.K.; Kirkland, J.L.; Corkey, B.E.; Hamilton, J.A. Esterification of free fatty acids in adipocytes: A comparison between octanoate and oleate. Biochem. J. 2000, 349, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Bederman, I.R.; Foy, S.; Chandramouli, V.; Alexander, J.C.; Previs, S.F. Triglyceride synthesis in epididymal adipose tissue: Contribution of glucose and non-glucose carbon sources. J. Biol. Chem. 2009, 284, 6101–6108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duarte, J.A.; Carvalho, F.; Pearson, M.; Horton, J.D.; Browning, J.D.; Jones, J.G.; Burgess, S.C. A high-fat diet suppresses de novo lipogenesis and desaturation but not elongation and triglyceride synthesis in mice. J. Lipid Res. 2014, 55, 2541–2553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, J.C.P.; Marques, C.; Martins, F.O.; Viegas, I.; Tavares, L.; Macedo, M.P.; Jones, J.G. Determining contributions of exogenous glucose and fructose to de novo fatty acid and glycerol synthesis in liver and adipose tissue. Metab. Eng. 2019, 56, 69–76. [Google Scholar] [CrossRef]
- Aguayo, J.B.; Gamcsik, M.P.; Dick, J.D. High Resolution Deuterium NMR Studies of Bacterial Metabolism. J. Biol. Chem. 1987, 263, 19552–19557. [Google Scholar] [CrossRef]
- Lisanti, S.; Tavecchio, M.; Chae, Y.C.; Liu, Q.; Brice, A.K.; Thakur, M.L.; Languino, L.R.; Altieri, D.C. Deletion of the mitochondrial chaperone TRAP-1 uncovers global reprogramming of metabolic networks. Cell Rep. 2014, 8, 671–677. [Google Scholar] [CrossRef]
- Zhang, H.; Ding, L.; Fang, X.; Shi, Z.; Zhang, Y.; Chen, H.; Yan, X.; Dai, J. Biological responses to perfluorododecanoic acid exposure in rat kidneys as determined by integrated proteomic and metabonomic studies. PLoS ONE 2011, 6, e20862. [Google Scholar] [CrossRef] [Green Version]
- Fan, T.W.M.; Lane, A.N. Assignment Strategies for Nuclear Magnetic Resonances in Metabolomic Research. In Methodologies for Metabolomics: Experimental Strategies and Techniques; Sweedler, J.V., Lutz, N.W., Wevers, R.A., Eds.; Cambridge University Press: Cambridge, UK, 2013; pp. 525–584. [Google Scholar] [CrossRef]
- National Lung Screening Trial Research Team. Reduced Lung-Cancer Mortality with Low-Dose Computed Tomographic Screening. N. Engl. J. Med. 2011, 365, 395–409. [Google Scholar] [CrossRef] [Green Version]
- Yoo, H.; Antoniewicz, M.R.; Stephanopoulos, G.; Kelleher, J.K. Quantifying reductive carboxylation flux of glutamine to lipid in a brown adipocyte cell line. J. Biol. Chem. 2008, 283, 20621–20627. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Shestov, A.A.; Swain, P.; Yang, C.; Parker, S.J.; Wang, Q.A.; Terada, L.S.; Adams, N.D.; McCabe, M.T.; Pietrak, B.; et al. Reductive carboxylation supports redox homeostasis during anchorage-independent growth. Nature 2016, 532, 255–258. [Google Scholar] [CrossRef]
- Grassian, A.R.; Parker, S.J.; Davidson, S.M.; Divakaruni, A.S.; Green, C.R.; Zhang, X.M.; Slocum, K.L.; Pu, M.Y.; Lin, F.; Vickers, C.; et al. IDH1 Mutations Alter Citric Acid Cycle Metabolism and Increase Dependence on Oxidative Mitochondrial Metabolism. Cancer Res. 2014, 74, 3317–3331. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.; Kamphorst, J.J.; Rabinowitz, J.D.; Shlomi, T. Fatty Acid Labeling from Glutamine in Hypoxia Can Be Explained by Isotope Exchange without Net Reductive Isocitrate Dehydrogenase (IDH) Flux. J. Biol. Chem. 2013, 288, 31363–31369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieva-Echevarría, B.; Goicoechea, E.; Manzanos, M.J.; Guillén, M.D. A method based on 1H NMR spectral data useful to evaluate the hydrolysis level in complex lipid mixtures. Food Res. Int. 2014, 66, 379–387. [Google Scholar] [CrossRef] [Green Version]
- Nieva-Echevarria, B.; Goicoechea, E.; Manzanos, M.J.; Guillen, M.D. Usefulness of (1)H NMR in assessing the extent of lipid digestion. Food Chem 2015, 179, 182–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Veen, J.N.; Kennelly, J.P.; Wan, S.; Vance, J.E.; Vance, D.E.; Jacobs, R.L. The critical role of phosphatidylcholine and phosphatidylethanolamine metabolism in health and disease. Biochim. Biophys. Acta (BBA) Biomembr. 2017, 1859, 1558–1572. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.; Parton, R.G. Lipid droplets: A unified view of a dynamic organelle. Nat. Rev. Mol. Cell Biol. 2006, 7, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Szlasa, W.; Zendran, I.; Zalesińska, A.; Tarek, M.; Kulbacka, J. Lipid composition of the cancer cell membrane. J. Bioenerg. Biomembr. 2020, 20, 321–342. [Google Scholar] [CrossRef] [PubMed]
- Mullen, A.R.; Wheaton, W.W.; Jin, E.S.; Chen, P.-H.; Sullivan, L.B.; Cheng, T.; Yang, Y.; Linehan, W.M.; Chandel, N.S.; DeBerardinis, R.J. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 2011, 481, 385–388. [Google Scholar] [CrossRef] [Green Version]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2011, 481, 380–384. [Google Scholar] [CrossRef] [Green Version]
- Michalopoulou, E.; Bulusu, V.; Kamphorst, J.J. Metabolic scavenging by cancer cells: When the going gets tough, the tough keep eating. Br. J. Cancer 2016, 115, 635–640. [Google Scholar] [CrossRef]
- Kamphorst, J.J.; Cross, J.R.; Fan, J.; de Stanchina, E.; Mathew, R.; White, E.P.; Thompson, C.B.; Rabinowitz, J.D. Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc. Natl. Acad. Sci. USA 2013, 110, 8882–8887. [Google Scholar] [CrossRef] [Green Version]
- Kuksis, A.; Roberts, A.; Thompson, J.S.; Myher, J.J.; Geher, K. Plasma phosphatidylcholine/free cholesterol ratio as an indicator for atherosclerosis. Arteriosclerosis 1983, 3, 389–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Agellon, L.B.; Allen, T.M.; Umeda, M.; Jewell, L.; Mason, A.; Vance, D.E. The ratio of phosphatidylcholine to phosphatidylethanolamine influences membrane integrity and steatohepatitis. Cell Metab. 2006, 3, 321–331. [Google Scholar] [CrossRef] [Green Version]
- Crooks, D.R.; Fan, T.W.-M.; Linehan, W.M. Metabolic Labeling of Cultured Mammalian Cells for Stable Isotope-Resolved Metabolomics: Practical Aspects of Tissue Culture and Sample Extraction. Methods Mol. Biol. 2019, 1928, 1–27. [Google Scholar] [PubMed]
- Sun, R.C.; Fan, T.W.M.; Deng, P.; Higashi, R.M.; Lane, A.N.; Le, A.-T.; Scott, T.L.; Sun, Q.; Warmoes, M.O.; Yang, Y. Noninvasive liquid diet delivery of stable isotopes into mouse models for deep metabolic network tracing. Nat. Commun. 2017, 8, 1646. [Google Scholar] [CrossRef] [PubMed]
- Lane, A.N.; Fan, T.W. Quantification and identification of isotopomer distributions of metabolites in crude cell extracts using 1H TOCSY. Metabolomics 2007, 3, 79–86. [Google Scholar] [CrossRef]
- Sellers, K.; Fox, M.P.; Bousamra, M.; Slone, S.; Higashi, R.M.; Miller, D.M.; Wang, Y.; Yan, J.; Yuneva, M.O.; Deshpande, R.; et al. Pyruvate carboxylase is critical for non-small-cell lung cancer proliferation. J. Clin. Investig. 2015, 125, 687–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, T.W.-M.; Lane, A.N.; Higashi, R.M.; Yan, J. Stable Isotope Resolved Metabolomics of Lung Cancer in a SCID Mouse Model. Metabolomics 2011, 7, 257–269. [Google Scholar] [CrossRef] [Green Version]
- Fan, T.W.M.; Bruntz, R.C.; Yang, Y.; Song, H.; Chernyavskaya, Y.; Deng, P.; Zhang, Y.; Shah, P.P.; Beverly, L.J.; Qi, Z.; et al. De novo synthesis of serine and glycine fuels purine nucleotide biosynthesis in human lung cancer tissues. J. Biol. Chem. 2019, 294, 13464–13477. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | UMUC 3 | PC3 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Glc | Gln | Unlabeled | Glc | Gln | Unlabeled | |||||||
| WT | KO | WT | KO | WT | KO | WT | KO | WT | KO | WT | KO | |
| Acyl CH2 | 2.7 ± 0.43 | 3.0 ± 0.60 | 2.8 ± 0.37 | 3.9 ± 0.39 | 1.73 | 1.28 | 3.8 ± 0.24 | 5.4 ± 0.29 | 1.8 ± 0.20 | 1.45 ± 0.07 | 1.0 | 0.86 |
|
Methyl– CH2–CH3 | 3.1 ± 0.15 | 2.45 ± 0.16 | 2.7 ± 0.33 | 2.3 ± 0.29 | 1.81 | 2.18 | 2.6 ± 0.07 | 3.1 ± 0.07 | 1.5 ± 0.01 | 1.5 ± 0.11 | 0.94 | 1.08 |
|
Acyl CH2–CO | 5.2 ±0.26 | 4.9 ± 0.42 | 3.9 ± 0.21 | 3.7 ± 0.18 | 0.81 | 1.34 | 7.4 ±0.28 | 8.5 ± 0.52 | 3.2 ± 0.03 | 3.1 ±0.07 | 0.94 | 1.08 |
|
Acyl CO–CH2–CH2 | 3.1 ± 0.19 | 2.5 ± 0.05 | 2.3 ± 0.09 | 2.5 ± 0.33 | 1.24 | 0.39 | 3.9 ± 0.14 | 5.5 ± 0.22 | 2.0 ± 0.06 | 2.1 ± 0.20 | 0.95 | 0.91 |
| Glyceryl–C1H | 59.3 ± 0.90 | 52.8 ± 1.28 | nd a | nd | nd | nd | 62.3 ± 1.17 | 65.2 ± 0.88 | nd | nd | 0.80 | nd |
| Glyceryl–C2H | 41. 9 ± 0.55 | 33.4 ± 0.41 | nd | nd | nd | nd | 48.9 ± 1.70 | 53.0 ± 2.93 | nd | nd | 1.45 | nd |
| Glyceryl–C3H | 61.5 ± 6.81 | 50.4 ± 3.22 | 1.84 | 2.17 | 1.84 | 1.1 | 67.1 ± 0.41 | 72.8 ± 1.63 | 2 ± 0.08 | 2.1 ± 0.41 | 1.76 | 1.87 |
|
Acyl =CH– | 1.2 ± 0.11 | 1.3 ± 0.03 | 1.1 ± 0.07 | 0.91 ± 0.07 | 0.88 | 1.04 | 0.90 ± 0.08 | 0.99 ± 0.06 | 0.88 ± 0.04 | 0.97 ± 0.03 | 0.83 | 0.79 |
|
Acyl =CH–CH2–CH= | 0.97 ± 0.18 | 1.11 ± 0.14 | 1.03 ± 0.22 | 1.08 ± 0.14 | 0.97 | 0.89 | 0.77 ± 0.05 | 0.94 ± 0.11 | 0.78 ± 0.04 | 0.85 ± 0.05 | 0.89 | 0.70 |
|
Acyl CH2–CH2–CH= | 2.2 ± 0.07 | 1.5 ± 0.03 | 1.8 ± 0.15 | 1.5 ± 0.06 | 0.74 | 0.45 | 1.8 ± 0.54 | 2.0 ± 0.06 | 0.96 ± 0.04 | 1.10 ± 0.06 | 0.93 | 0.90 |
| Chol–18 | 5.5 ± 0.33 | 4.6 ± 0.04 | 4.4 ± 0.69 | 4.0 ± 0.36 | 1.46 | nd | 3.6 ± 0.08 | 3.2 ± 0.23 | 2.1 ± 0.10 | 1.9 ± 0.09 | 1.54 | 1.38 |
| Chol–19 | 3.8 ± 0.24 | 3.6 ± 0.11 | 2.6 ± 0.14 | 3.3 ± 0.15 | 0.68 | nd | 2.7 ± 0.04 | 2.1 ± 0.07 | 1.7 ± 0.08 | 1.4 ± 0.16 | 0.85 | 1.36 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, P.; Dai, L.; Crooks, D.R.; Neckers, L.M.; Higashi, R.M.; Fan, T.W.-M.; Lane, A.N. NMR Methods for Determining Lipid Turnover via Stable Isotope Resolved Metabolomics. Metabolites 2021, 11, 202. https://doi.org/10.3390/metabo11040202
Lin P, Dai L, Crooks DR, Neckers LM, Higashi RM, Fan TW-M, Lane AN. NMR Methods for Determining Lipid Turnover via Stable Isotope Resolved Metabolomics. Metabolites. 2021; 11(4):202. https://doi.org/10.3390/metabo11040202
Chicago/Turabian StyleLin, Penghui, Li Dai, Daniel R. Crooks, Leonard M. Neckers, Richard M. Higashi, Teresa W-M. Fan, and Andrew N. Lane. 2021. "NMR Methods for Determining Lipid Turnover via Stable Isotope Resolved Metabolomics" Metabolites 11, no. 4: 202. https://doi.org/10.3390/metabo11040202
APA StyleLin, P., Dai, L., Crooks, D. R., Neckers, L. M., Higashi, R. M., Fan, T. W. -M., & Lane, A. N. (2021). NMR Methods for Determining Lipid Turnover via Stable Isotope Resolved Metabolomics. Metabolites, 11(4), 202. https://doi.org/10.3390/metabo11040202