
Role of Steroid Hormones in the Pathogenesis of Nonalcoholic Fatty Liver Disease



Abstract
:1. Introduction
2. Steroid Hormones and Cognate Receptors
2.1. Steroid Hormones
2.1.1. Estrogens
2.1.2. Androgens
2.1.3. Progestogens
2.1.4. Glucocorticoids
2.1.5. Mineralocorticoids
2.1.6. Vitamin D
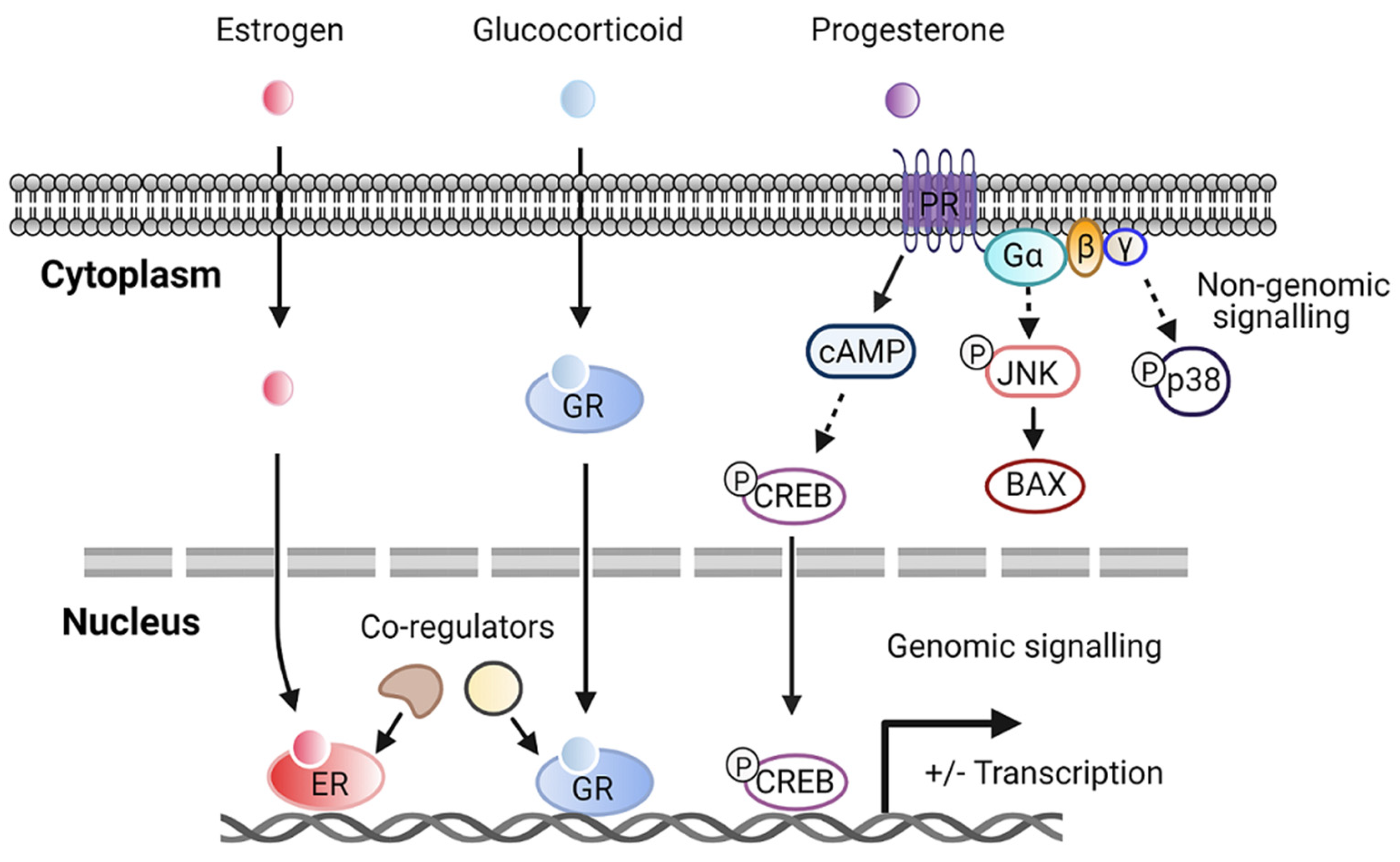
2.2. Steroid Hormone Receptors
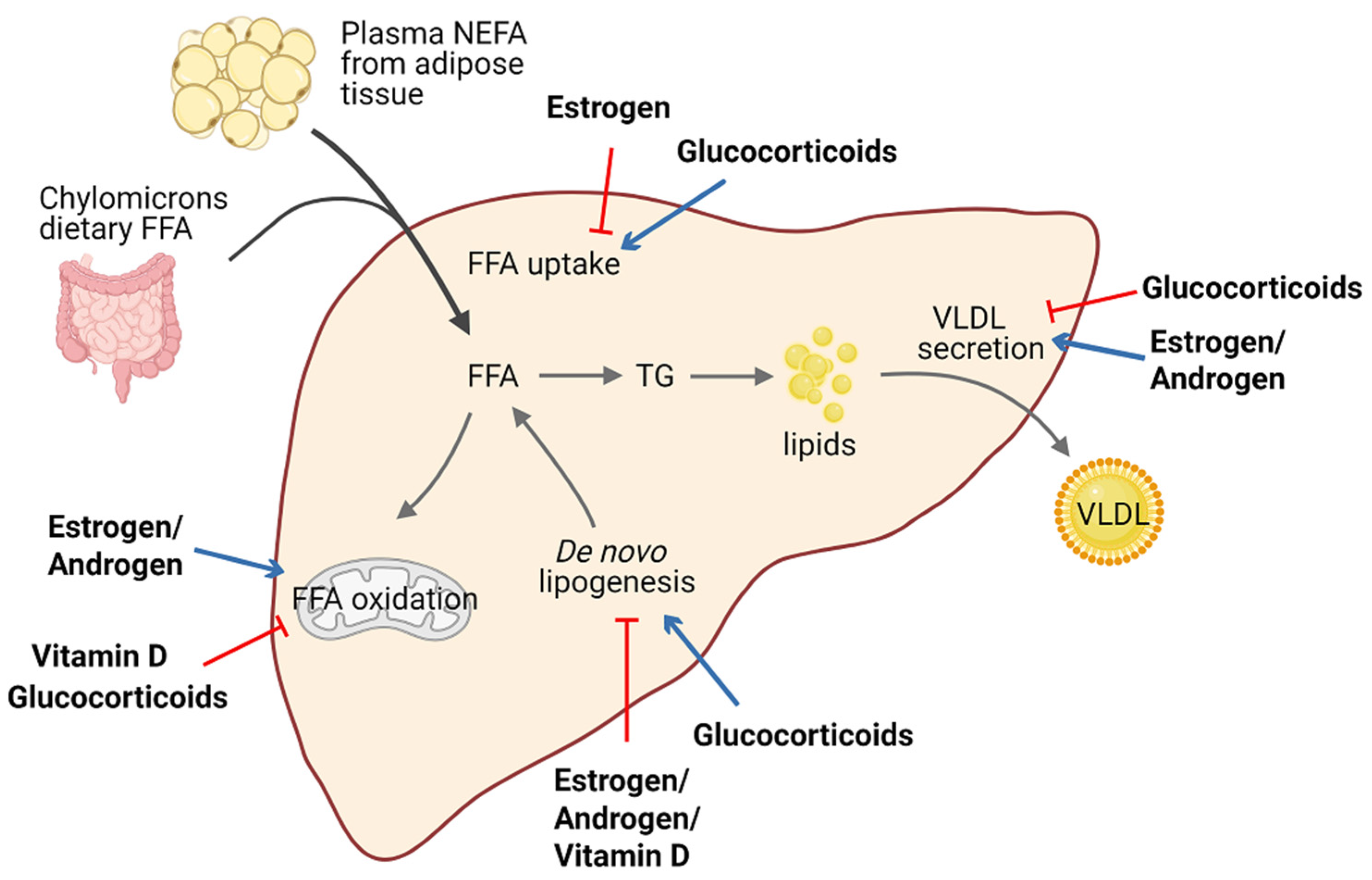
3. Role of Steroid Hormones in Hepatic Steatosis and Metabolism

{kind=link}
{kind=link}
{kind=link}
| Steroid Hormones | Model(s) Used | Major Phenotypes Examined |
|---|---|---|
| Estrogen | - Female ERα-deficient mice fed HFD for 10 weeks [71] - Male hepatic ERα-deficient mice fed HFD for 12 weeks [72] - OVX mice treated with E2 and fed HFD for 6 weeks [74] | - Liver weight, hepatic steatosis, and ALT level ↑ - Hepatic steatosis and insulin resistance ↑ - Hepatic steatosis and insulin- mediated suppression of VLDL secretion ↓ |
| Androgen | - Male hepatic AR-deficient mice fed HFD for 8 weeks [76] | - Body weight, hepatic steatosis, and insulin resistance ↑ |
| Glucocorticoid | - db/db mice treated with GC shRNA for 14 days [77] - SD rats treated with exogenous corticosterone and fed HFD for 16 days [78] | - Hepatic steatosis and genes critical for lipid storage and transport ↓ - Hepatic steatosis, uptake of FA into liver, and ALT level ↑ |
| Mineralocorticoid | - Myeloid MR-deficient ob/ob mice [79] - Aldosterone synthase-deficient mice fed HFD for 12 weeks [80] | - Hepatic steatosis, lipogenesis, and insulin resistance ↓ - HFD-feeding-induced hepatic steatosis ↓ |
| Vitamin D | - C57BL6 mice fed a high-fat/ high-sucrose diet followed by treatment with vitamin D for 15 weeks [81] - SD rats fed HFD followed by treatment with vitamin D for 16 weeks [82] | - Hepatic steatosis and hepatic de novo lipogenesis ↓ - Liver weight, hepatic steatosis, and ALT level ↓ |
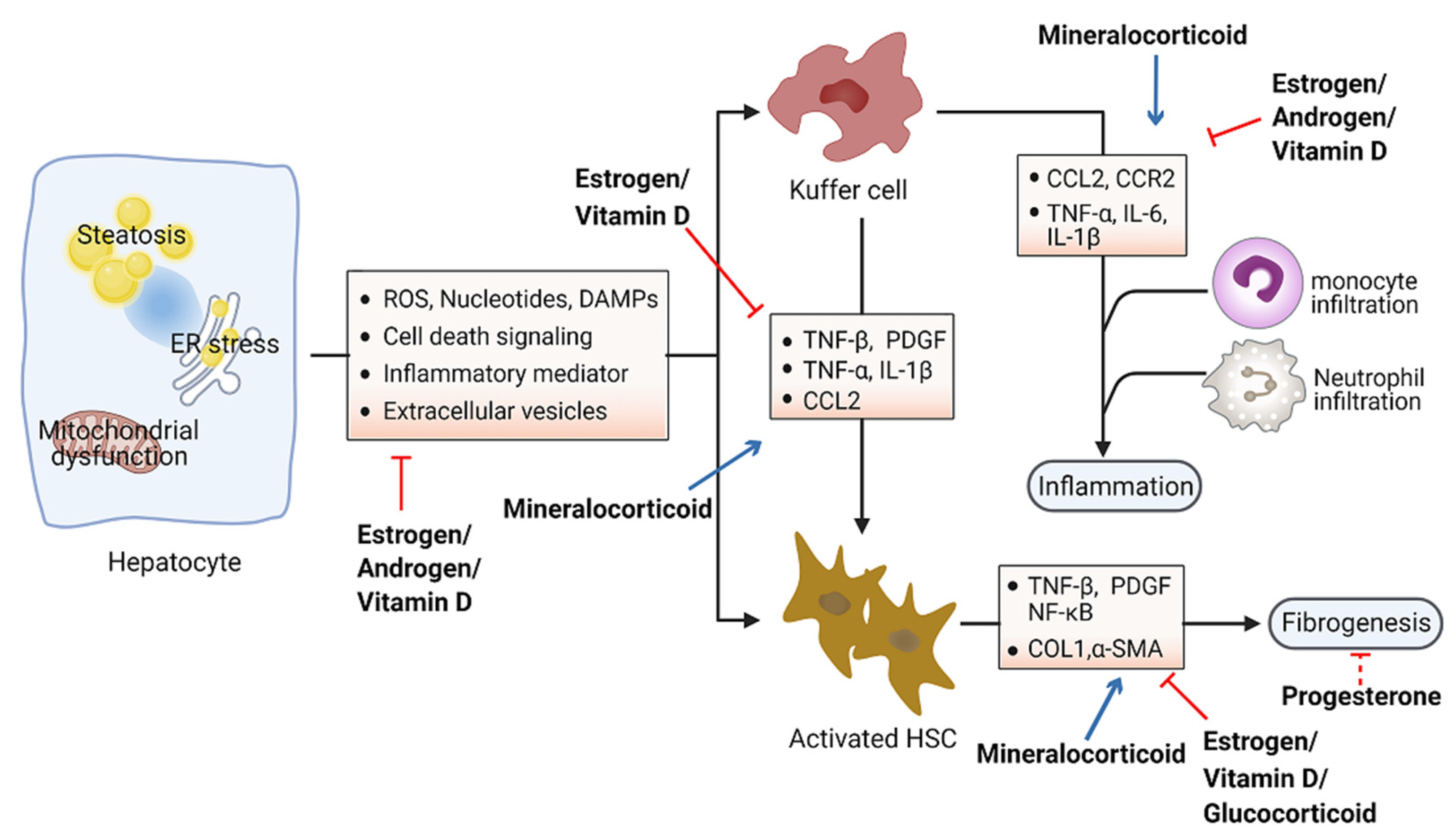
4. Role of Steroid Hormones in Hepatic Inflammation and Fibrosis
| Steroid Hormones | Model (s) Used | Major Phenotypes Examined |
|---|---|---|
| Estrogen | - OVX mice fed HFD and high-fructose water for 12 weeks [111] - OVX mice fed a high-fat and high-cholic-acid diet for 6 weeks [109] - Old female zebrafish fed a high-calorie diet for 24 weeks [112] - Orchidectomized C57/BL6 mice treated with estradiol benzoate-fed MCD for 4 weeks [110] - Male C57BL6 mice treated with β-LGND2 and fed HFD for 10 weeks [115] | - Hepatic inflammation and fibrosis, ALT level and ballooning degeneration ↑ - Liver fibrosis, inflammation, and hepatocyte ballooning degeneration ↑ - Liver fibrosis, IL-6, and TNF-β ↑ - Hepatic inflammation, MyD88, and IL-6 ↓ - Hepatic steatosis and insulin resistance ↓ |
| Androgen | - Orchidectomized male SD rats treated with dihydrotestosterone and fed HFD for 75 days [117] | - Portal inflammation, TNF-α, and IL-6 ↓ |
| Progesterone | - Hepatic fibrosis model of New Zealand male rabbits treated with progesterone for 180 days [119] | - Liver fibrosis, fat metamorphosis, and inflammatory infiltrate ↓ |
| Glucocorticoid | - Immune cell-specific GR-knockout mice treated with CCl4 and dexamethasone for 6 weeks [120] - HSC-specific GR-knockout mice treated with CCl4 and dexamethasone for 6 weeks [120] | - Inflammation and monocyte recruitment ↓ - Hepatic fibrosis and fibrotic gene expression ↓ |
| Mineralocorticoid | - C57BL6 mice fed HFFD mixed with eplerenone for 12 weeks [121] - Male C57BL6 mice fed a CDAA diet for 22 weeks with eplerenone [122] - Male SD rats treated with aldosterone for 4 weeks [123] | - Lipid accumulation, lobular inflammation, and collagen deposition ↓ - Hepatic fibrosis, steatosis, and inflammation ↓ - Hepatic fibrosis, oxidative stress, and DNA double-strand breaks ↑ |
| Vitamin D | - Vitamin D-deficient SD rats fed WD for 10 weeks [124] - CDAA diet-induced rat NASH model with phototherapy for 6 or 12 weeks [125] | - Foci of lobular inflammation and ballooning degeneration ↑ - Collagen fibrosis, insulin and leptin resistance, inflammation, and HSC activation ↓ |
5. Conclusions and Perspective
Funding
Conflicts of Interest
References
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global Burden of Nafld and Nash: Trends, Predictions, Risk Factors and Prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Baffy, G.; Brunt, E.M.; Caldwell, S.H. Hepatocellular Carcinoma in Non-Alcoholic Fatty Liver Disease: An Emerging Menace. J. Hepatol. 2012, 56, 1384–1391. [Google Scholar] [CrossRef] [Green Version]
- Jou, J.; Choi, S.S.; Diehl, A.M. Mechanisms of Disease Progression in Nonalcoholic Fatty Liver Disease. Semin. Liver Dis. 2008, 28, 370–379. [Google Scholar] [CrossRef]
- Williams, C.D.; Stengel, J.; Asike, M.I.; Torres, D.M.; Shaw, J.; Contreras, M.; Landt, C.L.; Harrison, S.A. Prevalence of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis among a Largely Middle-Aged Population Utilizing Ultrasound and Liver Biopsy: A Prospective Study. Gastroenterology 2011, 140, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, M.J.; Adams, L.A.; Canbay, A.; Syn, W.K. Extrahepatic Complications of Nonalcoholic Fatty Liver Disease. Hepatology 2014, 59, 1174–1197. [Google Scholar] [CrossRef]
- Cusi, K. Role of Obesity and Lipotoxicity in the Development of Nonalcoholic Steatohepatitis: Pathophysiology and Clinical Implications. Gastroenterology 2012, 142, 711–725. [Google Scholar] [CrossRef]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The Multiple-Hit Pathogenesis of Non-Alcoholic Fatty Liver Disease (Nafld). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Haas, J.T.; Francque, S.; Staels, B. Pathophysiology and Mechanisms of Nonalcoholic Fatty Liver Disease. Annu. Rev. Physiol. 2016, 78, 181–205. [Google Scholar] [CrossRef] [PubMed]
- Gaggini, M.; Morelli, M.; Buzzigoli, E.; DeFronzo, R.A.; Bugianesi, E.; Gastaldelli, A. Non-Alcoholic Fatty Liver Disease (Nafld) and Its Connection with Insulin Resistance, Dyslipidemia, Atherosclerosis and Coronary Heart Disease. Nutrients 2013, 5, 1544–1560. [Google Scholar] [CrossRef]
- Utzschneider, K.M.; Kahn, S.E. Review: The Role of Insulin Resistance in Nonalcoholic Fatty Liver Disease. J. Clin. Endocrinol. Metab. 2006, 91, 4753–4761. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Zhang, M.; Liu, Q.; Xu, T.; Huang, T.; Yao, D.; Wong, C.W.; Liu, J.; Guan, M. 18beta-Glycyrrhetinic Acid Acts through Hepatocyte Nuclear Factor 4 Alpha to Modulate Lipid and Carbohydrate Metabolism. Pharmacol. Res. 2020, 157, 104840. [Google Scholar] [CrossRef]
- Guan, M.; Qu, L.; Tan, W.; Chen, L.; Wong, C.W. Hepatocyte Nuclear Factor-4 Alpha Regulates Liver Triglyceride Metabolism in Part through Secreted Phospholipase a(2) Gxiib. Hepatology 2011, 53, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Gusdon, A.M.; Song, K.X.; Qu, S. Nonalcoholic Fatty Liver Disease: Pathogenesis and Therapeutics from a Mitochondria-Centric Perspective. Oxid. Med. Cell. Longev. 2014, 2014, 637027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Yang, M.; Wang, N.; Liu, Q.; Wang, B.; Huang, T.; Tong, Y.; Ming, Y.; Wong, C.W.; Liu, J.; et al. Andrographolide Modulates Hnf4alpha Activity Imparting on Hepatic Metabolism. Mol. Cell. Endocrinol. 2020, 513, 110867. [Google Scholar] [CrossRef] [PubMed]
- Dowman, J.K.; Tomlinson, J.W.; Newsome, P.N. Pathogenesis of Non-Alcoholic Fatty Liver Disease. QJM 2010, 103, 71–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caligiuri, A.; Gentilini, A.; Marra, F. Molecular Pathogenesis of Nash. Int. J. Mol. Sci. 2016, 17, 1575. [Google Scholar] [CrossRef] [Green Version]
- Handa, P.; Maliken, B.D.; Nelson, J.E.; Morgan-Stevenson, V.; Messner, D.J.; Dhillon, B.K.; Klintworth, H.M.; Beauchamp, M.; Yeh, M.M.; Elfers, C.T.; et al. Reduced Adiponectin Signaling Due to Weight Gain Results in Nonalcoholic Steatohepatitis through Impaired Mitochondrial Biogenesis. Hepatology 2014, 60, 133–145. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Gramlich, T.; Liu, Y.C.; Matteoni, C.; Petrelli, M.; Goldblum, J.; Rybicki, L.; McCullough, A.J. Nonalcoholic Fatty Liver Disease: Assessment of Variability in Pathologic Interpretations. Mod. Pathol. 1998, 11, 560–565. [Google Scholar]
- Beste, L.A.; Leipertz, S.L.; Green, P.K.; Dominitz, J.A.; Ross, D.; Ioannou, G.N. Trends in Burden of Cirrhosis and Hepatocellular Carcinoma by Underlying Liver Disease in Us Veterans, 2001-2013. Gastroenterology 2015, 149, 1471–1482.e5. [Google Scholar] [CrossRef] [Green Version]
- Ashraf, N.U.; Sheikh, T.A. Endoplasmic Reticulum Stress and Oxidative Stress in the Pathogenesis of Non-Alcoholic Fatty Liver Disease. Free Radic. Res. 2015, 49, 1405–1418. [Google Scholar] [CrossRef]
- Tarantino, G.; Finelli, C. Pathogenesis of Hepatic Steatosis: The Link between Hypercortisolism and Non-Alcoholic Fatty Liver Disease. World J. Gastroenterol. 2013, 19, 6735–6743. [Google Scholar] [CrossRef]
- Charni-Natan, M.; Aloni-Grinstein, R.; Osher, E.; Rotter, V. Liver and Steroid Hormones-Can a Touch of P53 Make a Difference? Front. Endocrinol. 2019, 10, 374. [Google Scholar] [CrossRef] [Green Version]
- Morris, E.M.; Fletcher, J.A.; Thyfault, J.P.; Rector, R.S. The Role of Angiotensin Ii in Nonalcoholic Steatohepatitis. Mol. Cell. Endocrinol. 2013, 378, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.A.; Lacin, S.; Abdel-Wahab, R.; Uemura, M.; Hassan, M.; Rashid, A.; Duda, D.G.; Kaseb, A.O. Nonalcoholic Steatohepatitis-Related Hepatocellular Carcinoma: Is There a Role for the Androgen Receptor Pathway? Onco Targets Ther. 2017, 10, 1403–1412. [Google Scholar] [CrossRef] [Green Version]
- Miller, W.L.; Auchus, R.J. The Molecular Biology, Biochemistry, and Physiology of Human Steroidogenesis and Its Disorders. Endocr. Rev. 2011, 32, 81–151. [Google Scholar] [CrossRef] [Green Version]
- Schneider, A.E.; Karpati, E.; Schuszter, K.; Toth, E.A.; Kiss, E.; Kulcsar, M.; Laszlo, G.; Matko, J. A Dynamic Network of Estrogen Receptors in Murine Lymphocytes: Fine-Tuning the Immune Response. J. Leukoc. Biol. 2014, 96, 857–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thakur, M.K.; Paramanik, V. Role of Steroid Hormone Coregulators in Health and Disease. Horm. Res. 2009, 71, 194–200. [Google Scholar] [CrossRef]
- McLachlan, J.A.; Newbold, R.R. Estrogens and Development. Environ Health Perspect 1987, 75, 25–27. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Shen, Y.; Li, R. Estrogen Synthesis and Signaling Pathways During Aging: From Periphery to Brain. Trends Mol. Med. 2013, 19, 197–209. [Google Scholar] [CrossRef] [Green Version]
- Lobo, R.A.; Pickar, J.H.; Stevenson, J.C.; Mack, W.J.; Hodis, H.N. Back to the Future: Hormone Replacement Therapy as Part of a Prevention Strategy for Women at the Onset of Menopause. Atherosclerosis 2016, 254, 282–290. [Google Scholar] [CrossRef]
- Trabert, B.; Wentzensen, N.; Yang, H.P.; Sherman, M.E.; Hollenbeck, A.R.; Park, Y.; Brinton, L.A. Is Estrogen Plus Progestin Menopausal Hormone Therapy Safe with Respect to Endometrial Cancer Risk? Int. J. Cancer 2013, 132, 417–426. [Google Scholar] [CrossRef] [Green Version]
- Pierard-Franchimont, C.; Pierard, G.E. Postmenopausal Aging of the Sebaceous Follicle: A Comparison between Women Receiving Hormone Replacement Therapy or Not. Dermatology 2002, 204, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Camporez, J.P.; Lyu, K.; Goldberg, E.L.; Zhang, D.; Cline, G.W.; Jurczak, M.J.; Dixit, V.D.; Petersen, K.F.; Shulman, G.I. Anti-Inflammatory Effects of Oestrogen Mediate the Sexual Dimorphic Response to Lipid-Induced Insulin Resistance. J. Physiol. 2019, 597, 3885–3903. [Google Scholar] [CrossRef]
- Palmisano, B.T.; Zhu, L.; Stafford, J.M. Role of Estrogens in the Regulation of Liver Lipid Metabolism. Adv. Exp. Med. Biol. 2017, 1043, 227–256. [Google Scholar] [PubMed] [Green Version]
- Shen, M.; Kumar, S.P.; Shi, H. Estradiol Regulates Insulin Signaling and Inflammation in Adipose Tissue. Horm. Mol. Biol. Clin. Investig. 2014, 17, 99–107. [Google Scholar] [CrossRef]
- Wilson, J.D.; Griffin, J.E.; Leshin, M.; George, F.W. Role of Gonadal Hormones in Development of the Sexual Phenotypes. Hum. Genet. 1981, 58, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Quigley, C.A.; De Bellis, A.; Marschke, K.B.; el-Awady, M.K.; Wilson, E.M.; French, F.S. Androgen Receptor Defects: Historical, Clinical, and Molecular Perspectives. Endocr. Rev. 1995, 16, 271–321. [Google Scholar] [CrossRef]
- Mooradian, A.D.; Morley, J.E.; Korenman, S.G. Biological Actions of Androgens. Endocr. Rev. 1987, 8, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wu, Y.; Zhao, J.; Wang, H.; Tan, J.; Yang, M.; Li, Y.; Deng, S.; Gao, S.; Li, H.; et al. Distinct Cardiac Energy Metabolism and Oxidative Stress Adaptations between Obese and Non-Obese Type 2 Diabetes Mellitus. Theranostics 2020, 10, 2675–2695. [Google Scholar] [CrossRef]
- Pikler, G.M.; Webster, R.A.; Spelsberg, T.C. Nuclear Binding of Progesterone in Hen Oviduct. Binding to Multiple Sites in Vitro. Biochem. J. 1976, 156, 399–408. [Google Scholar] [CrossRef] [Green Version]
- Oettel, M.; Mukhopadhyay, A.K. Progesterone: The Forgotten Hormone in Men? Aging Male 2004, 7, 236–257. [Google Scholar] [CrossRef] [PubMed]
- Manyonda, I.; Talaulikar, V.S.; Pirhadi, R.; Onwude, J. Progestogens Are the Problem in Hormone Replacement Therapy: Time to Reappraise Their Use. Post Reprod. Health 2020, 26, 26–31. [Google Scholar] [CrossRef]
- Adcock, I.M.; Mumby, S. Glucocorticoids. Handb. Exp. Pharmacol. 2017, 237, 171–196. [Google Scholar]
- Vandewalle, J.; Luypaert, A.; De Bosscher, K.; Libert, C. Therapeutic Mechanisms of Glucocorticoids. Trends Endocrinol. Metab. 2018, 29, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Rhen, T.; Cidlowski, J.A. Antiinflammatory Action of Glucocorticoids--New Mechanisms for Old Drugs. N. Engl. J. Med. 2005, 353, 1711–1723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pazirandeh, A.; Xue, Y.; Prestegaard, T.; Jondal, M.; Okret, S. Effects of Altered Glucocorticoid Sensitivity in the T Cell Lineage on Thymocyte and T Cell Homeostasis. FASEB J. 2002, 16, 727–729. [Google Scholar] [CrossRef]
- Scheller, K.; Sekeris, C.E. The Effects of Steroid Hormones on the Transcription of Genes Encoding Enzymes of Oxidative Phosphorylation. Exp. Physiol. 2003, 88, 129–140. [Google Scholar] [CrossRef] [Green Version]
- Allan, E.H.; Chisholm, A.B.; Titheradge, M.A. The Stimulation of Hepatic Oxidative Phosphorylation Following Dexamethasone Treatment of Rats. Biochim. Biophys. Acta 1983, 725, 71–76. [Google Scholar] [CrossRef]
- Briet, M.; Schiffrin, E.L. Aldosterone: Effects on the Kidney and Cardiovascular System. Nat. Rev. Nephrol. 2010, 6, 261–273. [Google Scholar] [CrossRef]
- Pearce, D.; Bhargava, A.; Cole, T.J. Aldosterone: Its Receptor, Target Genes, and Actions. Vitam. Horm. 2003, 66, 29–76. [Google Scholar]
- Weinberger, M.H. Mineralocorticoids and Blood Pressure. Curr. Opin. Nephrol. Hypertens. 1994, 3, 550–553. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.J. Contribution of Aldosterone to Cardiovascular and Renal Inflammation and Fibrosis. Nat. Rev. Nephrol. 2013, 9, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Bender, S.B.; McGraw, A.P.; Jaffe, I.Z.; Sowers, J.R. Mineralocorticoid Receptor-Mediated Vascular Insulin Resistance: An Early Contributor to Diabetes-Related Vascular Disease? Diabetes 2013, 62, 313–319. [Google Scholar] [CrossRef] [Green Version]
- Feraco, A.; Armani, A.; Mammi, C.; Fabbri, A.; Rosano, G.M.; Caprio, M. Role of Mineralocorticoid Receptor and Renin-Angiotensin-Aldosterone System in Adipocyte Dysfunction and Obesity. J. Steroid Biochem. Mol. Biol. 2013, 137, 99–106. [Google Scholar] [CrossRef]
- Garg, R.; Adler, G.K. Role of Mineralocorticoid Receptor in Insulin Resistance. Curr. Opin. Endocrinol. Diabetes Obes. 2012, 19, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Bikle, D.D. Vitamin D Metabolism, Mechanism of Action, and Clinical Applications. Chem. Biol. 2014, 21, 319–329. [Google Scholar] [CrossRef] [Green Version]
- Calvo, M.S.; Whiting, S.J.; Barton, C.N. Vitamin D Intake: A Global Perspective of Current Status. J. Nutr. 2005, 135, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Norman, A.W. From Vitamin D to Hormone D: Fundamentals of the Vitamin D Endocrine System Essential for Good Health. Am. J. Clin. Nutr. 2008, 88, 491S–499S. [Google Scholar] [CrossRef] [Green Version]
- Alkharfy, K.M.; Al-Daghri, N.M.; Yakout, S.M.; Ahmed, M. Calcitriol Attenuates Weight-Related Systemic Inflammation and Ultrastructural Changes in the Liver in a Rodent Model. Basic Clin. Pharmacol. Toxicol. 2013, 112, 42–49. [Google Scholar] [CrossRef]
- Kumar, R.; Litwack, G. Structural and Functional Relationships of the Steroid Hormone Receptors’ N-Terminal Transactivation Domain. Steroids 2009, 74, 877–883. [Google Scholar] [CrossRef] [Green Version]
- Weigel, N.L. Steroid Hormone Receptors and Their Regulation by Phosphorylation. Biochem. J. 1996, 319 (Pt. 3), 657–667. [Google Scholar] [CrossRef] [Green Version]
- Robinson-Rechavi, M.; Escriva Garcia, H.; Laudet, V. The Nuclear Receptor Superfamily. J. Cell Sci. 2003, 116, 585–586. [Google Scholar] [CrossRef] [Green Version]
- Liao, R.S.; Ma, S.; Miao, L.; Li, R.; Yin, Y.; Raj, G.V. Androgen Receptor-Mediated Non-Genomic Regulation of Prostate Cancer Cell Proliferation. Transl. Androl. Urol. 2013, 2, 187–196. [Google Scholar]
- Pupo, M.; Maggiolini, M.; Musti, A.M. Gper Mediates Non-Genomic Effects of Estrogen. Methods Mol. Biol. 2016, 1366, 471–488. [Google Scholar] [PubMed]
- Yasar, P.; Ayaz, G.; User, S.D.; Gupur, G.; Muyan, M. Molecular Mechanism of Estrogen-Estrogen Receptor Signaling. Reprod. Med. Biol. 2017, 16, 4–20. [Google Scholar] [CrossRef] [PubMed]
- Scheller, K.; Seibel, P.; Sekeris, C.E. Glucocorticoid and Thyroid Hormone Receptors in Mitochondria of Animal Cells. Int. Rev. Cytol. 2003, 222, 1–61. [Google Scholar] [PubMed]
- Valadez-Cosmes, P.; Vazquez-Martinez, E.R.; Cerbon, M.; Camacho-Arroyo, I. Membrane Progesterone Receptors in Reproduction and Cancer. Mol. Cell. Endocrinol. 2016, 434, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Dressing, G.E.; Goldberg, J.E.; Charles, N.J.; Schwertfeger, K.L.; Lange, C.A. Membrane Progesterone Receptor Expression in Mammalian Tissues: A Review of Regulation and Physiological Implications. Steroids 2011, 76, 11–17. [Google Scholar] [CrossRef] [Green Version]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular Mechanisms of Hepatic Lipid Accumulation in Non-Alcoholic Fatty Liver Disease. Cell. Mol. Life Sci. 2018, 75, 3313–3327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.L.; Madak-Erdogan, Z. Estrogens and Female Liver Health. Steroids 2018, 133, 38–43. [Google Scholar] [CrossRef]
- Hart-Unger, S.; Arao, Y.; Hamilton, K.J.; Lierz, S.L.; Malarkey, D.E.; Hewitt, S.C.; Freemark, M.; Korach, K.S. Hormone Signaling and Fatty Liver in Females: Analysis of Estrogen Receptor Alpha Mutant Mice. Int. J. Obes. 2017, 41, 945–954. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Martinez, M.N.; Emfinger, C.H.; Palmisano, B.T.; Stafford, J.M. Estrogen Signaling Prevents Diet-Induced Hepatic Insulin Resistance in Male Mice with Obesity. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E1188–E1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.; Liu, Q.; Huang, T.; Tan, W.; Qu, L.; Chen, T.; Pan, H.; Chen, L.; Liu, J.; Wong, C.W.; et al. Dysfunction of Estrogen-Related Receptor Alpha-Dependent Hepatic Vldl Secretion Contributes to Sex Disparity in Nafld/Nash Development. Theranostics 2020, 10, 10874–10891. [Google Scholar] [CrossRef]
- Della Torre, S. Non-Alcoholic Fatty Liver Disease as a Canonical Example of Metabolic Inflammatory-Based Liver Disease Showing a Sex-Specific Prevalence: Relevance of Estrogen Signaling. Front. Endocrinol. 2020, 11, 572490. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.T.; Pan, H.J.; Lee, C.H. Prevention of Tamoxifen-Related Nonalcoholic Fatty Liver Disease in Breast Cancer Patients. Clin. Breast Cancer 2018, 18, e677–e685. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Yu, I.C.; Wang, R.S.; Chen, Y.T.; Liu, N.C.; Altuwaijri, S.; Hsu, C.L.; Ma, W.L.; Jokinen, J.; Sparks, J.D.; et al. Increased Hepatic Steatosis and Insulin Resistance in Mice Lacking Hepatic Androgen Receptor. Hepatology 2008, 47, 1924–1935. [Google Scholar] [CrossRef] [PubMed]
- Lemke, U.; Krones-Herzig, A.; Berriel Diaz, M.; Narvekar, P.; Ziegler, A.; Vegiopoulos, A.; Cato, A.C.; Bohl, S.; Klingmuller, U.; Screaton, R.A.; et al. The Glucocorticoid Receptor Controls Hepatic Dyslipidemia through Hes1. Cell Metab. 2008, 8, 212–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Souza, A.M.; Beaudry, J.L.; Szigiato, A.A.; Trumble, S.J.; Snook, L.A.; Bonen, A.; Giacca, A.; Riddell, M.C. Consumption of a High-Fat Diet Rapidly Exacerbates the Development of Fatty Liver Disease That Occurs with Chronically Elevated Glucocorticoids. Am. J. Physiol Gastrointest. Liver Physiol. 2012, 302, G850–G863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.Y.; Li, C.; Yao, G.F.; Du, L.J.; Liu, Y.; Zheng, X.J.; Yan, S.; Sun, J.Y.; Liu, Y.; Liu, M.Z.; et al. Deletion of Macrophage Mineralocorticoid Receptor Protects Hepatic Steatosis and Insulin Resistance through Eralpha/Hgf/Met Pathway. Diabetes 2017, 66, 1535–1547. [Google Scholar] [CrossRef] [Green Version]
- Luo, P.; Dematteo, A.; Wang, Z.; Zhu, L.; Wang, A.; Kim, H.S.; Pozzi, A.; Stafford, J.M.; Luther, J.M. Aldosterone Deficiency Prevents High-Fat-Feeding-Induced Hyperglycaemia and Adipocyte Dysfunction in Mice. Diabetologia 2013, 56, 901–910. [Google Scholar] [CrossRef] [Green Version]
- Marziou, A.; Philouze, C.; Couturier, C.; Astier, J.; Obert, P.; Landrier, J.F.; Riva, C. Vitamin D Supplementation Improves Adipose Tissue Inflammation and Reduces Hepatic Steatosis in Obese C57bl/6j Mice. Nutrients 2020, 12, 342. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.G.; Liu, Y.X.; Wang, H.; Wang, B.P.; Qu, H.Q.; Wang, B.L.; Zhu, M. Active Form of Vitamin D Ameliorates Non-Alcoholic Fatty Liver Disease by Alleviating Oxidative Stress in a High-Fat Diet Rat Model. Endocr. J. 2017, 64, 663–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaruvongvanich, V.; Sanguankeo, A.; Riangwiwat, T.; Upala, S. Testosterone, Sex Hormone-Binding Globulin and Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Ann. Hepatol. 2017, 16, 382–394. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kwon, H.; Park, J.H.; Cho, B.; Kim, D.; Oh, S.W.; Lee, C.M.; Choi, H.C. A Low Level of Serum Total Testosterone Is Independently Associated with Nonalcoholic Fatty Liver Disease. BMC Gastroenterol. 2012, 12, 69. [Google Scholar] [CrossRef] [Green Version]
- Eguchi, Y.; Eguchi, T.; Mizuta, T.; Ide, Y.; Yasutake, T.; Iwakiri, R.; Hisatomi, A.; Ozaki, I.; Yamamoto, K.; Kitajima, Y.; et al. Visceral Fat Accumulation and Insulin Resistance Are Important Factors in Nonalcoholic Fatty Liver Disease. J. Gastroenterol. 2006, 41, 462–469. [Google Scholar] [CrossRef]
- Ma, W.L.; Lai, H.C.; Yeh, S.; Cai, X.; Chang, C. Androgen Receptor Roles in Hepatocellular Carcinoma, Fatty Liver, Cirrhosis and Hepatitis. Endocr. Relat. Cancer 2014, 21, R165–R182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.C.; Gray, N.E.; Kuo, T.; Harris, C.A. Regulation of Triglyceride Metabolism by Glucocorticoid Receptor. Cell Biosci. 2012, 2, 19. [Google Scholar] [CrossRef] [Green Version]
- Sorensen, H.N.; Gautik, K.M.; Bremer, J.; Spydevold, O. Induction of the Three Peroxisomal Beta-Oxidation Enzymes Is Synergistically Regulated by Dexamethasone and Fatty Acids, and Counteracted by Insulin in Morris 7800c1 Hepatoma Cells in Culture. Eur. J. Biochem. 1992, 208, 705–711. [Google Scholar] [CrossRef]
- Andrews, R.C.; Walker, B.R. Glucocorticoids and Insulin Resistance: Old Hormones, New Targets. Clin. Sci. 1999, 96, 513–523. [Google Scholar] [CrossRef]
- Rockall, A.G.; Sohaib, S.A.; Evans, D.; Kaltsas, G.; Isidori, A.M.; Monson, J.P.; Besser, G.M.; Grossman, A.B.; Reznek, R.H. Hepatic Steatosis in Cushing’s Syndrome: A Radiological Assessment Using Computed Tomography. Eur. J. Endocrinol. 2003, 149, 543–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belden, Z.; Deiuliis, J.A.; Dobre, M.; Rajagopalan, S. The Role of the Mineralocorticoid Receptor in Inflammation: Focus on Kidney and Vasculature. Am. J. Nephrol. 2017, 46, 298–314. [Google Scholar] [CrossRef] [PubMed]
- Barchetta, I.; Carotti, S.; Labbadia, G.; Gentilucci, U.V.; Muda, A.O.; Angelico, F.; Silecchia, G.; Leonetti, F.; Fraioli, A.; Picardi, A.; et al. Liver Vitamin D Receptor, Cyp2r1, and Cyp27a1 Expression: Relationship with Liver Histology and Vitamin D3 Levels in Patients with Nonalcoholic Steatohepatitis or Hepatitis C Virus. Hepatology 2012, 56, 2180–2187. [Google Scholar] [CrossRef] [PubMed]
- Barchetta, I.; Cimini, F.A.; Cavallo, M.G. Vitamin D and Metabolic Dysfunction-Associated Fatty Liver Disease (Mafld): An Update. Nutrients 2020, 12, 3302. [Google Scholar] [CrossRef]
- Kim, K.H.; Lee, M.S. Pathogenesis of Nonalcoholic Steatohepatitis and Hormone-Based Therapeutic Approaches. Front. Endocrinol. 2018, 9, 485. [Google Scholar] [CrossRef] [PubMed]
- Parthasarathy, G.; Revelo, X.; Malhi, H. Pathogenesis of Nonalcoholic Steatohepatitis: An Overview. Hepatol. Commun. 2020, 4, 478–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broom, L.J.; Kogut, M.H. Inflammation: Friend or Foe for Animal Production? Poult. Sci. 2018, 97, 510–514. [Google Scholar] [CrossRef]
- Medzhitov, R. Origin and Physiological Roles of Inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef]
- Kamei, Y.; Xu, L.; Heinzel, T.; Torchia, J.; Kurokawa, R.; Gloss, B.; Lin, S.C.; Heyman, R.A.; Rose, D.W.; Glass, C.K.; et al. A Cbp Integrator Complex Mediates Transcriptional Activation and Ap-1 Inhibition by Nuclear Receptors. Cell 1996, 85, 403–414. [Google Scholar] [CrossRef] [Green Version]
- Koyama, Y.; Brenner, D.A. Liver Inflammation and Fibrosis. J. Clin. Investig. 2017, 127, 55–64. [Google Scholar] [CrossRef]
- Parola, M.; Pinzani, M. Liver Fibrosis: Pathophysiology, Pathogenetic Targets and Clinical Issues. Mol. Aspects Med. 2019, 65, 37–55. [Google Scholar] [CrossRef]
- Tsuchida, T.; Friedman, S.L. Mechanisms of Hepatic Stellate Cell Activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Higashi, T.; Friedman, S.L.; Hoshida, Y. Hepatic Stellate Cells as Key Target in Liver Fibrosis. Adv. Drug Deliv. Rev. 2017, 121, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Mechanisms of Hepatic Fibrogenesis. Gastroenterology 2008, 134, 1655–1669. [Google Scholar] [CrossRef] [Green Version]
- Browning, J.D.; Horton, J.D. Molecular Mediators of Hepatic Steatosis and Liver Injury. J. Clin. Investig. 2004, 114, 147–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunt, E.M.; Kleiner, D.E.; Wilson, L.A.; Belt, P.; Neuschwander-Tetri, B.A.; Network, N.C.R. Nonalcoholic Fatty Liver Disease (Nafld) Activity Score and the Histopathologic Diagnosis in Nafld: Distinct Clinicopathologic Meanings. Hepatology 2011, 53, 810–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Florentino, G.S.; Cotrim, H.P.; Vilar, C.P.; Florentino, A.V.; Guimaraes, G.M.; Barreto, V.S. Nonalcoholic Fatty Liver Disease in Menopausal Women. Arq. Gastroenterol. 2013, 50, 180–185. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, A.; Abdelmalek, M.F. Nonalcoholic Fatty Liver Disease in Women. Womens Health 2009, 5, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Galmes-Pascual, B.M.; Martinez-Cignoni, M.R.; Moran-Costoya, A.; Bauza-Thorbrugge, M.; Sbert-Roig, M.; Valle, A.; Proenza, A.M.; Llado, I.; Gianotti, M. 17beta-Estradiol Ameliorates Lipotoxicity-Induced Hepatic Mitochondrial Oxidative Stress and Insulin Resistance. Free Radic. Biol. Med. 2020, 150, 148–160. [Google Scholar] [CrossRef]
- Kamada, Y.; Kiso, S.; Yoshida, Y.; Chatani, N.; Kizu, T.; Hamano, M.; Tsubakio, M.; Takemura, T.; Ezaki, H.; Hayashi, N.; et al. Estrogen Deficiency Worsens Steatohepatitis in Mice Fed High-Fat and High-Cholesterol Diet. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G1031–G1043. [Google Scholar] [CrossRef] [Green Version]
- Xin, G.; Qin, S.; Wang, S.; Wang, X.; Zhang, Y.; Wang, J. Sex Hormone Affects the Severity of Non-Alcoholic Steatohepatitis through the Myd88-Dependent Il-6 Signaling Pathway. Exp. Biol. Med. 2015, 240, 1279–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohashi, T.; Kato, M.; Yamasaki, A.; Kuwano, A.; Suzuki, H.; Kohjima, M.; Ogawa, Y. Effects of High Fructose Intake on Liver Injury Progression in High Fat Diet Induced Fatty Liver Disease in Ovariectomized Female Mice. Food Chem. Toxicol. 2018, 118, 190–197. [Google Scholar] [CrossRef]
- Turola, E.; Petta, S.; Vanni, E.; Milosa, F.; Valenti, L.; Critelli, R.; Miele, L.; Maccio, L.; Calvaruso, V.; Fracanzani, A.L.; et al. Ovarian Senescence Increases Liver Fibrosis in Humans and Zebrafish with Steatosis. Dis. Model. Mech. 2015, 8, 1037–1046. [Google Scholar] [CrossRef] [Green Version]
- Klair, J.S.; Yang, J.D.; Abdelmalek, M.F.; Guy, C.D.; Gill, R.M.; Yates, K.; Unalp-Arida, A.; Lavine, J.E.; Clark, J.M.; Diehl, A.M.; et al. A Longer Duration of Estrogen Deficiency Increases Fibrosis Risk among Postmenopausal Women with Nonalcoholic Fatty Liver Disease. Hepatology 2016, 64, 85–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, G.; Shimizu, I.; Cui, X.; Itonaga, M.; Tamaki, K.; Fukuno, H.; Inoue, H.; Honda, H.; Ito, S. Antioxidant and Antiapoptotic Activities of Idoxifene and Estradiol in Hepatic Fibrosis in Rats. Life Sci. 2004, 74, 897–907. [Google Scholar] [CrossRef]
- Ponnusamy, S.; Tran, Q.T.; Thiyagarajan, T.; Miller, D.D.; Bridges, D.; Narayanan, R. An Estrogen Receptor Beta-Selective Agonist Inhibits Non-Alcoholic Steatohepatitis in Preclinical Models by Regulating Bile Acid and Xenobiotic Receptors. Exp. Biol. Med. 2017, 242, 606–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamad, N.V.; Wong, S.K.; Wan Hasan, W.N.; Jolly, J.J.; Nur-Farhana, M.F.; Ima-Nirwana, S.; Chin, K.Y. The Relationship between Circulating Testosterone and Inflammatory Cytokines in Men. Aging Male 2019, 22, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, Y.; Wang, L.; Li, Z.; Zhang, H.; Wu, J.; Rahman, N.; Guo, Y.; Li, D.; Li, N.; et al. Differential Effects of Estrogen/Androgen on the Prevention of Nonalcoholic Fatty Liver Disease in the Male Rat. J. Lipid Res. 2013, 54, 345–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morita, A.; Ishigaki, Y. Gender-Difference in Diabetes Mellitus. Nihon Rinsho 2015, 73, 606–610. [Google Scholar]
- Lanari, A.; Garegnani, T.C.; Heinrichs, G.; Castresana, M.P. [Hepatic Fibrosis and Progesterone]. Acta Gastroenterol. Latinoam. 1988, 18, 161–171. [Google Scholar]
- Kim, K.H.; Lee, J.M.; Zhou, Y.; Harpavat, S.; Moore, D.D. Glucocorticoids Have Opposing Effects on Liver Fibrosis in Hepatic Stellate and Immune Cells. Mol. Endocrinol. 2016, 30, 905–916. [Google Scholar] [CrossRef]
- Wada, T.; Miyashita, Y.; Sasaki, M.; Aruga, Y.; Nakamura, Y.; Ishii, Y.; Sasahara, M.; Kanasaki, K.; Kitada, M.; Koya, D.; et al. Eplerenone Ameliorates the Phenotypes of Metabolic Syndrome with Nash in Liver-Specific Srebp-1c Tg Mice Fed High-Fat and High-Fructose Diet. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E1415–E1425. [Google Scholar] [CrossRef] [Green Version]
- Pizarro, M.; Solis, N.; Quintero, P.; Barrera, F.; Cabrera, D.; Rojas-de Santiago, P.; Arab, J.P.; Padilla, O.; Roa, J.C.; Moshage, H.; et al. Beneficial Effects of Mineralocorticoid Receptor Blockade in Experimental Non-Alcoholic Steatohepatitis. Liver Int. 2015, 35, 2129–2138. [Google Scholar] [CrossRef] [Green Version]
- Queisser, N.; Happ, K.; Link, S.; Jahn, D.; Zimnol, A.; Geier, A.; Schupp, N. Aldosterone Induces Fibrosis, Oxidative Stress and DNA Damage in Livers of Male Rats Independent of Blood Pressure Changes. Toxicol. Appl. Pharmacol. 2014, 280, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Roth, C.L.; Elfers, C.T.; Figlewicz, D.P.; Melhorn, S.J.; Morton, G.J.; Hoofnagle, A.; Yeh, M.M.; Nelson, J.E.; Kowdley, K.V. Vitamin D Deficiency in Obese Rats Exacerbates Nonalcoholic Fatty Liver Disease and Increases Hepatic Resistin and Toll-Like Receptor Activation. Hepatology 2012, 55, 1103–1111. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Cheng, Y.F.; Lai, C.Y.; Hsu, L.W.; Chang, Y.C.; Deng, J.Y.; Huang, Y.Z.; Honda, H.; Chen, K.D.; Wang, C.C.; et al. Impact of Artificial Sunlight Therapy on the Progress of Non-Alcoholic Fatty Liver Disease in Rats. J. Hepatol. 2011, 55, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Nolly, M.B.; Caldiz, C.I.; Yeves, A.M.; Villa-Abrille, M.C.; Morgan, P.E.; Amado Mondaca, N.; Portiansky, E.L.; Chiappe de Cingolani, G.E.; Cingolani, H.E.; Ennis, I.L. The Signaling Pathway for Aldosterone-Induced Mitochondrial Production of Superoxide Anion in the Myocardium. J. Mol. Cell. Cardiol. 2014, 67, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Artunc, F.; Lang, F. Mineralocorticoid and Sgk1-Sensitive Inflammation and Tissue Fibrosis. Nephron Physiol. 2014, 128, 35–39. [Google Scholar] [CrossRef]
- Eliades, M.; Spyrou, E. Vitamin D: A New Player in Non-Alcoholic Fatty Liver Disease? World J. Gastroenterol. 2015, 21, 1718–1727. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.B.; Chen, Y.H.; Zhang, C.; Shi, C.E.; Hu, K.F.; Zhou, J.; Xu, D.X.; Chen, X. Low Vitamin D Status Is Associated with Advanced Liver Fibrosis in Patients with Nonalcoholic Fatty Liver Disease. Endocrine 2017, 55, 582–590. [Google Scholar] [CrossRef]
- Breitkopf, K.; Godoy, P.; Ciuclan, L.; Singer, M.V.; Dooley, S. Tgf-Beta/Smad Signaling in the Injured Liver. Z. Gastroenterol. 2006, 44, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Barchetta, I.; Cimini, F.A.; Cavallo, M.G. Vitamin D Supplementation and Non-Alcoholic Fatty Liver Disease: Present and Future. Nutrients 2017, 9, 1015. [Google Scholar] [CrossRef] [Green Version]
- Beilfuss, A.; Sowa, J.P.; Sydor, S.; Beste, M.; Bechmann, L.P.; Schlattjan, M.; Syn, W.K.; Wedemeyer, I.; Mathe, Z.; Jochum, C.; et al. Vitamin D Counteracts Fibrogenic Tgf-Beta Signalling in Human Hepatic Stellate Cells Both Receptor-Dependently and Independently. Gut 2015, 64, 791–799. [Google Scholar] [CrossRef] [PubMed]
- Hamaguchi, M.; Kojima, T.; Ohbora, A.; Takeda, N.; Fukui, M.; Kato, T. Aging Is a Risk Factor of Nonalcoholic Fatty Liver Disease in Premenopausal Women. World J. Gastroenterol. 2012, 18, 237–243. [Google Scholar] [CrossRef]
- Lonardo, A.; Nascimbeni, F.; Ballestri, S.; Fairweather, D.; Win, S.; Than, T.A.; Abdelmalek, M.F.; Suzuki, A. Sex Differences in Nonalcoholic Fatty Liver Disease: State of the Art and Identification of Research Gaps. Hepatology 2019, 70, 1457–1469. [Google Scholar] [CrossRef]
- Wang, Z.; Xu, M.; Hu, Z.; Shrestha, U.K. Prevalence of Nonalcoholic Fatty Liver Disease and Its Metabolic Risk Factors in Women of Different Ages and Body Mass Index. Menopause 2015, 22, 667–673. [Google Scholar] [CrossRef]
- Caballeria, L.; Pera, G.; Auladell, M.A.; Toran, P.; Munoz, L.; Miranda, D.; Aluma, A.; Casas, J.D.; Sanchez, C.; Gil, D.; et al. Prevalence and Factors Associated with the Presence of Nonalcoholic Fatty Liver Disease in an Adult Population in Spain. Eur. J. Gastroenterol. Hepatol. 2010, 22, 24–32. [Google Scholar] [CrossRef]
- Camhi, S.M.; Bray, G.A.; Bouchard, C.; Greenway, F.L.; Johnson, W.D.; Newton, R.L.; Ravussin, E.; Ryan, D.H.; Smith, S.R.; Katzmarzyk, P.T. The Relationship of Waist Circumference and Bmi to Visceral, Subcutaneous, and Total Body Fat: Sex and Race Differences. Obesity 2011, 19, 402–408. [Google Scholar] [CrossRef]
- Younossi, Z.M. Non-Alcoholic Fatty Liver Disease—A Global Public Health Perspective. J. Hepatol. 2019, 70, 531–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Lu, Y.; Wang, E.; Zhang, Z.; Xiong, X.; Zhang, H.; Lu, J.; Zheng, S.; Yang, J.; Xia, X.; et al. Hepatic Estrogen Receptor Alpha Improves Hepatosteatosis through Upregulation of Small Heterodimer Partner. J. Hepatol. 2015, 63, 183–190. [Google Scholar] [CrossRef]
- Pan, J.J.; Fallon, M.B. Gender and Racial Differences in Nonalcoholic Fatty Liver Disease. World J. Hepatol. 2014, 6, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Summart, U.; Thinkhamrop, B.; Chamadol, N.; Khuntikeo, N.; Songthamwat, M.; Kim, C.S. Gender Differences in the Prevalence of Nonalcoholic Fatty Liver Disease in the Northeast of Thailand: A Population-Based Cross-Sectional Study. F1000Research 2017, 6, 1630. [Google Scholar] [CrossRef] [PubMed]
- Rao, P.K.; Boulet, S.L.; Mehta, A.; Hotaling, J.; Eisenberg, M.L.; Honig, S.C.; Warner, L.; Kissin, D.M.; Nangia, A.K.; Ross, L.S. Trends in Testosterone Replacement Therapy Use from 2003 to 2013 among Reproductive-Age Men in the United States. J. Urol. 2017, 197, 1121–1126. [Google Scholar] [CrossRef]
- Phan, H.; Richard, A.; Lazo, M.; Nelson, W.G.; Denmeade, S.R.; Groopman, J.; Kanarek, N.; Platz, E.A.; Rohrmann, S. The Association of Sex Steroid Hormone Concentrations with Non-Alcoholic Fatty Liver Disease and Liver Enzymes in Us Men. Liver Int. 2021, 41, 300–310. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Wang, S.; Meng, Y.; Yu, Q.; Wang, Q.; Xu, H.; Yuan, H.; Li, X.; Chen, L. Effects of Vitamin D Supplementation in Patients with Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Int. J. Endocrinol. Metab. 2020, 18, e97205. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, M.; Ma, F.; Guan, M. Role of Steroid Hormones in the Pathogenesis of Nonalcoholic Fatty Liver Disease. Metabolites 2021, 11, 320. https://doi.org/10.3390/metabo11050320
Yang M, Ma F, Guan M. Role of Steroid Hormones in the Pathogenesis of Nonalcoholic Fatty Liver Disease. Metabolites. 2021; 11(5):320. https://doi.org/10.3390/metabo11050320
Chicago/Turabian StyleYang, Meng, Feng Ma, and Min Guan. 2021. "Role of Steroid Hormones in the Pathogenesis of Nonalcoholic Fatty Liver Disease" Metabolites 11, no. 5: 320. https://doi.org/10.3390/metabo11050320
APA StyleYang, M., Ma, F., & Guan, M. (2021). Role of Steroid Hormones in the Pathogenesis of Nonalcoholic Fatty Liver Disease. Metabolites, 11(5), 320. https://doi.org/10.3390/metabo11050320