
Mitochondrial Respiration in Response to Iron Deficiency Anemia: Comparison of Peripheral Blood Mononuclear Cells and Liver

 , , ,
, , ,  and
and
Abstract
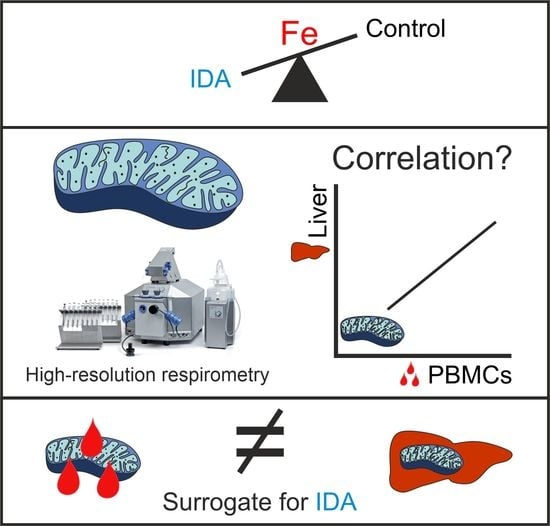
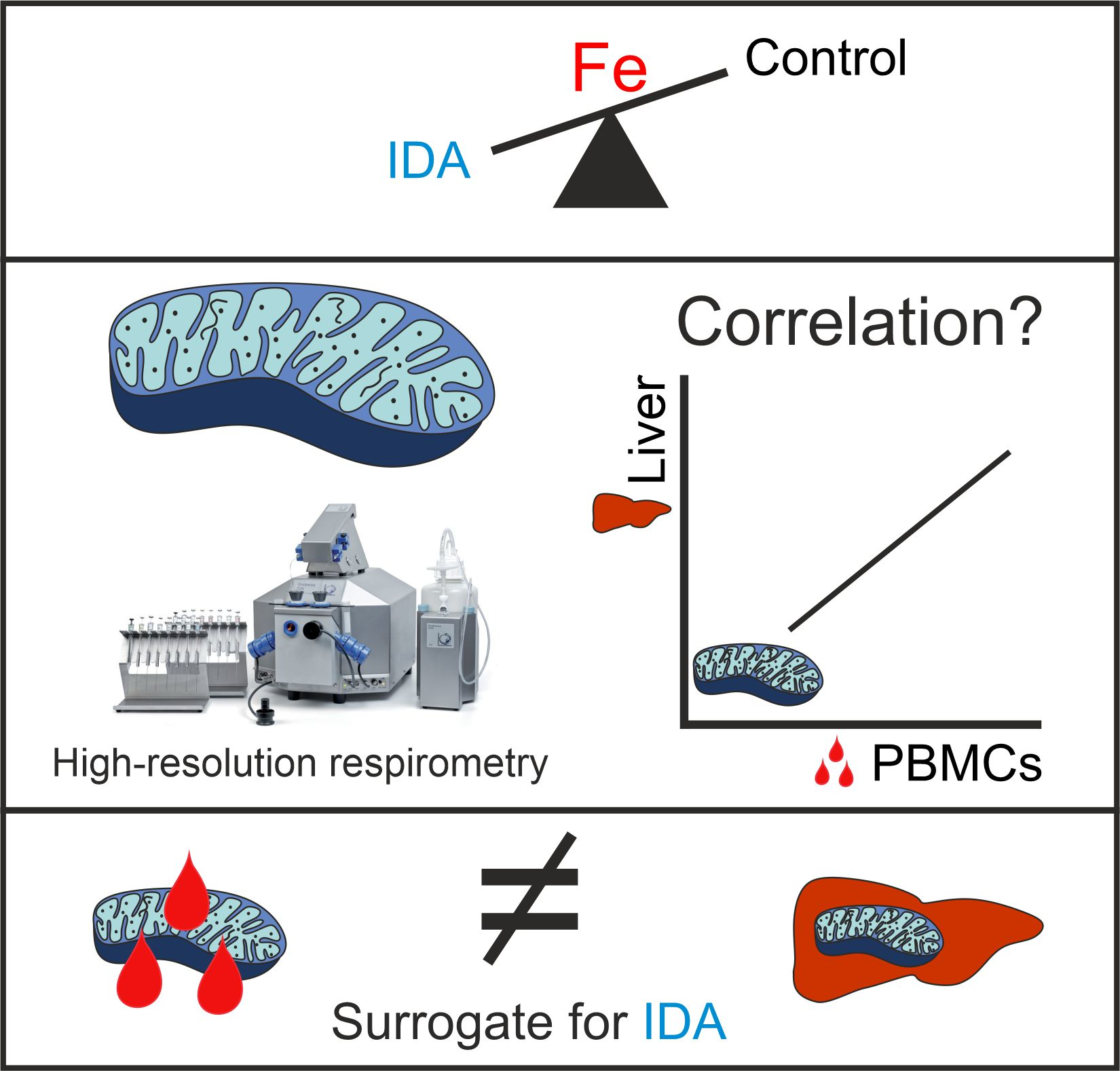
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
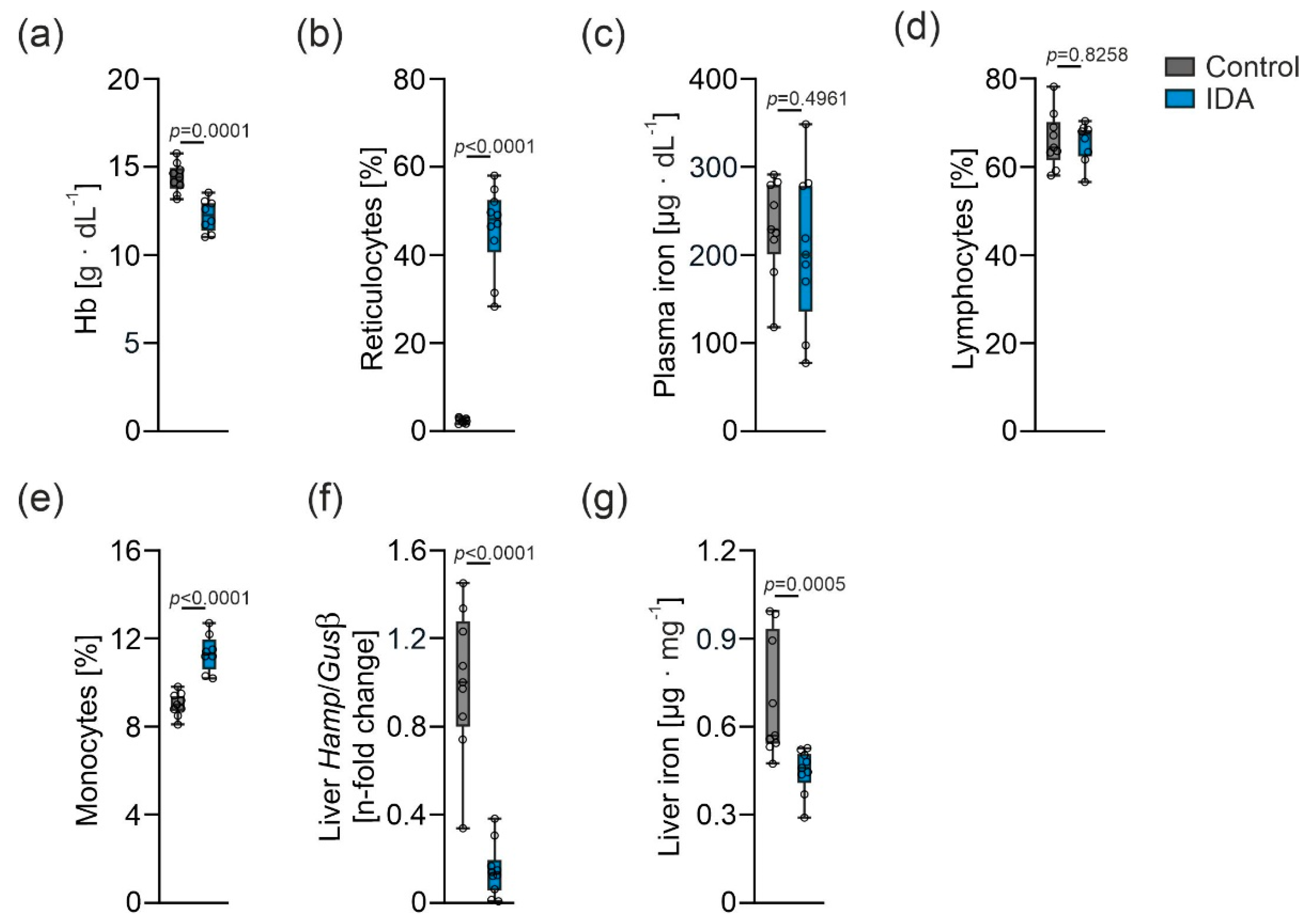
2.1. Effects of IDA on Hematological and Iron Parameters
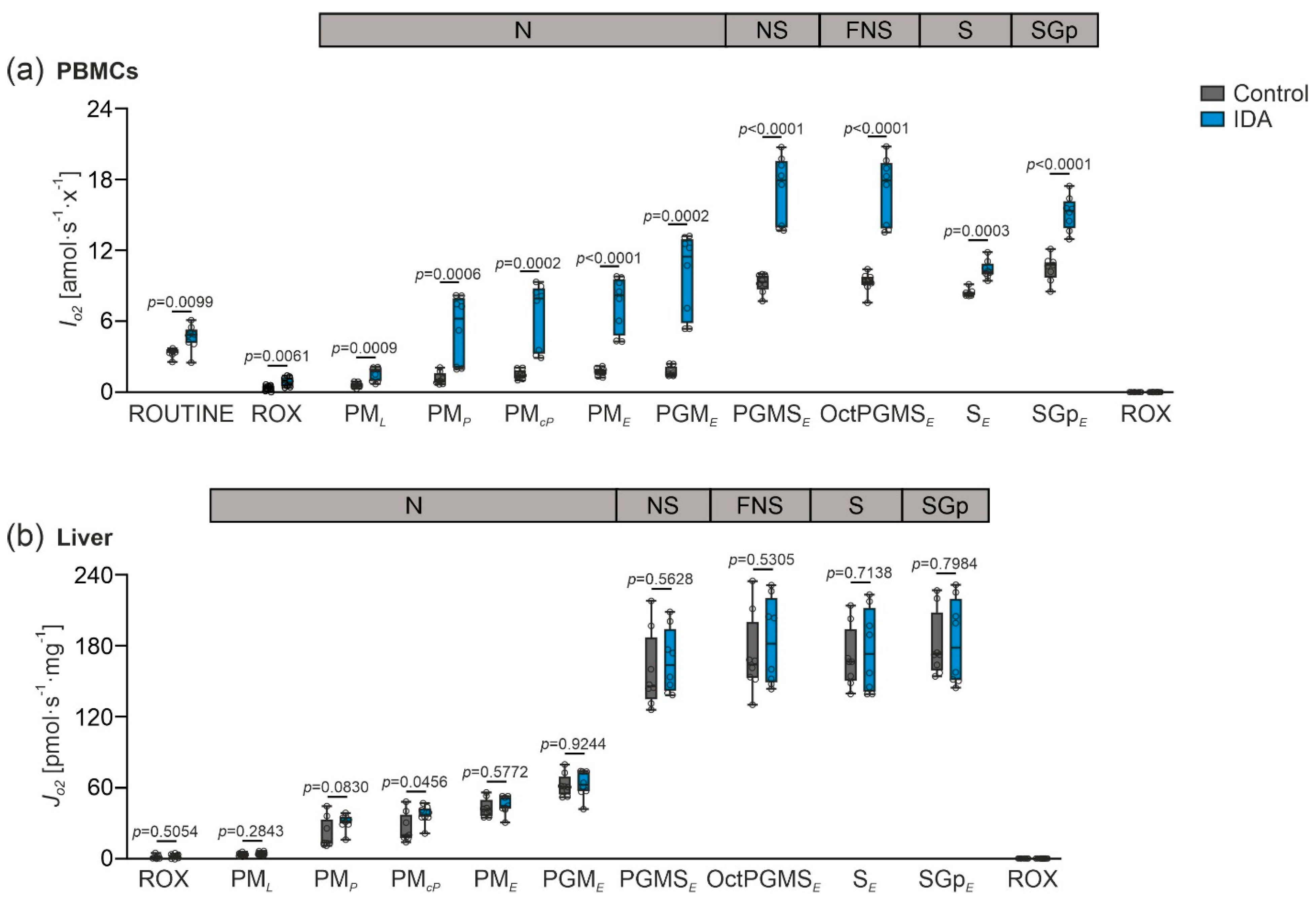
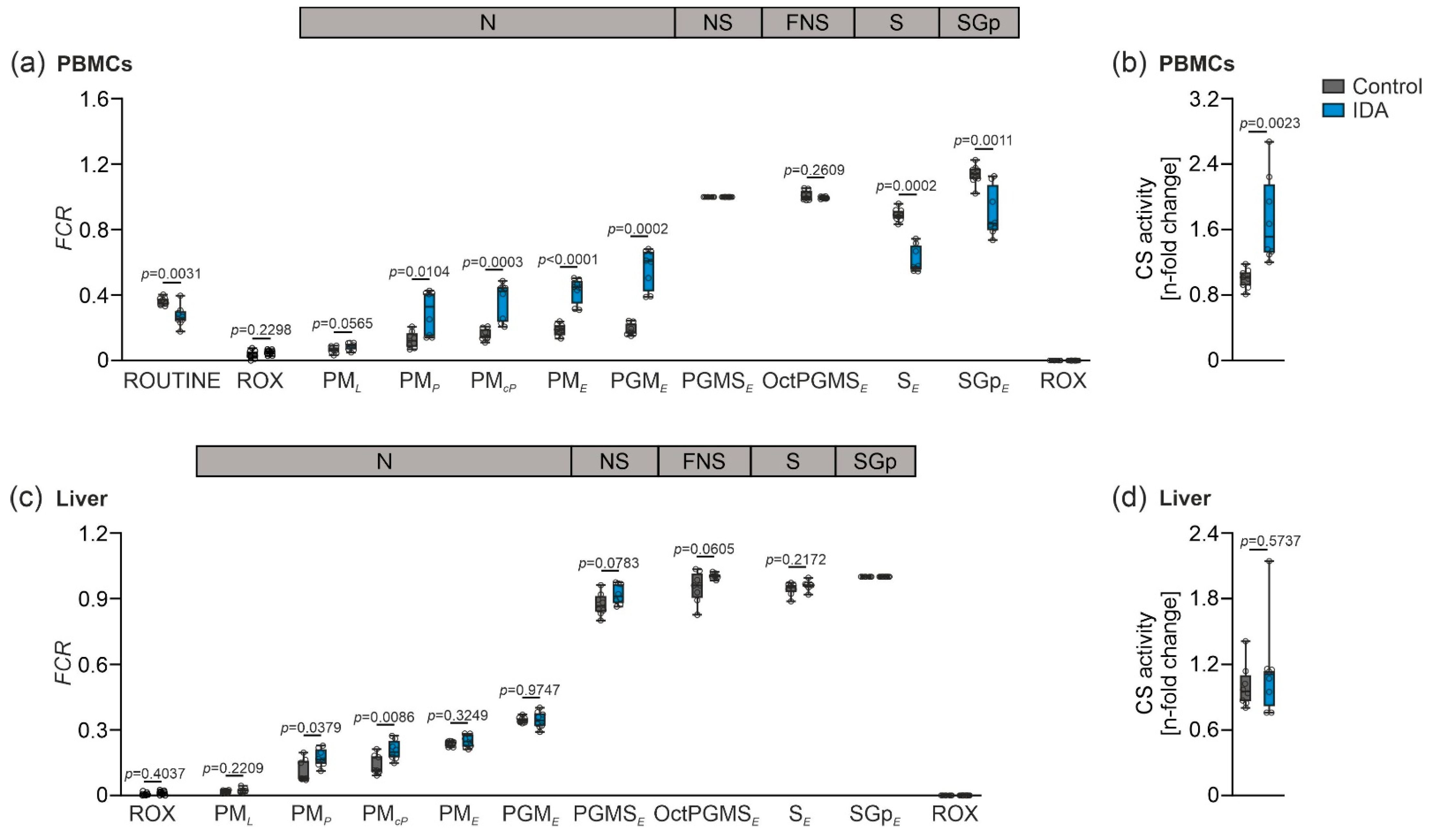
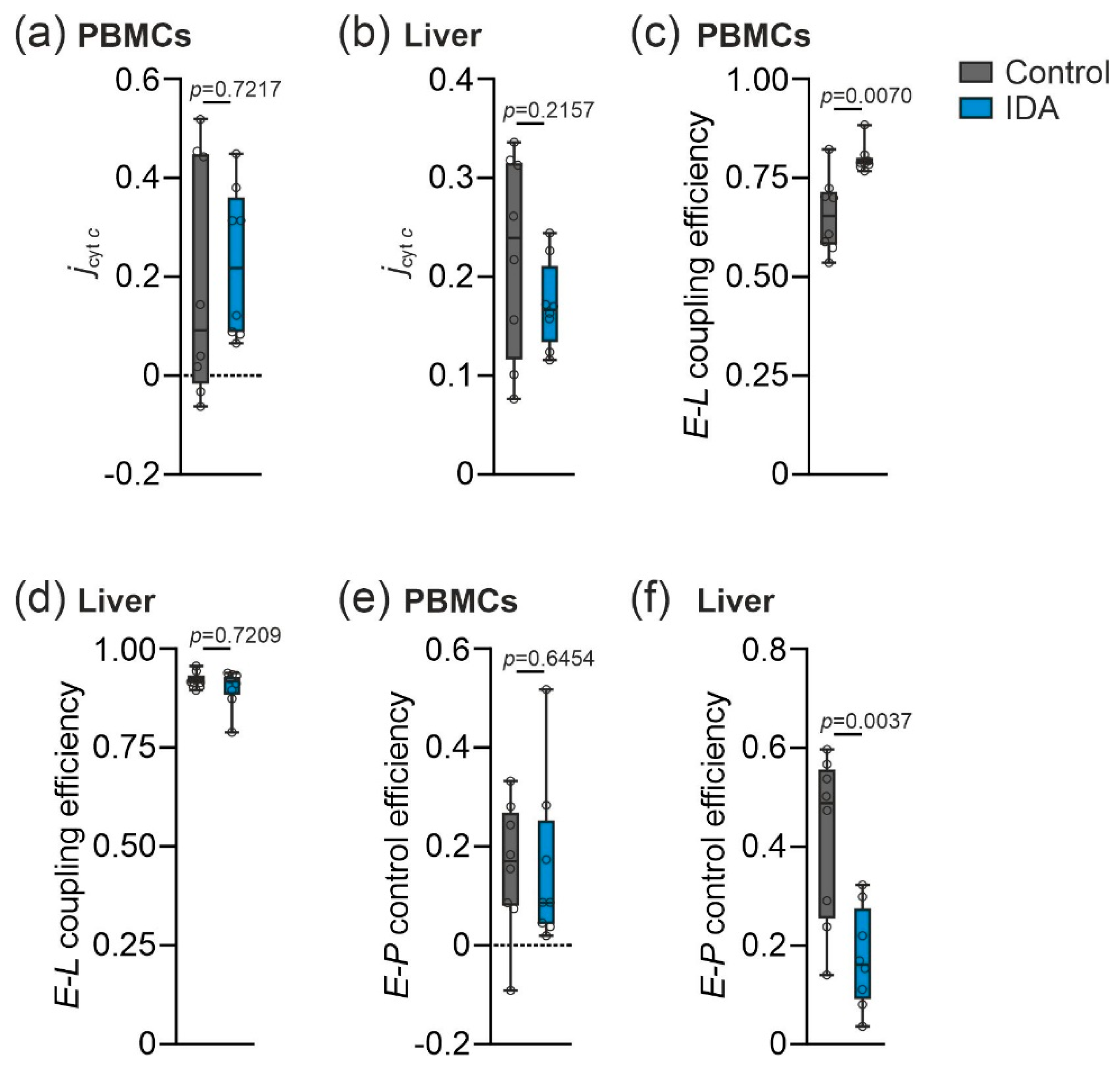
2.2. Mitochondrial Respiration Differs in PBMCs and Livers in Response to IDA
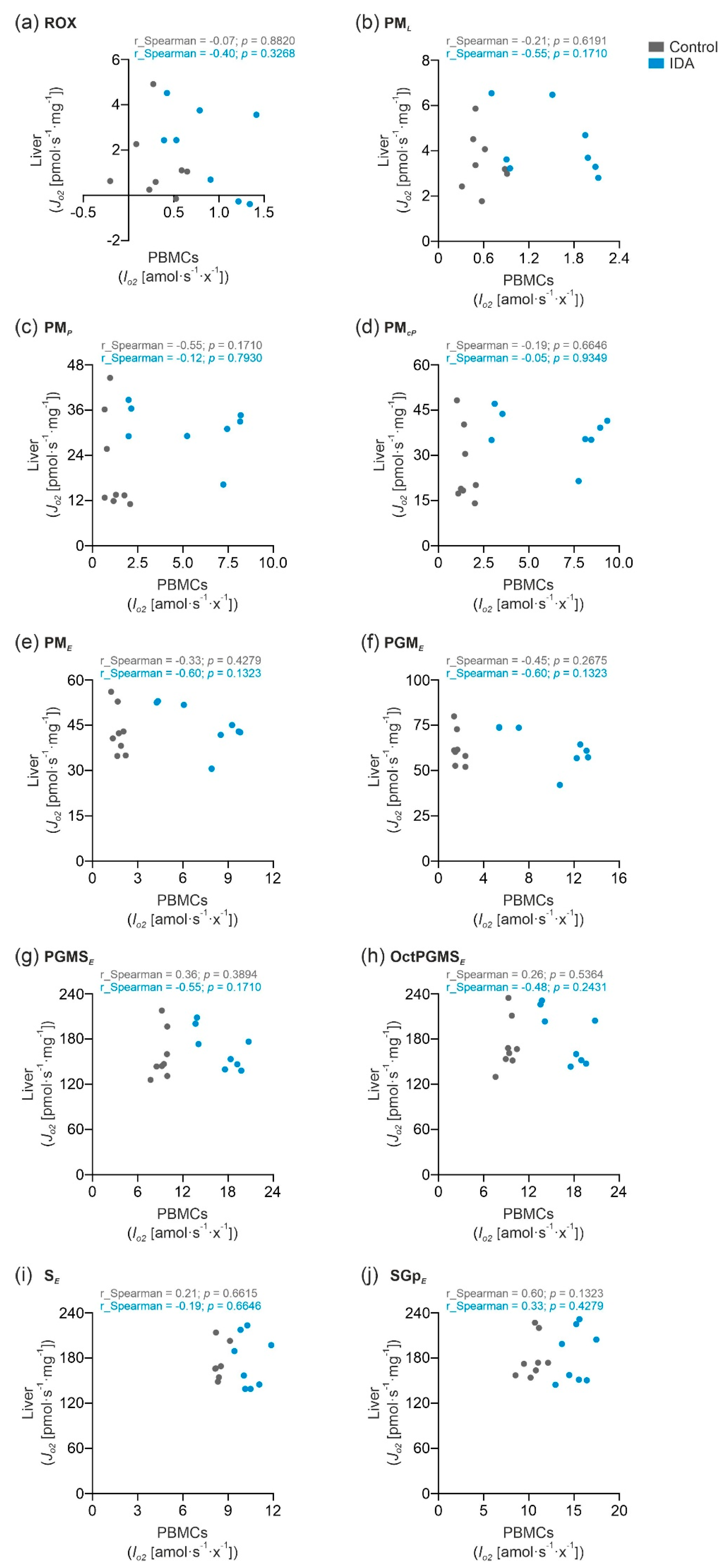
2.3. PBMC Mitochondrial Respiration was Not Correlated with Liver Mitochondrial Respiration
3. Discussion
4. Materials and Methods
4.1. Animal Experiments
4.2. Reticulocyte Quantification
4.3. Isolation of PBMCs
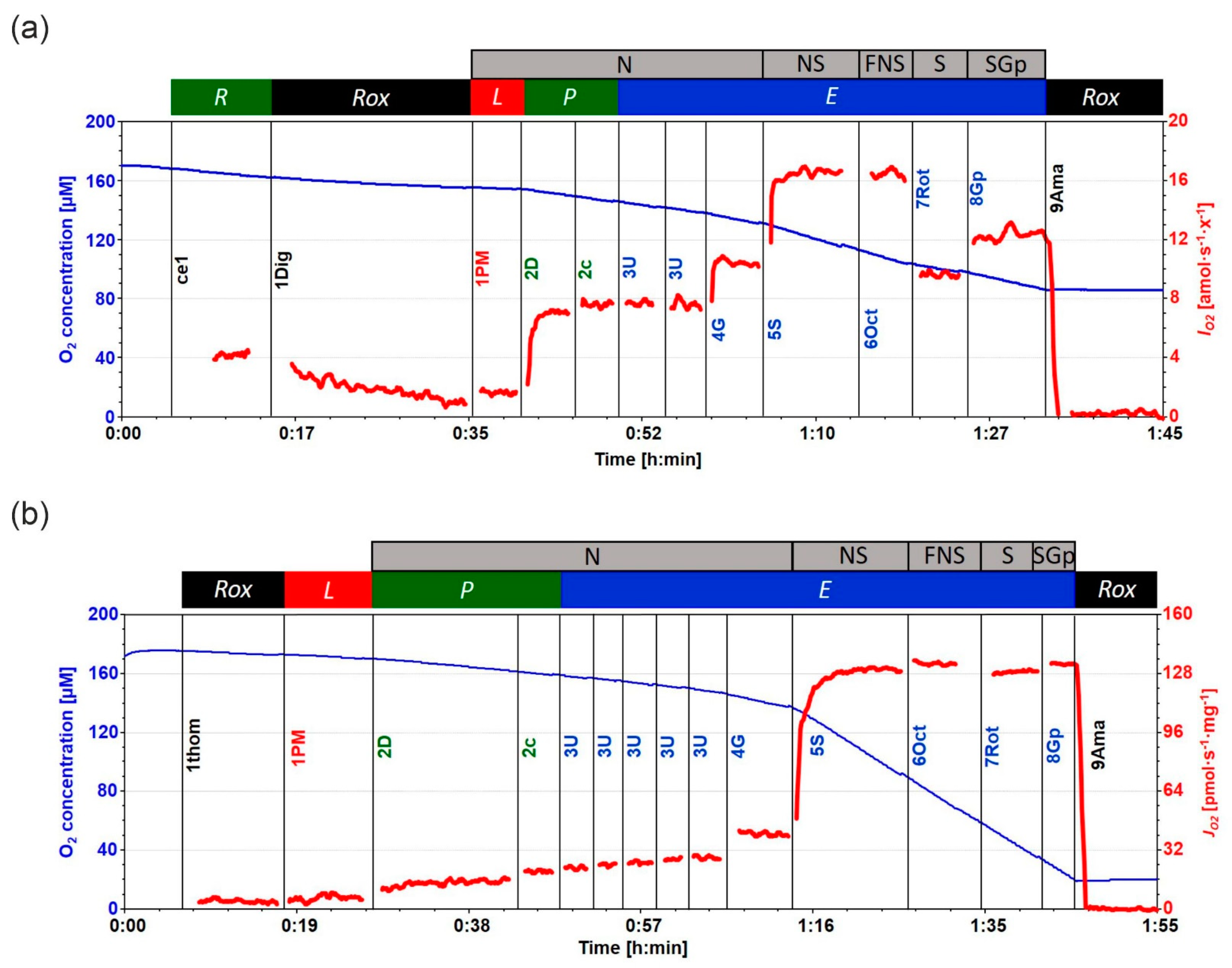
4.4. High-Resolution Respirometry

4.5. Plasma and Total Tissue Iron
4.6. RNA Extraction and Quantitative Real-Time PCR
4.7. CS Activity
4.8. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Camaschella, C. Iron-Deficiency Anemia. N. Engl. J. Med. 2015, 372, 1832–1843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kassebaum, N.J. The Global Burden of Anemia. Hematol. Oncol. Clin. N. Am. 2016, 30, 247–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanser, L.; Burkert, F.R.; Bellmann-Weiler, R.; Schroll, A.; Wildner, S.; Fritsche, G.; Weiss, G. Dynamics in Anemia Development and Dysregulation of Iron Homeostasis in Hospitalized Patients with COVID-19. Metabolites 2021, 11, 653. [Google Scholar] [CrossRef] [PubMed]
- Sachdev, H.P.S.; Gera, T.; Nestel, P. Effect of iron supplementation on mental and motor development in children: Systematic review of randomised controlled trials. Public Health Nutr. 2005, 8, 117–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasricha, S.-R.; Tye-Din, J.; Muckenthaler, M.U.; Swinkels, D.W. Iron deficiency. Lancet 2021, 397, 233–248. [Google Scholar] [CrossRef]
- Muckenthaler, M.U.; Rivella, S.; Hentze, M.W.; Galy, B. A Red Carpet for Iron Metabolism. Cell 2017, 168, 344–361. [Google Scholar] [CrossRef] [Green Version]
- Katsarou, A.; Pantopoulos, K. Basics and principles of cellular and systemic iron homeostasis. Mol. Asp. Med. 2020, 75, 100866. [Google Scholar] [CrossRef]
- Gnaiger, E. Mitochondrial pathways and respiratory control. An introduction to OXPHOS analysis. 5th ed. Bioenerg. Commun. 2020, 2020, 112. [Google Scholar] [CrossRef]
- Gnaiger, E. MitoEAGLE Task Group. Mitochondrial physiology. Bioenerg. Commun. 2020, 2020, 1. [Google Scholar] [CrossRef]
- Lill, R. Function and biogenesis of iron–sulphur proteins. Nature 2009, 460, 831–838. [Google Scholar] [CrossRef]
- Oexle, H.; Gnaiger, E.; Weiss, G. Iron-dependent changes in cellular energy metabolism: Influence on citric acid cycle and oxidative phosphorylation. Biochim. Biophys. Acta (BBA)-Bioenerg. 1999, 1413, 99–107. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Pantopoulos, K. Regulation of cellular iron metabolism. Biochem. J. 2011, 434, 365–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemeth, E.; Ganz, T. Hepcidin-Ferroportin Interaction Controls Systemic Iron Homeostasis. Int. J. Mol. Sci. 2021, 22, 6493. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, V.W.; Jacobus, W.E. Substrate-dependent functional defects and altered mitochondrial respiratory capacity in hearts from guinea pigs with iron deficiency anemia. Biochim. Biophys. Acta (BBA)-Bioenerg. 1987, 891, 103–114. [Google Scholar] [CrossRef]
- Hoes, M.F.; Grote Beverborg, N.; Kijlstra, J.D.; Kuipers, J.; Swinkels, D.W.; Giepmans, B.N.G.; Rodenburg, R.J.; van Veldhuisen, D.J.; de Boer, R.A.; van der Meer, P. Iron deficiency impairs contractility of human cardiomyocytes through decreased mitochondrial function. Eur. J. Heart Fail. 2018, 20, 910–919. [Google Scholar] [CrossRef] [Green Version]
- Cartier, L.J.; Ohira, Y.; Chen, M.; Cuddihee, R.W.; Holloszy, J.O. Perturbation of mitochondrial composition in muscle by iron deficiency. Implications regarding regulation of mitochondrial assembly. J. Biol. Chem. 1986, 261, 13827–13832. [Google Scholar] [CrossRef]
- Rineau, E.; Gaillard, T.; Gueguen, N.; Procaccio, V.; Henrion, D.; Prunier, F.; Lasocki, S. Iron deficiency without anemia is responsible for decreased left ventricular function and reduced mitochondrial complex I activity in a mouse model. Int. J. Cardiol. 2018, 266, 206–212. [Google Scholar] [CrossRef]
- McKay, R.H.; Higuchi, D.A.; Winder, W.W.; Fell, R.D. and Brown, E.B. Tissue effects of iron deficiency in the rat. Biochim. Biophys. Acta (BBA)-Gen. Subj. 1983, 757, 352–358. [Google Scholar] [CrossRef]
- Dallman, P.R.; Goodman, J.R. Enlargement of Mitochondrial Compartment in Iron and Copper Deficiency. Blood 1970, 35, 496–505. [Google Scholar] [CrossRef] [Green Version]
- Dallman, P.R.; Goodman, J.R. The effects of iron deficiency on the hepatocyte: A biochemical and ultrastructural study. J. Cell Biol. 1971, 48, 79–90. [Google Scholar] [CrossRef] [Green Version]
- Haschka, D.; Volani, C.; Stefani, A.; Tymoszuk, P.; Mitterling, T.; Holzknecht, E.; Heidbreder, A.; Coassin, S.; Sumbalova, Z.; Seifert, M.; et al. Association of mitochondrial iron deficiency and dysfunction with idiopathic restless legs syndrome. Mov. Disord. 2019, 34, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Ajaz, S.; McPhail, M.J.; Gnudi, L.; Trovato, F.M.; Mujib, S.; Napoli, S.; Carey, I.; Agarwal, K. Mitochondrial dysfunction as a mechanistic biomarker in patients with non-alcoholic fatty liver disease (NAFLD). Mitochondrion 2021, 57, 119–130. [Google Scholar] [CrossRef]
- Belikova, I.; Lukaszewicz, A.C.; Faivre, V.; Damoisel, C.; Singer, M.; Payen, D. Oxygen consumption of human peripheral blood mononuclear cells in severe human sepsis. Crit. Care Med. 2007, 35, 2702–2708. [Google Scholar] [CrossRef] [PubMed]
- Japiassú, A.M.; Santiago, A.P.; d’Avila, J.C.; Garcia-Souza, L.F.; Galina, A.; Castro Faria-Neto, H.C.; Bozza, F.A.; Oliveira, M.F. Bioenergetic failure of human peripheral blood monocytes in patients with septic shock is mediated by reduced F1Fo adenosine-5′-triphosphate synthase activity. Crit. Care Med. 2011, 39, 1056–1063. [Google Scholar] [CrossRef] [PubMed]
- Theurl, I.; Aigner, E.; Theurl, M.; Nairz, M.; Seifert, M.; Schroll, A.; Sonnweber, T.; Eberwein, L.; Witcher, D.R.; Murphy, A.T.; et al. Regulation of iron homeostasis in anemia of chronic disease and iron deficiency anemia: Diagnostic and therapeutic implications. Blood 2009, 113, 5277–5286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doerrier, C.; Garcia-Souza, L.F.; Krumschnabel, G.; Wohlfarter, Y.; Mészáros, A.T.; Gnaiger, E. High-Resolution FluoRespirometry and OXPHOS Protocols for Human Cells, Permeabilized Fibers from Small Biopsies of Muscle, and Isolated Mitochondria. In Mitochondrial Bioenergetics: Methods and Protocols; Palmeira, C.M., Moreno, A.J., Eds.; Springer: New York, NY, USA, 2018; pp. 31–70. [Google Scholar]
- Volani, C.; Doerrier, C.; Demetz, E.; Haschka, D.; Paglia, G.; Lavdas, A.A.; Gnaiger, E.; Weiss, G. Dietary iron loading negatively affects liver mitochondrial function. Metallomics 2017, 9, 1634–1644. [Google Scholar] [CrossRef] [PubMed]
- Holloszy, J.O.; Oscai, L.B.; Don, I.J.; Molé, P.A. Mitochondrial citric acid cycle and related enzymes: Adaptive response to exercise. Biochem. Biophys. Res. Commun. 1970, 40, 1368–1373. [Google Scholar] [CrossRef]
- Hood, D.A.; Zak, R.; Pette, D. Chronic stimulation of rat skeletal muscle induces coordinate increases in mitochondrial and nuclear mRNAs of cytochrome-c-oxidase subunits. Eur. J. Biochem. 1989, 179, 275–280. [Google Scholar] [CrossRef]
- Williams, R.S.; Salmons, S.; Newsholme, E.A.; Kaufman, R.E.; Mellor, J. Regulation of nuclear and mitochondrial gene expression by contractile activity in skeletal muscle. J. Biol. Chem. 1986, 261, 376–380. [Google Scholar] [CrossRef]
- Gnaiger, E.; Boushel, R.; Søndergaard, H.; Munch-Andersen, T.; Damsgaard, R.; Hagen, C.; Díez-Sánchez, C.; Ara, I.; Wright-Paradis, C.; Schrauwen, P.; et al. Mitochondrial coupling and capacity of oxidative phosphorylation in skeletal muscle of Inuit and Caucasians in the arctic winter. Scand. J. Med. Sci. Sports 2015, 25, 126–134. [Google Scholar] [CrossRef]
- Distante, S.; Eikeland, J.; Pawar, T.; Skinnes, R.; Høie, K.; You, P.; Mørkrid, L.; Eide, L. Blood removal therapy in hereditary hemochromatosis induces a stress response resulting in improved genome integrity. Transfusion 2016, 56, 1435–1441. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, J.H.; Jacobs, A. Morphological abnormalities in lymphocyte mitochondria associated with iron-deficiency anaemia. J. Clin. Pathol. 1974, 27, 973–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theurl, I.; Schroll, A.; Nairz, M.; Seifert, M.; Theurl, M.; Sonnweber, T.; Kulaksiz, H.; Weiss, G. Pathways for the regulation of hepcidin expression in anemia of chronic disease and iron deficiency anemia in vivo. Haematologica 2011, 96, 1761–1769. [Google Scholar] [CrossRef] [PubMed]
- Döhla, J.; Kuuluvainen, E.; Gebert, N.; Amaral, A.; Englund, J.I.; Gopalakrishnan, S.; Konovalova, S.; Nieminen, A.I.; Salminen, E.S.; Torregrosa Muñumer, R.; et al. Metabolic determination of cell fate through selective inheritance of mitochondria. Nat. Cell Biol. 2022, 24, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Fischer, C.; Volani, C.; Komlódi, T.; Seifert, M.; Demetz, E.; Valente de Souza, L.; Auer, K.; Petzer, V.; von Raffay, L.; Moser, P.; et al. Dietary Iron Overload and Hfe−/− Related Hemochromatosis Alter Hepatic Mitochondrial Function. Antioxidants 2021, 10, 1818. [Google Scholar] [CrossRef]
- Volani, C.; Paglia, G.; Smarason, S.V.; Pramstaller, P.P.; Demetz, E.; Pfeifhofer-Obermair, C.; Weiss, G. Metabolic Signature of Dietary Iron Overload in a Mouse Model. Cells 2018, 7, 264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- SUIT-001 O2 ce-pce D003. Available online: https://www.bioblast.at/index.php/SUIT-001_O2_ce-pce_D003 (accessed on 23 February 2022).
- SUIT-001 O2 mt D001. Available online: https://www.bioblast.at/index.php/SUIT-001_O2_mt_D001 (accessed on 23 February 2022).
- Sonnweber, T.; Ress, C.; Nairz, M.; Theurl, I.; Schroll, A.; Murphy, A.T.; Wroblewski, V.; Witcher, D.R.; Moser, P.; Ebenbichler, C.F.; et al. High-fat diet causes iron deficiency via hepcidin-independent reduction of duodenal iron absorption. J. Nutr. Biochem. 2012, 23, 1600–1608. [Google Scholar] [CrossRef]
- Brigo, N.; Pfeifhofer-Obermair, C.; Tymoszuk, P.; Demetz, E.; Engl, S.; Barros-Pinkelnig, M.; Dichtl, S.; Fischer, C.; De Souza, V.L.; Petzer, V.; et al. Cytokine-Mediated Regulation of ARG1 in Macrophages and Its Impact on the Control of Salmonella enterica Serovar Typhimurium Infection. Cells 2021, 10, 1823. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fischer, C.; Valente de Souza, L.; Komlódi, T.; Garcia-Souza, L.F.; Volani, C.; Tymoszuk, P.; Demetz, E.; Seifert, M.; Auer, K.; Hilbe, R.; et al. Mitochondrial Respiration in Response to Iron Deficiency Anemia: Comparison of Peripheral Blood Mononuclear Cells and Liver. Metabolites 2022, 12, 270. https://doi.org/10.3390/metabo12030270
Fischer C, Valente de Souza L, Komlódi T, Garcia-Souza LF, Volani C, Tymoszuk P, Demetz E, Seifert M, Auer K, Hilbe R, et al. Mitochondrial Respiration in Response to Iron Deficiency Anemia: Comparison of Peripheral Blood Mononuclear Cells and Liver. Metabolites. 2022; 12(3):270. https://doi.org/10.3390/metabo12030270
Chicago/Turabian StyleFischer, Christine, Lara Valente de Souza, Timea Komlódi, Luiz F. Garcia-Souza, Chiara Volani, Piotr Tymoszuk, Egon Demetz, Markus Seifert, Kristina Auer, Richard Hilbe, and et al. 2022. "Mitochondrial Respiration in Response to Iron Deficiency Anemia: Comparison of Peripheral Blood Mononuclear Cells and Liver" Metabolites 12, no. 3: 270. https://doi.org/10.3390/metabo12030270
APA StyleFischer, C., Valente de Souza, L., Komlódi, T., Garcia-Souza, L. F., Volani, C., Tymoszuk, P., Demetz, E., Seifert, M., Auer, K., Hilbe, R., Brigo, N., Petzer, V., Asshoff, M., Gnaiger, E., & Weiss, G. (2022). Mitochondrial Respiration in Response to Iron Deficiency Anemia: Comparison of Peripheral Blood Mononuclear Cells and Liver. Metabolites, 12(3), 270. https://doi.org/10.3390/metabo12030270