
Transcriptome Profile Reveals Genetic and Metabolic Mechanisms Related to Essential Fatty Acid Content of Intramuscular Longissimus thoracis in Nellore Cattle


 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. Differentially Expressed Genes Analysis
2.2. Differential Co-Expression Analysis (PCIT—DH)
2.3. Differential Co-Expression Analysis (PCIT—RIF)
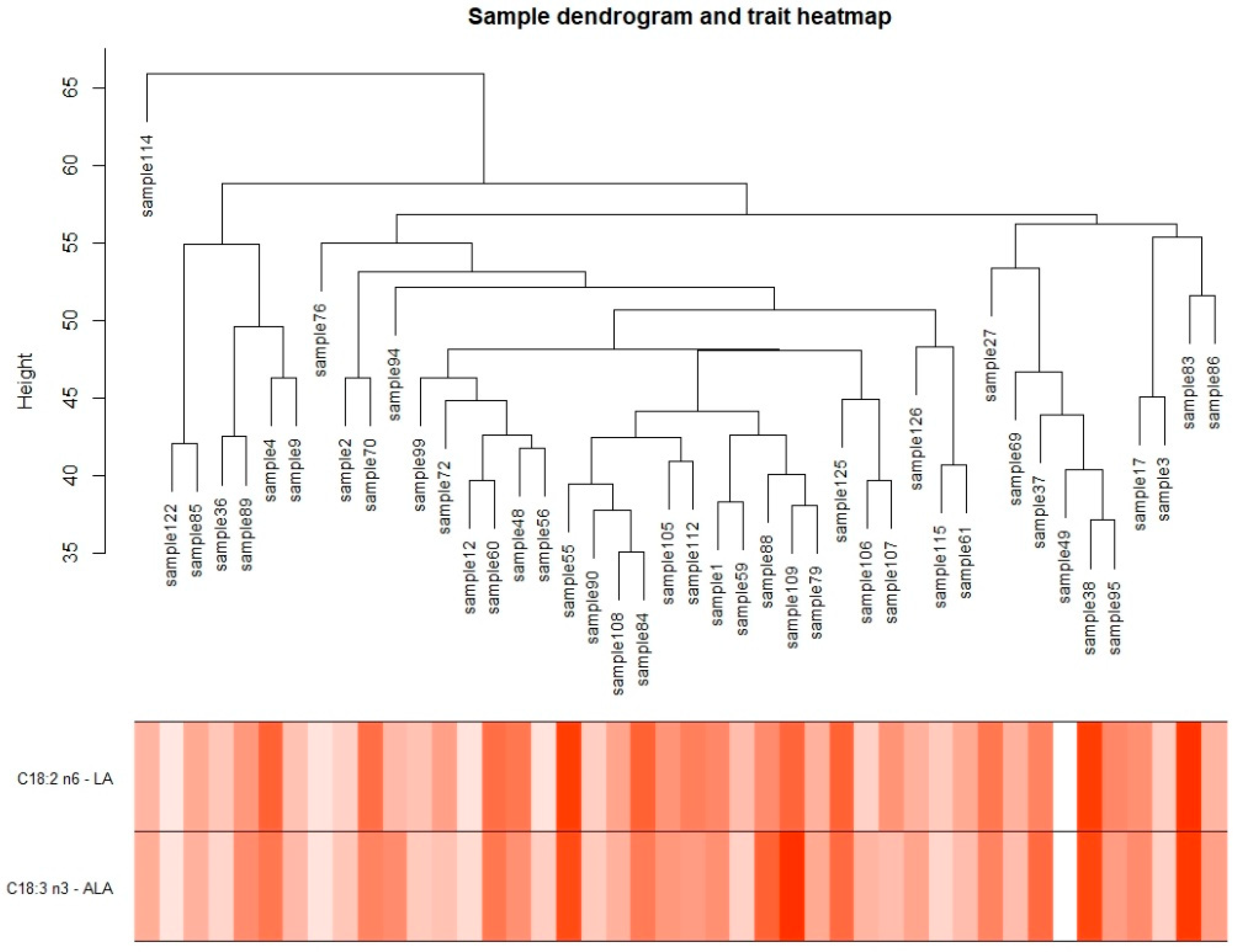
2.4. Differential Co-Expression Analysis (WGCNA)
3. Discussion
3.1. Differentially Expressed Genes
3.2. Differential Co-Expression Analysis (PCIT—DH)
3.3. Differential Co-Expression Analysis (PCIT—RIF)
3.4. Differential Co-Expression Analysis (WGCNA)
4. Materials and Methods
4.1. Animals and Sampling
4.2. Lipid Extraction and Quantification
4.3. Fatty Acid Profile Identification
4.4. RNA Extraction
4.5. Read Alignment and Gene Count
4.6. Differentially Expressed Genes Analysis
4.7. Gene Co-Expresion Analysis: Partial Correlation and Information Theory (PCIT)
4.8. Gene Co-Expresion Analysis: Weighted Gene Co-Expression Network Analysis (WGCNA)
4.9. Functional Enrichment
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Biesalski, H.K. Meat as a Component of a Healthy Diet-Are There Any Risks or Benefits If Meat is Avoided in the Diet? Meat Sci. 2005, 70, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Eilander, A.; Harika, R.K.; Zock, P.L. Intake and Sources of Dietary Fatty Acids in Europe: Are Current Population Intakes of Fats Aligned with Dietary Recommendations? Eur. J. Lipid Sci. Technol. 2015, 117, 1370–1377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, M.; Alves, S.P.; Cappucci, A.; Cook, S.R.; Duarte, A.; Caldeira, R.M.; McAllister, T.A.; Bessa, R.J.B. Effects of Condensed and Hydrolyzable Tannins on Rumen Metabolism with Emphasis on the Biohydrogenation of Unsaturated Fatty Acids. J. Agric. Food Chem. 2018, 66, 3367–3377. [Google Scholar] [CrossRef] [PubMed]
- da Costa, A.S.; Pires, V.M.; Fontes, C.M.; Prates, J.A.M. Expression of Genes Controlling Fat Deposition in Two Genetically Diverse Beef Cattle Breeds Fed High or Low Silage Diets. BMC Vet. Res. 2013, 9, 118. [Google Scholar] [CrossRef] [Green Version]
- Fiorentini, G.; Lage, J.F.; Carvalho, I.P.C.; Messana, J.D.; Canesin, R.C.; Reis, R.A.; Berchielli, T.T. Lipid Sources with Different Fatty Acid Profile Alters the Fatty Acid Profile and Quality of Beef from Confined Nellore Steers. Asian-Australas. J. Anim. Sci. 2015, 28, 976–986. [Google Scholar] [CrossRef] [Green Version]
- Osorio, J.S.; Moisa, S.J. Gene Regulation in Ruminants: A Nutritional Perspective. In Gene Expression and Control; IntechOpen: London, UK, 2019; pp. 1–27. [Google Scholar]
- Aboujaoude, C.; Pereira, A.S.C.; Feitosa, F.L.B.; De Lemos, M.V.A.; Chiaia, H.L.J.; Berton, M.P.; Peripolli, E.; Silva, R.M.D.O.; Ferrinho, A.M.; Mueller, L.F.; et al. Genetic Parameters for Fatty Acids in Intramuscular Fat from Feedlot-Finished Nelore Carcasses. Anim. Prod. Sci. 2016, 58, 234–243. [Google Scholar] [CrossRef] [Green Version]
- Chiaia, H.L.J.; Peripoli, E.; de Oliveira Silva, R.M.; Aboujaoude, C.; Feitosa, F.L.B.; de Lemos, M.V.A.; Berton, M.P.; Olivieri, B.F.; Espigolan, R.; Tonussi, R.L.; et al. Genomic Prediction for Beef Fatty Acid Profile in Nellore Cattle. Meat Sci. 2017, 128, 60–67. [Google Scholar] [CrossRef] [Green Version]
- Cesar, A.S.M.; Regitano, L.C.A.; Mourão, G.B.; Tullio, R.R.; Lanna, D.P.D.; Nassu, R.T.; Mudado, M.A.; Oliveira, P.S.N.; do Nascimento, M.L.; Chaves, A.S.; et al. Genome-Wide Association Study for Intramuscular Fat Deposition and Composition in Nellore Cattle. BMC Genet. 2014, 15, 39. [Google Scholar] [CrossRef] [Green Version]
- Fonseca, L.F.S.; dos Santos Silva, D.B.; Gimenez, D.F.J.; Baldi, F.; Ferro, J.A.; Chardulo, L.A.L.; de Albuquerque, L.G. Gene Expression Profiling and Identification of Hub Genes in Nellore Cattle with Different Marbling Score Levels. Genomics 2020, 112, 873–879. [Google Scholar] [CrossRef]
- Lemos, M.V.A.; Chiaia, H.L.J.; Berton, M.P.; Feitosa, F.L.B.; Aboujaoud, C.; Camargo, G.M.F.; Pereira, A.S.C.; Albuquerque, L.G.; Ferrinho, A.M.; Mueller, L.F.; et al. Genome-Wide Association between Single Nucleotide Polymorphisms with Beef Fatty Acid Profile in Nellore Cattle Using the Single Step Procedure. BMC Genom. 2016, 17, 213. [Google Scholar] [CrossRef] [Green Version]
- Hudson, N.J.; Dalrymple, B.P.; Reverter, A. Beyond Differential Expression: The Quest for Causal Mutations and Effector Molecules Keywords Why Skeletal Muscle ? BMC Genom. 2012, 13, 356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chowdhury, H.A.; Bhattacharyya, D.K.; Kalita, J.K. (Differential) Co-Expression Analysis of Gene Expression: A Survey of Best Practices. IEEE/ACM Trans. Comput. Biol. Bioinform. 2019, 17, 1154–1173. [Google Scholar] [CrossRef]
- Singh, A.J.; Ramsey, S.A.; Filtz, T.M.; Kioussi, C. Differential Gene Regulatory Networks in Development and Disease. Cell. Mol. Life Sci. 2018, 75, 1013–1025. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R Package for Weighted Correlation Network Analysis Peter. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reverter, A.; Chan, E.K.F. Combining Partial Correlation and an Information Theory Approach to the Reversed Engineering of Gene Co-Expression Networks. Bioinformatics 2008, 24, 2491–2497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, T.; Liang, A.; Liang, S.; Ma, X.; Lu, X.; Duan, A.; Pang, C.; Hua, G.; Liu, S.; Campanile, G.; et al. Integrative Analysis of Transcriptome and GWAS Data to Identify the Hub Genes Associated with Milk Yield Trait in Buffalo. Front. Genet. 2019, 10, 36. [Google Scholar] [CrossRef] [Green Version]
- Yan, Z.; Huang, H.; Freebern, E.; Santos, D.J.A.; Dai, D.; Si, J.; Ma, C.; Cao, J.; Guo, G.; Liu, G.E.; et al. Integrating RNA-Seq with GWAS Reveals Novel Insights into the Molecular Mechanism Underpinning Ketosis in Cattle. BMC Genom. 2020, 21, 489. [Google Scholar] [CrossRef]
- Lim, D.; Chai, H.-H.; Lee, S.-H.; Cho, Y.-M.; Choi, J.-W.; Kim, N.-K. Gene Expression Patterns Associated with Peroxisome Proliferator-Activated Receptor (PPAR) Signaling in the Longissimus Dorsi of Hanwoo (Korean Cattle). Asian-Australas. J. Anim. Sci. 2015, 28, 1075–1083. [Google Scholar] [CrossRef] [Green Version]
- Houten, S.M.; Wanders, R.J.A. A General Introduction to the Biochemistry of Mitochondrial Fatty Acid β-Oxidation. J. Inherit. Metab. Dis. 2010, 33, 469–477. [Google Scholar] [CrossRef] [Green Version]
- Schulz, H. Oxidation of Fatty Acids in Eukaryotes. In Biochemistry of Lipids, Lipoproteins and Membranes; Elsevier: Amsterdam, The Netherlands, 2002; pp. 131–154. ISBN 9780444532190. [Google Scholar]
- Rutter, J.; Winge, D.R.; Schiffman, J.D. Succinate Dehydrogenase—Assembly, Regulation and Role in Human Disease. Mitochondrion 2010, 10, 393–401. [Google Scholar] [CrossRef] [Green Version]
- Jeong, J.; Bong, J.; Kim, G.D.; Joo, S.T.; Lee, H.-J.; Baik, M. Transcriptome Changes Favoring Intramuscular Fat Deposition in the Longissimus Muscle Following Castration of Bulls1. J. Anim. Sci. 2013, 91, 4692–4704. [Google Scholar] [CrossRef] [PubMed]
- Rakhshandehroo, M.; Knoch, B.; Müller, M.; Kersten, S. Peroxisome Proliferator-Activated Receptor Alpha Target Genes. PPAR Res. 2010, 2010, 612089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, S.; Xue-Feng, Y.; Hao-Lei, L.; Nian, F.; Yan, O.; Kai, Q. Long-Chain Acyl-CoA Synthetase in Fatty Acid Metabolism Involved in Liver and Other Diseases: An Update. World J. Gastroenterol. 2015, 21, 3492. [Google Scholar] [CrossRef] [PubMed]
- Gross, D.A.; Zhan, C.; Silver, D.L. Direct Binding of Triglyceride to Fat Storage-Inducing Transmembrane Proteins 1 and 2 Is Important for Lipid Droplet Formation. Proc. Natl. Acad. Sci. USA 2011, 108, 19581–19586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadereit, B.; Kumar, P.; Wang, W.-J.; Miranda, D.; Snapp, E.L.; Severina, N.; Torregroza, I.; Evans, T.; Silver, D.L. Evolutionarily Conserved Gene Family Important for Fat Storage. Proc. Natl. Acad. Sci. USA 2008, 105, 94–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bi, X.; Xu, Y.; Li, T.; Li, X.; Li, W.; Shao, W.; Wang, K.; Zhan, G.; Wu, Z.; Liu, W.; et al. RNA Targets Ribogenesis Factor WDR43 to Chromatin for Transcription and Pluripotency Control. Mol. Cell 2019, 75, 102–116.e9. [Google Scholar] [CrossRef]
- Oliveira, P.S.N.; Coutinho, L.L.; Cesar, A.S.M.; Da Silva Diniz, W.J.; De Souza, M.M.; Andrade, B.G.; Koltes, J.E.; Mourão, G.B.; Zerlotini, A.; Reecy, J.M.; et al. Co-Expression Networks Reveal Potential Regulatory Roles of MiRNAs in Fatty Acid Composition of Nelore Cattle. Front. Genet. 2019, 10, 651. [Google Scholar] [CrossRef] [Green Version]
- Lim, D.; Lee, S.H.; Kim, N.K.; Cho, Y.M.; Chai, H.H.; Seong, H.H.; Kim, H. Gene Co-Expression Analysis to Characterize Genes Related to Marbling Trait in Hanwoo (Korean) Cattle. Asian-Australas. J. Anim. Sci. 2013, 26, 19–29. [Google Scholar] [CrossRef]
- Jensen, J.H.; Conley, L.N.; Hedegaard, J.; Nielsen, M.; Young, J.F.; Oksbjerg, N.; Hornshøj, H.; Bendixen, C.; Thomsen, B. Gene Expression Profiling of Porcine Skeletal Muscle in the Early Recovery Phase Following Acute Physical Activity. Exp. Physiol. 2012, 97, 833–848. [Google Scholar] [CrossRef]
- Wang, Y.H.; Bower, N.I.; Reverter, A.; Tan, S.H.; De Jager, N.; Wang, R.; McWilliam, S.M.; Cafe, L.M.; Greenwood, P.L.; Lehnert, S.A. Gene Expression Patterns during Intramuscular Fat Development in Cattle. J. Anim. Sci. 2009, 87, 119–130. [Google Scholar] [CrossRef]
- Muñoz, M.; García-Casco, J.M.; Caraballo, C.; Fernández-Barroso, M.Á.; Sánchez-Esquiliche, F.; Gómez, F.; del Carmen Rodríguez, M.; Silió, L. Identification of Candidate Genes and Regulatory Factors Underlying Intramuscular Fat Content Through Longissimus Dorsi Transcriptome Analyses in Heavy Iberian Pigs. Front. Genet. 2018, 9, 608. [Google Scholar] [CrossRef] [PubMed]
- Howe, V.; Sharpe, L.J.; Alexopoulos, S.J.; Kunze, S.V.; Chua, N.K.; Li, D.; Brown, A.J. Cholesterol Homeostasis: How Do Cells Sense Sterol Excess? Chem. Phys. Lipids 2016, 199, 170–178. [Google Scholar] [CrossRef]
- Huber, M.D.; Vesely, P.W.; Datta, K.; Gerace, L. Erlins Restrict SREBP Activation in the ER and Regulate Cellular Cholesterol Homeostasis. J. Cell Biol. 2013, 203, 427–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, S.P. The Origin Recognition Complex: From Simple Origins to Complex Functions. Genes Dev. 2002, 16, 659–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fluge, Ø.; Bruland, O.; Akslen, L.A.; Varhaug, J.E.; Lillehaug, J.R. NATH, a Novel Gene Overexpressed in Papillary Thyroid Carcinomas. Oncogene 2002, 21, 5056–5068. [Google Scholar] [CrossRef] [Green Version]
- Carmeli, E.; Moas, M.; Reznick, A.Z.; Coleman, R. Matrix Metalloproteinases and Skeletal Muscle: A Brief Review. Muscle Nerve 2004, 29, 191–197. [Google Scholar] [CrossRef]
- Urs, S.; Smith, C.; Campbell, B.; Saxton, A.M.; Taylor, J.; Zhang, B.; Snoddy, J.; Voy, B.J.; Moustaid-Moussa, N. Gene Expression Profiling in Human Preadipocytes and Adipocytes by Microarray Analysis. J. Nutr. 2004, 134, 762–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- dos Santos Silva, D.B.; Fonseca, L.F.S.; Magalhães, A.F.B.; Muniz, M.M.M.; Baldi, F.; Ferro, J.A.; Chardulo, L.A.L.; Pinheiro, D.G.; de Albuquerque, L.G. Transcriptome Profiling of Muscle in Nelore Cattle Phenotypically Divergent for the Ribeye Muscle Area. Genomics 2020, 112, 1257–1263. [Google Scholar] [CrossRef]
- Flanagan, J.N.; Linder, K.; Mejhert, N.; Dungner, E.; Wahlen, K.; Decaunes, P.; Rydén, M.; Bjoörklund, P.; Arver, S.; Bhasin, S.; et al. Role of Follistatin in Promoting Adipogenesis in Women. J. Clin. Endocrinol. Metab. 2009, 94, 3003–3009. [Google Scholar] [CrossRef] [Green Version]
- Tao, W.; Moore, R.; Meng, Y.; Yeasky, T.M.; Smith, E.R.; Xu, X.-X. Disabled-2 Determines Commitment of a Pre-Adipocyte Population in Juvenile Mice. Sci. Rep. 2016, 6, 35947. [Google Scholar] [CrossRef] [Green Version]
- Terrand, J.; Bruban, V.; Zhou, L.; Gong, W.; El Asmar, Z.; May, P.; Zurhove, K.; Haffner, P.; Philippe, C.; Woldt, E.; et al. LRP1 Controls Intracellular Cholesterol Storage and Fatty Acid Synthesis through Modulation of Wnt Signaling. J. Biol. Chem. 2009, 284, 381–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, W.; Moore, R.; Meng, Y.; Smith, E.R.; Xu, X.-X. Endocytic Adaptors Arh and Dab2 Control Homeostasis of Circulatory Cholesterol. J. Lipid Res. 2016, 57, 809–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masson, O.; Chavey, C.; Dray, C.; Meulle, A.; Daviaud, D.; Quilliot, D.; Muller, C.; Valet, P.; Liaudet-Coopman, E. LRP1 Receptor Controls Adipogenesis and is Up-Regulated In Human and Mouse Obese Adipose Tissue. PLoS ONE 2009, 4, e7422. [Google Scholar] [CrossRef] [Green Version]
- Yi, H.-C.; Liu, Y.-L.; You, P.; Pan, J.-S.; Zhou, J.-Y.; Liu, Z.-J.; Zhang, Z.-Y. Overexpression of DEK Gene Is Correlated with Poor Prognosis in Hepatocellular Carcinoma. Mol. Med. Rep. 2015, 11, 1318–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Novera, W.; Zhang, Y.; Deng, L.-W. MLL5 (KMT2E): Structure, Function, and Clinical Relevance. Cell. Mol. Life Sci. 2017, 74, 2333–2344. [Google Scholar] [CrossRef] [PubMed]
- Daguenet, E.; Dujardin, G.; Valcárcel, J. The Pathogenicity of Splicing Defects: Mechanistic Insights into Pre-mRNA Processing Inform Novel Therapeutic Approaches. EMBO Rep. 2015, 16, 1640–1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakkaraju, A.K.K.; Thankappan, R.; Mary, C.; Garrison, J.L.; Taunton, J.; Strub, K. Efficient Secretion of Small Proteins in Mammalian Cells Relies on Sec62-Dependent Posttranslational Translocation. Mol. Biol. Cell 2012, 23, 2712–2722. [Google Scholar] [CrossRef]
- Niendorf, S.; Oksche, A.; Kisser, A.; Loöhler, J.; Prinz, M.; Schorle, H.; Feller, S.; Lewitzky, M.; Horak, I.; Knobeloch, K.-P. Essential Role of Ubiquitin-Specific Protease 8 for Receptor Tyrosine Kinase Stability and Endocytic Trafficking In Vivo. Mol. Cell. Biol. 2007, 27, 5029–5039. [Google Scholar] [CrossRef] [Green Version]
- Waki, H.; Nakamura, M.; Yamauchi, T.; Wakabayashi, K.; Yu, J.; Hirose-Yotsuya, L.; Take, K.; Sun, W.; Iwabu, M.; Okada-Iwabu, M.; et al. Global Mapping of Cell Type–Specific Open Chromatin by FAIRE-Seq Reveals the Regulatory Role of the NFI Family in Adipocyte Differentiation. PLoS Genet. 2011, 7, e1002311. [Google Scholar] [CrossRef]
- Hiraike, Y.; Waki, H.; Yu, J.; Nakamura, M.; Miyake, K.; Nagano, G.; Nakaki, R.; Suzuki, K.; Kobayashi, H.; Yamamoto, S.; et al. NFIA Co-Localizes with PPARγ and Transcriptionally Controls the Brown Fat Gene Program. Nat. Cell Biol. 2017, 19, 1081–1092. [Google Scholar] [CrossRef] [Green Version]
- Ramayo-Caldas, Y.; Mármol-Sánchez, E.; Ballester, M.; Sánchez, J.P.; González-Prendes, R.; Amills, M.; Quintanilla, R. Integrating Genome-Wide Co-Association and Gene Expression to Identify Putative Regulators and Predictors of Feed Efficiency in Pigs. Genet. Sel. Evol. 2019, 51, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandre, P.A.; Kogelman, L.J.A.; Santana, M.H.A.; Passarelli, D.; Pulz, L.H.; Fantinato-Neto, P.; Silva, P.L.; Leme, P.R.; Strefezzi, R.F.; Coutinho, L.L.; et al. Liver Transcriptomic Networks Reveal Main Biological Processes Associated with Feed Efficiency in Beef Cattle. BMC Genom. 2015, 16, 1073. [Google Scholar] [CrossRef] [Green Version]
- Brunes, L.C.; Baldi, F.; Lopes, F.B.; Lôbo, R.B.; Espigolan, R.; Costa, M.F.O.; Stafuzza, N.B.; Magnabosco, C.U. Weighted Single-step Genome-wide Association Study and Pathway Analyses for Feed Efficiency Traits in Nellore Cattle. J. Anim. Breed. Genet. 2020, 138, 23–44. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, J. The Novel Function of HINFP as a Co-Activator in Sterol-Regulated Transcription of PCSK9 in HepG2 Cells. Biochem. J. 2012, 443, 757–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishi-Tatsumi, M.; Yahagi, N.; Takeuchi, Y.; Toya, N.; Takarada, A.; Murayama, Y.; Aita, Y.; Sawada, Y.; Piao, X.; Oya, Y.; et al. A Key Role of Nuclear Factor Y in the Refeeding Response of Fatty Acid Synthase in Adipocytes. FEBS Lett. 2017, 591, 965–978. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.H.; An, S.M.; Ye, B.J.; Lee, J.H.; Yoo, E.J.; Jeong, G.W.; Kang, H.J.; Alfadda, A.A.; Lim, S.W.; Park, J.; et al. TonEBP/NFAT5 Promotes Obesity and Insulin Resistance by Epigenetic Suppression of White Adipose Tissue Beiging. Nat. Commun. 2019, 10, 3536. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Zhang, H.; Yuan, L.; Jing, H.; Thacker, P.; Li, D. CREBL2, Interacting with CREB, Induces Adipogenesis in 3T3-L1 Adipocytes. Biochem. J. 2011, 439, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Nishizuka, M.; Tsuchiya, T.; Nishihara, T.; Imagawa, M. Induction of Bach1 and ARA70 Gene Expression at an Early Stage of Adipocyte Differentiation of Mouse 3T3-L1 Cells. Biochem. J. 2002, 361, 629. [Google Scholar] [CrossRef]
- Laity, J.H.; Lee, B.M.; Wright, P.E. Zinc Finger Proteins: New Insights into Structural and Functional Diversity. Curr. Opin. Struct. Biol. 2001, 11, 39–46. [Google Scholar] [CrossRef]
- Vivas-García, Y.; Falletta, P.; Liebing, J.; Louphrasitthiphol, P.; Feng, Y.; Chauhan, J.; Scott, D.A.; Glodde, N.; Chocarro-Calvo, A.; Bonham, S.; et al. Lineage-Restricted Regulation of SCD and Fatty Acid Saturation by MITF Controls Melanoma Phenotypic Article Lineage-Restricted Regulation of SCD and Fatty Acid Saturation by MITF Controls Melanoma Phenotypic Plasticity. Mol. Cell 2020, 77, 120–137. [Google Scholar] [CrossRef]
- Lowe, C.E.; Dennis, R.J.; Obi, U.; O’Rahilly, S.; Rochford, J.J. Investigating the Involvement of the ATF6α Pathway of the Unfolded Protein Response in Adipogenesis. Int. J. Obes. 2012, 36, 1248–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, W.; Takagi, T.; Kanesashi, S.; Kurahashi, T.; Nomura, T.; Harada, J.; Ishii, S. Schnurri-2 Controls BMP-Dependent Adipogenesis via Interaction with Smad Proteins. Dev. Cell 2006, 10, 461–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, P.; Nixon, S.J.; Parton, R.G.; Muscat, G.E.O. RORa Regulates the Expression of Genes Involved in Lipid Homeostasis in Skeletal Muscle Cells. J. Biol. Chem. 2004, 279, 36828–36840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reith, W.; Durand, B.; Barras, E.; Mach, B. Function of Major Histocompatibility Complex Class II Promoters Requires Cooperative Binding between Factors RFX and NF-Y. Proc. Natl. Acad. Sci. USA 1994, 91, 554–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Júnior, G.A.F.; Costa, R.B.; de Camargo, G.M.F.; Carvalheiro, R.; Rosa, G.J.M.; Baldi, F.; Garcia, D.A.; Gordo, D.G.M.; Espigolan, R.; Takada, L.; et al. Genome Scan for Postmortem Carcass Traits in Nellore Cattle1. J. Anim. Sci. 2016, 94, 4087–4095. [Google Scholar] [CrossRef]
- Huang, L.; Yu, Z.; Zhang, T.; Zhao, X.; Huang, G. HSP40 Interacts with Pyruvate Kinase M2 and Regulates Glycolysis and Cell Proliferation in Tumor Cells. PLoS ONE 2014, 9, e92949. [Google Scholar] [CrossRef]
- Liang, F.; Li, Q.; Li, X.; Li, Z.; Gong, Z.; Deng, H.; Xiang, B.; Zhou, M.; Li, X.; Li, G.; et al. TSC22D2 Interacts with PKM2 and Inhibits Cell Growth in Colorectal Cancer. Int. J. Oncol. 2016, 49, 1046–1056. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.-B.; Shi, C.; Yang, B.; Lu, Z.-F.; Wu, Y.-L.; Zhang, R.-Y.; He, X.; Li, L.-M.; Hu, B.; Hu, Y.-W.; et al. Long Noncoding RNA ZNF800 Suppresses Proliferation and Migration of Vascular Smooth Muscle Cells by Upregulating PTEN and Inhibiting AKT/MTOR/HIF-1α Signaling. Atherosclerosis 2020, 312, 43–53. [Google Scholar] [CrossRef]
- Lei, D.; Hu, G.; Chen, Y.; Hao, T.; Gao, Y.; Luo, F. Forkhead Box S1 Inhibits the Progression of Hepatocellular Carcinoma. OncoTargets Ther. 2020, 13, 11839–11848. [Google Scholar] [CrossRef]
- Dambara, A.; Morinaga, T.; Fukuda, N.; Yamakawa, Y.; Kato, T.; Enomoto, A.; Asai, N.; Murakumo, Y.; Matsuo, S.; Takahashi, M. Nucleolin Modulates the Subcellular Localization of GDNF-Inducible Zinc Finger Protein 1 and Its Roles in Transcription and Cell Proliferation. Exp. Cell Res. 2007, 313, 3755–3766. [Google Scholar] [CrossRef]
- Borges, J.; TorÍo-Padrón, N.; Momeni, A.; Mueller, M.C.; Tegtmeier, F.T.; Stark, B.G. Adipose Precursor Cells (Preadipocytes) Induce Formation of New Vessels in Fibrin Glue on the Newly Developed Cylinder Chorioallantoic Membrane Model (CAM). Minim. Invasive Ther. Allied Technol. 2006, 15, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Kotarba, G.; Krzywinska, E.; Grabowska, A.I.; Taracha, A.; Wilanowski, T. TFCP2/TFCP2L1/UBP1 Transcription Factors in Cancer. Cancer Lett. 2018, 420, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Jack, B.H.A.; Crossley, M. GATA Proteins Work Together with Friend of GATA (FOG) and C-Terminal Binding Protein (CTBP) Co-Regulators to Control Adipogenesis. J. Biol. Chem. 2010, 285, 32405–32414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cesar, A.S.M.; Regitano, L.C.A.; Koltes, J.E.; Fritz-Waters, E.R.; Lanna, D.P.D.; Gasparin, G.; Mourão, G.B.; Oliveira, P.S.N.; Reecy, J.M.; Coutinho, L.L. Putative Regulatory Factors Associated with Intramuscular Fat Content. PLoS ONE 2015, 10, e0128350. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.-J.; Yang, D.-R.; Yang, G.; Lin, C.-Y.; Chang, H.-C.; Li, G.; Chang, C. TR2 and TR4 Orphan Nuclear Receptors. In Current Topics in Developmental Biology; Elsevier Inc.: Amsterdam, The Netherlands, 2017; Volume 125, pp. 357–373. [Google Scholar]
- Wenzel, J.J.; Piehler, A.; Kaminski, W.E. ABC A-Subclass Proteins: Gatekeepers of Cellular Phospho- and Sphingolipid Transport. Front. Biosci. 2007, 12, 3177. [Google Scholar] [CrossRef]
- Lefterova, M.I.; Lazar, M.A. New Developments in Adipogenesis. Trends Endocrinol. Metab. 2009, 20, 107–114. [Google Scholar] [CrossRef]
- Roh, S.G.; Hishikawa, D.; Hong, Y.H.; Sasaki, S. Control of Adipogenesis in Ruminants. Anim. Sci. J. 2006, 77, 472–477. [Google Scholar] [CrossRef]
- Sevane, N.; Armstrong, E.; Cortés, O.; Wiener, P.; Wong, R.P.; Dunner, S. Association of Bovine Meat Quality Traits with Genes Included in the PPARG and PPARGC1A Networks. Meat Sci. 2013, 94, 328–335. [Google Scholar] [CrossRef]
- Jeong, J.; Kwon, E.G.; Im, S.K.; Seo, K.S.; Baik, M. Expression of Fat Deposition and Fat Removal Genes Is Associated with Intramuscular Fat Content in Longissimus Dorsi Muscle of Korean Cattle Steers1. J. Anim. Sci. 2012, 90, 2044–2053. [Google Scholar] [CrossRef]
- Chen, L.; Ekine-Dzivenu, C.; Vinsky, M.; Basarab, J.; Aalhus, J.; Dugan, M.E.R.; Fitzsimmons, C.; Stothard, P.; Li, C. Genome-Wide Association and Genomic Prediction of Breeding Values for Fatty Acid Composition in Subcutaneous Adipose and Longissimus Lumborum Muscle of Beef Cattle. BMC Genet. 2015, 16, 135. [Google Scholar] [CrossRef] [Green Version]
- Ralston, J.C.; Matravadia, S.; Gaudio, N.; Holloway, G.P.; Mutch, D.M. Polyunsaturated Fatty Acid Regulation of Adipocyte FADS1 and FADS2 Expression and Function. Obesity 2015, 23, 725–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, H.; Nogi, T.; Tabuchi, I.; Oyama, K. The SNPs in the Promoter Regions of the Bovine FADS2 and FABP4 Genes Are Associated with Beef Quality Traits. Livest. Sci. 2014, 163, 34–40. [Google Scholar] [CrossRef]
- Leonard, A.E.; Bobik, E.G.; Dorado, J.; Kroeger, P.E.; Chuang, L.-T.; Thurmond, J.M.; Parker-Barnes, J.M.; Das, T.; Huang, Y.-S.; Mukerji, P. Cloning of a Human CDNA Encoding a Novel Enzyme Involved in the Elongation of Long-Chain Polyunsaturated Fatty Acids. Biochem. J. 2000, 350, 765. [Google Scholar] [CrossRef] [PubMed]
- Wakil, S.J.; Abu-Elheiga, L.A. Fatty Acid Metabolism: Target for Metabolic Syndrome. J. Lipid Res. 2009, 50, S138–S143. [Google Scholar] [CrossRef] [Green Version]
- Cotter, D.G.; Schugar, R.C.; Crawford, P.A. Ketone Body Metabolism and Cardiovascular Disease. Am. J. Physiol. Circ. Physiol. 2013, 304, H1060–H1076. [Google Scholar] [CrossRef] [Green Version]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef]
- Folch, J.; Less, M.; Stanley, G.H.S. A Simple Method for the Isolation and Purification of Total Lipides from Animal Tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [CrossRef]
- Kramer, J.K.G.; Fellner, V.; Dugan, M.E.R.; Sauer, F.D.; Mossoba, M.M.; Yurawecz, M.P. Evaluating Acid and Base Catalysts in the Methylation of Milk and Rumen Fatty Acids with Special Emphasis on Conjugated Dienes and Total Trans Fatty Acids. Lipids 1997, 32, 1219–1228. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python Framework to Work with High-Throughput Sequencing Data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Zabell, S.L. On Student’s 1908 Article “The Probable Error of a Mean”. J. Am. Stat. Assoc. 2008, 103, 1–7. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Watson-Haigh, N.S.; Kadarmideen, H.N.; Reverter, A. PCIT: An R Package for Weighted Gene Co-Expression Networks Based on Partial Correlation and Information Theory Approaches. Bioinformatics 2010, 26, 411–413. [Google Scholar] [CrossRef] [Green Version]
- Reverter, A.; Hudson, N.J.; Nagaraj, S.H.; Pérez-Enciso, M.; Dalrymple, B.P. Regulatory Impact Factors: Unraveling the Transcriptional Regulation of Complex Traits from Expression Data. Bioinformatics 2010, 26, 896–904. [Google Scholar] [CrossRef]
- Hu, H.; Miao, Y.-R.; Jia, L.-H.; Yu, Q.-Y.; Zhang, Q.; Guo, A.-Y. AnimalTFDB 3.0: A Comprehensive Resource for Annotation and Prediction of Animal Transcription Factors. Nucleic Acids Res. 2019, 47, D33–D38. [Google Scholar] [CrossRef]
- Zhang, B.; Horvath, S. A General Framework for Weighted Gene Co-Expression Network Analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4, 17. [Google Scholar] [CrossRef]
- Yip, A.M.; Horvath, S. Gene Network Interconnectedness and the Generalized Topological Overlap Measure. BMC Bioinform. 2007, 8, 22. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and Integrative Analysis of Large Gene Lists Using DAVID Bioinformatics Resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics Enrichment Tools: Paths toward the Comprehensive Functional Analysis of Large Gene Lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| Groups | Gene Symbol | DH | Groups | Gene Symbol | DH |
|---|---|---|---|---|---|
| WDR43 | 68 | ASB5 | 40 | ||
| ASB5 | 63 | TRAFD1 | 35 | ||
| LA-H | ORC4 | 60 | ALA-H | WDR43 | 31 |
| ERLIN1 | 57 | NAA15 | 30 | ||
| TRAFD1 | 50 | ERLIN1 | 30 | ||
| FSTL1 | −70 | DEK | −91 | ||
| FN1 | −66 | PRPF38B | −89 | ||
| LA-L | DAB2 | −63 | ALA-L | KMT2E | −77 |
| LRP1 | −62 | USP8 | −77 | ||
| MMP14 | −60 | SEC62 | −75 |
| Groups | Gene Symbol | RIF1 | RIF2 |
|---|---|---|---|
| RIF1 (+) | ZNF134 | 4.91 | −1.01 |
| NFIA | 4.50 | −0.39 | |
| NFYA | 4.07 | −0.84 | |
| NFAT5 | 4.02 | 0.78 | |
| ZNF584 | 3.54 | 0.07 | |
| RIF2 (+) | ZBTB43 | −0.46 | 2.95 |
| ZNF473 | −0.09 | 2.83 | |
| SLC2A4RG | −0.34 | 2.77 | |
| BACH1 | −0.17 | 2.73 | |
| NFYB | 2.87 | 2.68 | |
| RIF2 (−) | HINFP | 1.02 | −2.70 |
| WIZ | −0.63 | −2.65 |
| Groups | Gene Symbol | RIF1 | RIF2 |
|---|---|---|---|
| RIF1 (+) | NFIA | 7.43 | −1.05 |
| DNAJC1 | 5.11 | 0.67 | |
| RORA | 4.94 | 0.46 | |
| NFX1 | 4.80 | 2.27 | |
| MITF | 4.75 | 1.14 | |
| RIF1 (−) | FOXS1 | −3.18 | 0.15 |
| UBP1 | −2.81 | 0.14 | |
| GZF1 | −2.68 | 0.73 | |
| RIF2 (+) | TSC22D2 | −0.46 | 2.95 |
| ZNF800 | −0.09 | 2.83 | |
| ATF6 | −0.34 | 2.75 | |
| HIVEP2 | −0.17 | 2.73 | |
| ZNF473 | 2.87 | 2.68 | |
| RIF2 (−) | NR2C1 | 0.16 | −3.57 |
| HINFP | −0.32 | −2.68 | |
| ZFPM1 | 0.19 | −2.65 |
| Fatty Acid | Low Group (n = 15) | High Group (n = 15) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Min | Max | Mean | SD | Min | Max | Mean | SD | p-Value | |
| LA | 2.47 | 5.79 | 4.57 | 0.89 | 8.00 | 11.83 | 9.39 | 1.19 | * |
| ALA | 0.23 | 0.55 | 0.46 | 0.08 | 0.78 | 1.21 | 0.94 | 0.14 | * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schettini, G.P.; Peripolli, E.; Alexandre, P.A.; dos Santos, W.B.; Pereira, A.S.C.; de Albuquerque, L.G.; Baldi, F.; Curi, R.A. Transcriptome Profile Reveals Genetic and Metabolic Mechanisms Related to Essential Fatty Acid Content of Intramuscular Longissimus thoracis in Nellore Cattle. Metabolites 2022, 12, 471. https://doi.org/10.3390/metabo12050471
Schettini GP, Peripolli E, Alexandre PA, dos Santos WB, Pereira ASC, de Albuquerque LG, Baldi F, Curi RA. Transcriptome Profile Reveals Genetic and Metabolic Mechanisms Related to Essential Fatty Acid Content of Intramuscular Longissimus thoracis in Nellore Cattle. Metabolites. 2022; 12(5):471. https://doi.org/10.3390/metabo12050471
Chicago/Turabian StyleSchettini, Gustavo Pimenta, Elisa Peripolli, Pâmela Almeida Alexandre, Wellington Bizarria dos Santos, Angélica Simone Cravo Pereira, Lúcia Galvão de Albuquerque, Fernando Baldi, and Rogério Abdallah Curi. 2022. "Transcriptome Profile Reveals Genetic and Metabolic Mechanisms Related to Essential Fatty Acid Content of Intramuscular Longissimus thoracis in Nellore Cattle" Metabolites 12, no. 5: 471. https://doi.org/10.3390/metabo12050471
APA StyleSchettini, G. P., Peripolli, E., Alexandre, P. A., dos Santos, W. B., Pereira, A. S. C., de Albuquerque, L. G., Baldi, F., & Curi, R. A. (2022). Transcriptome Profile Reveals Genetic and Metabolic Mechanisms Related to Essential Fatty Acid Content of Intramuscular Longissimus thoracis in Nellore Cattle. Metabolites, 12(5), 471. https://doi.org/10.3390/metabo12050471