
Evolution of Natural Product Scaffolds as Potential Proteasome Inhibitors in Developing Cancer Therapeutics

 ,
,  , ,
, ,  ,
,  ,
,  and
and
Abstract
:1. Introduction
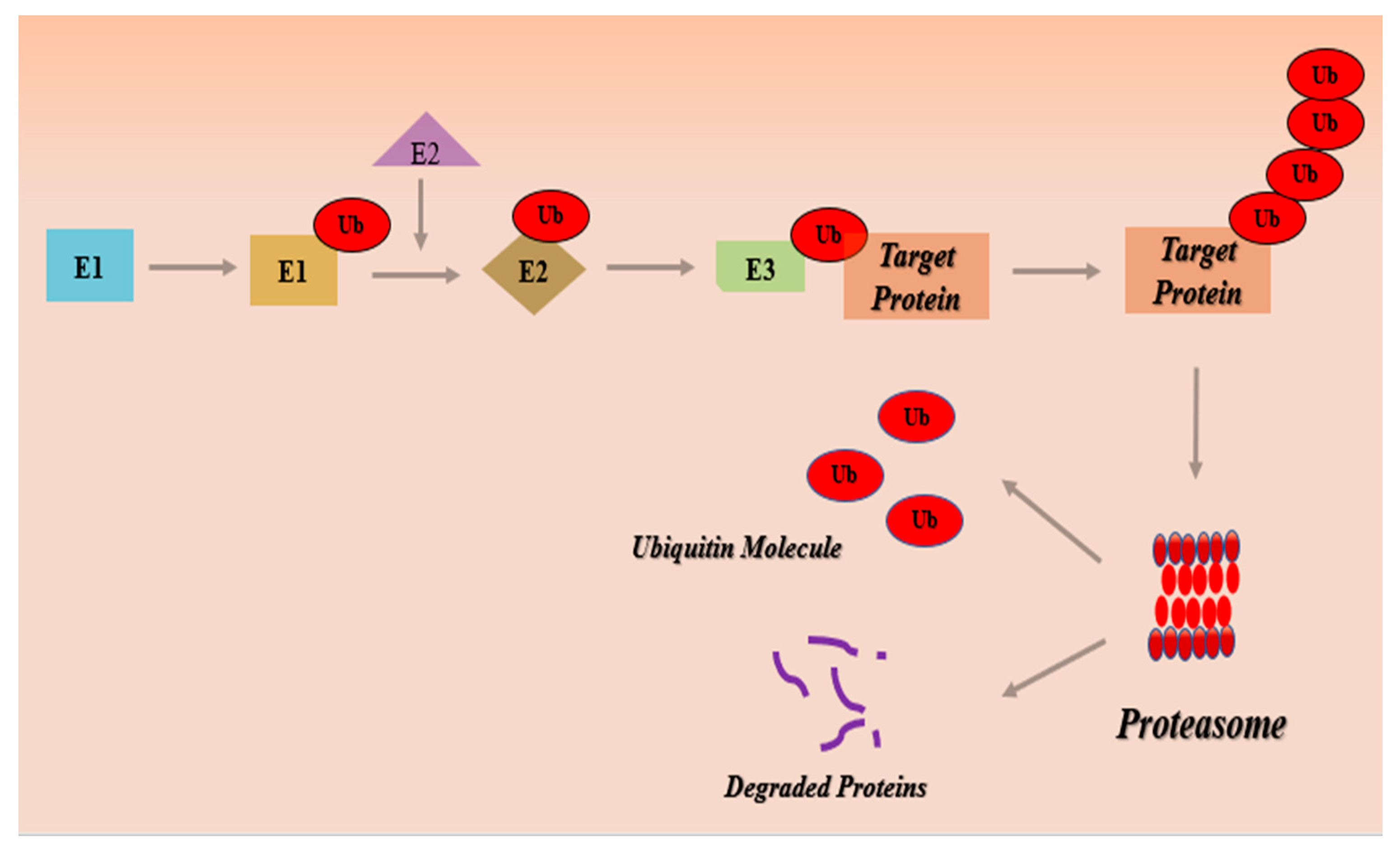
Ubiquitin-Proteasome Pathway (UPS)
2. Natural Product Scaffolds as Proteasome Inhibitors against Cancer
2.1. Plant-Derived Proteasome Inhibitors
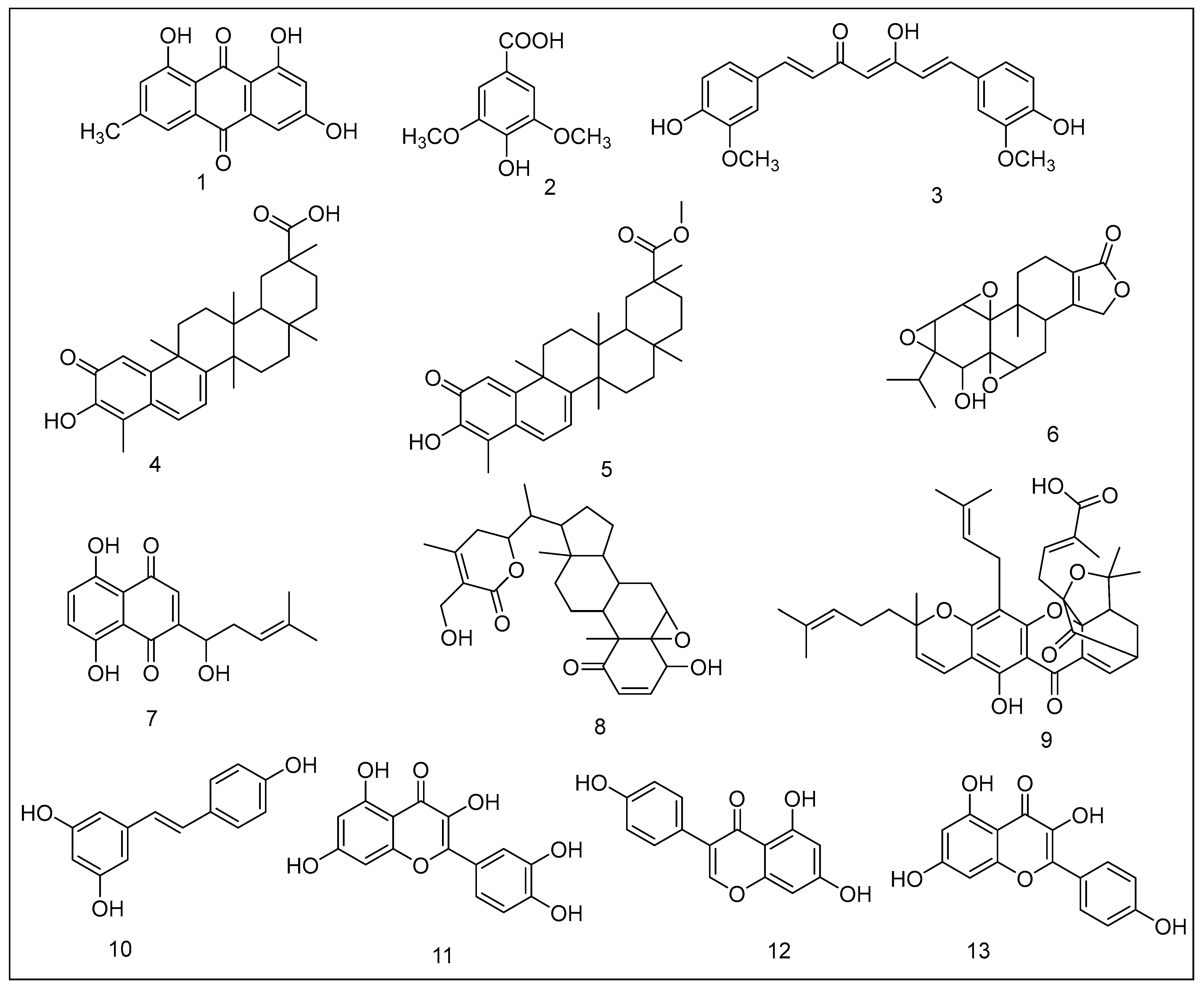
2.1.1. Emodin
2.1.2. Syringic Acid Derivatives
2.1.3. Curcumin
2.1.4. Celastrol
2.1.5. Pristimerin
2.1.6. Triptolide
2.1.7. Shikonin
2.1.8. Withaferin A
2.1.9. Gambogic Acid
2.1.10. Resveratrol
2.1.11. Quercetin
2.1.12. Genistein
2.1.13. Kaempferol
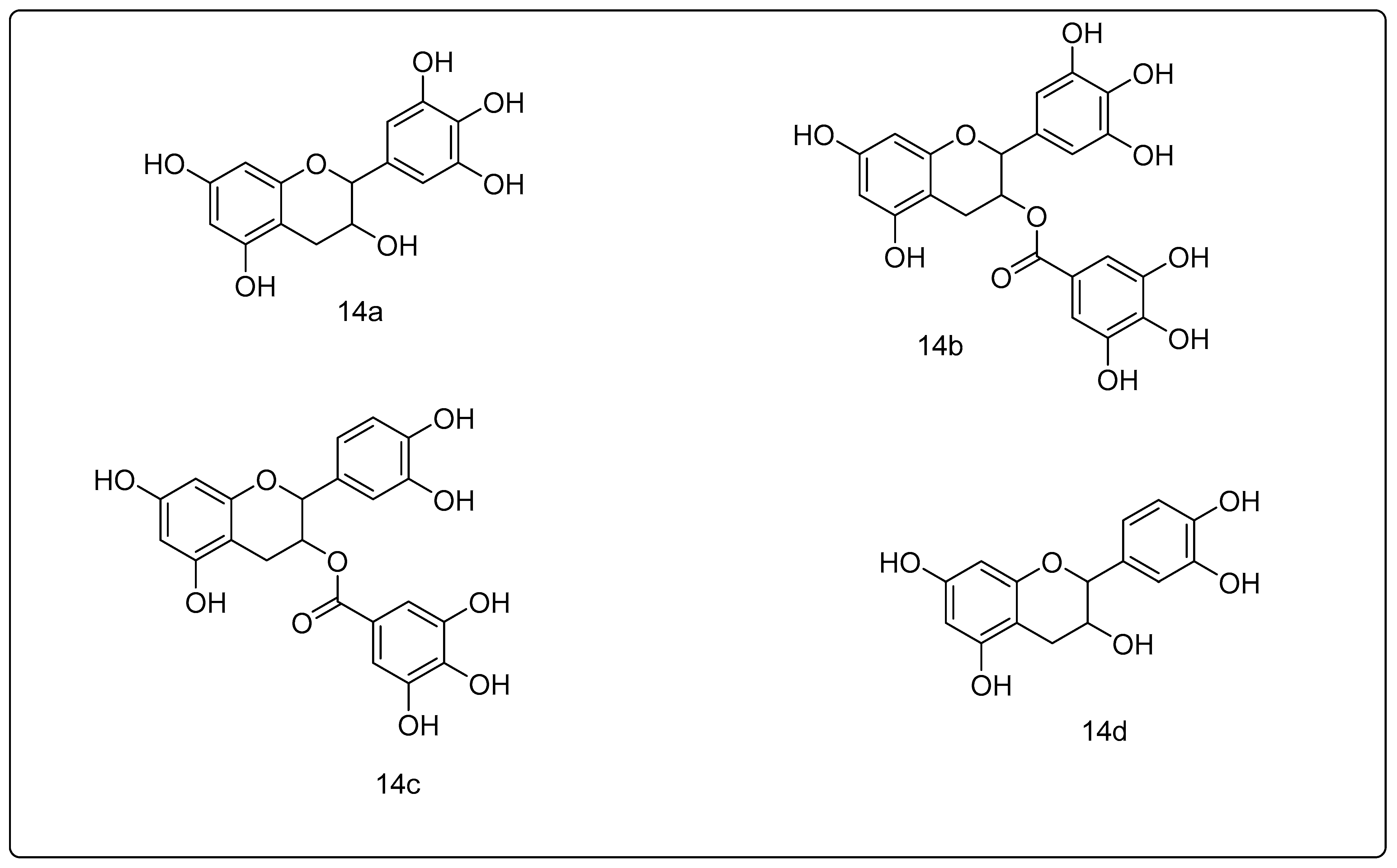
2.1.14. Green Tea Polyphenols
2.2. Marine-Derived Proteasome Inhibitors
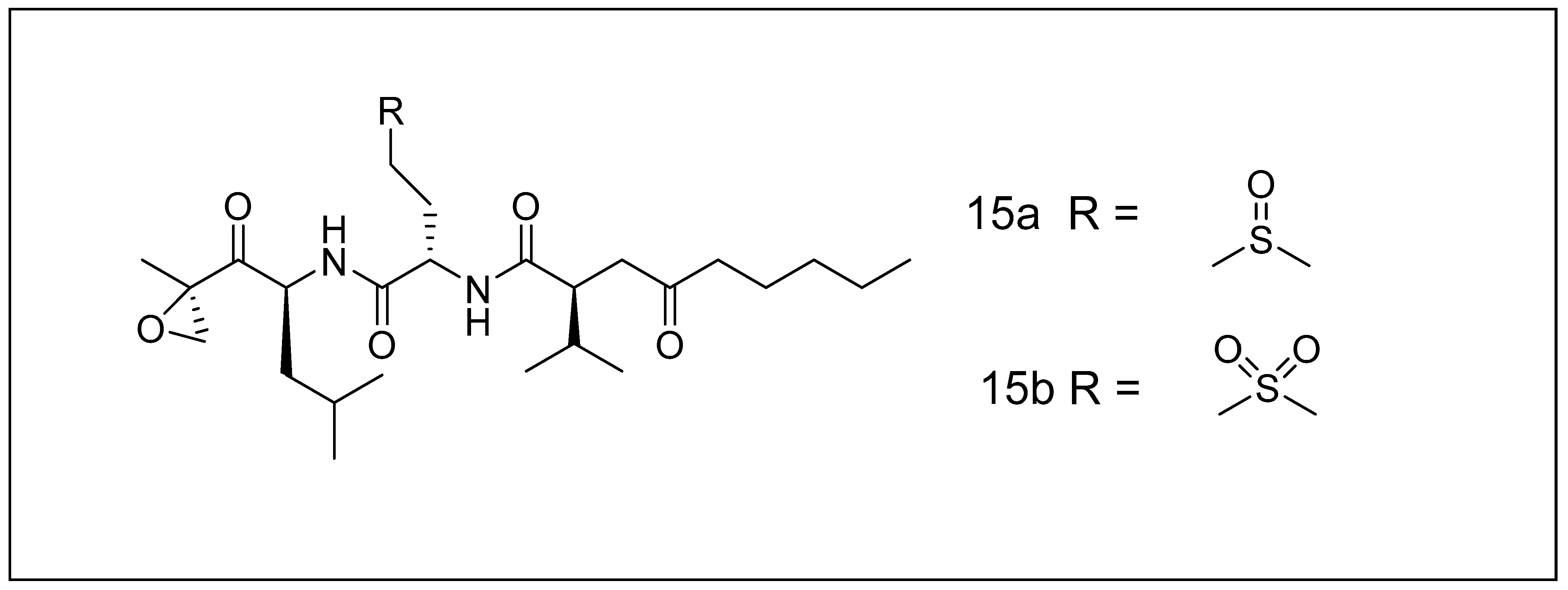
2.2.1. Carmaphycins
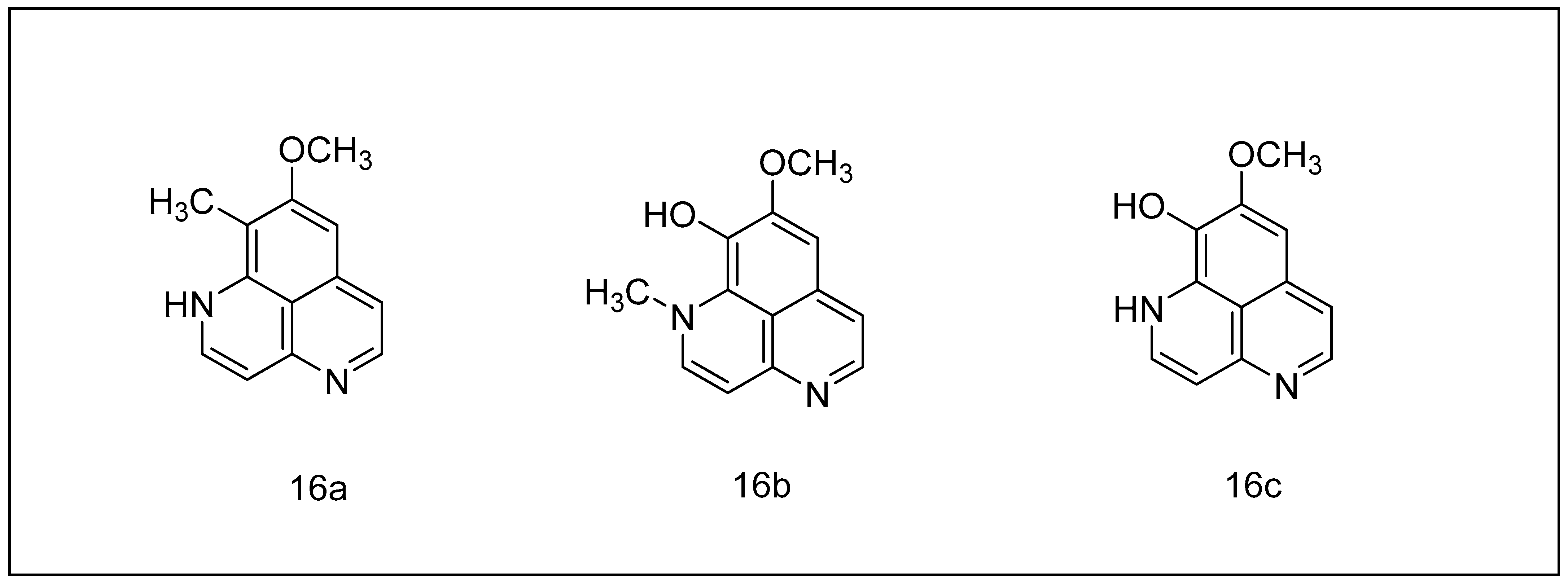
2.2.2. Aaptamine and Its Derivatives
2.2.3. Salinosporamides
2.3. Terrestrial-Derived Proteasome Inhibitors
2.3.1. Lactacystin
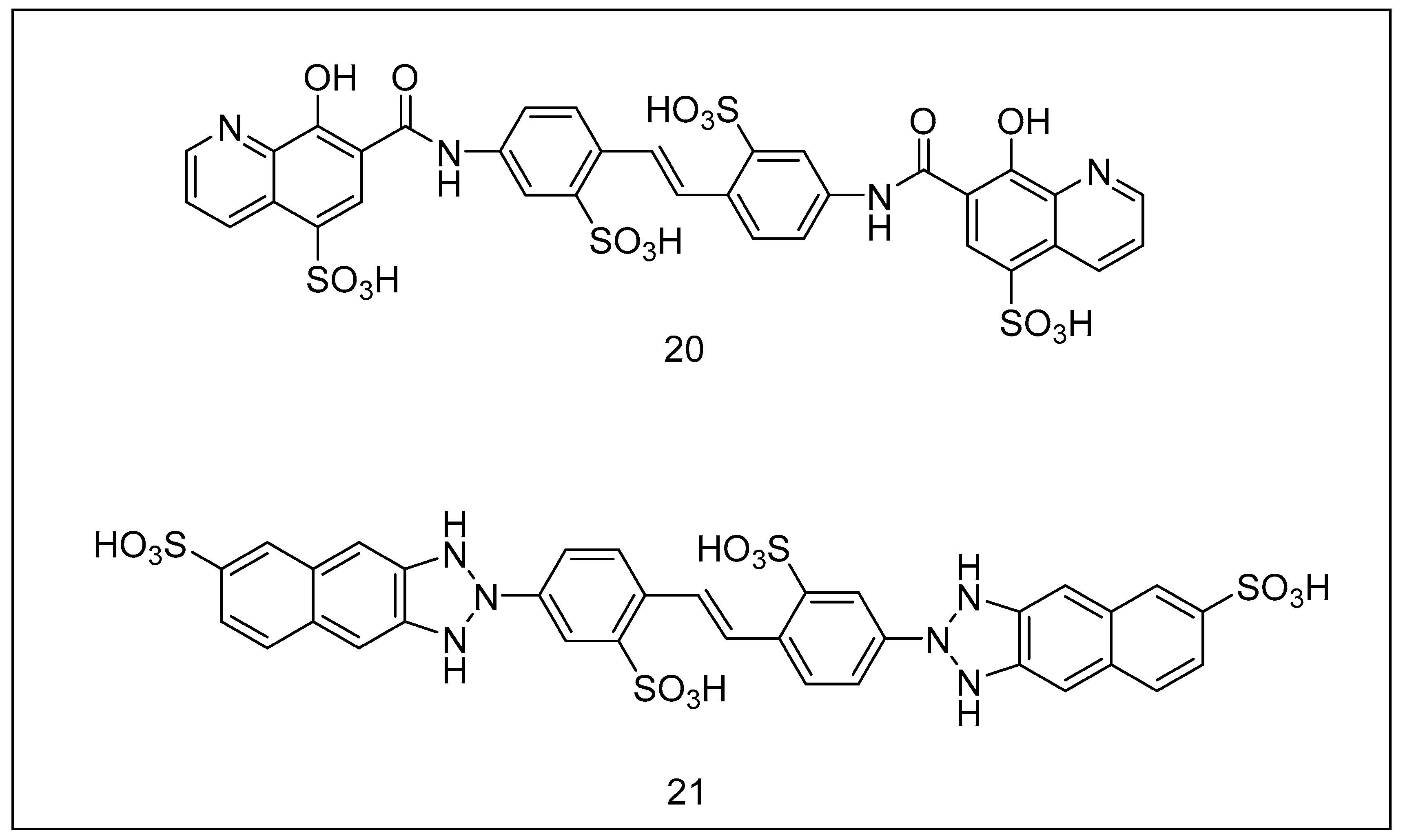
2.3.2. Ubistatins
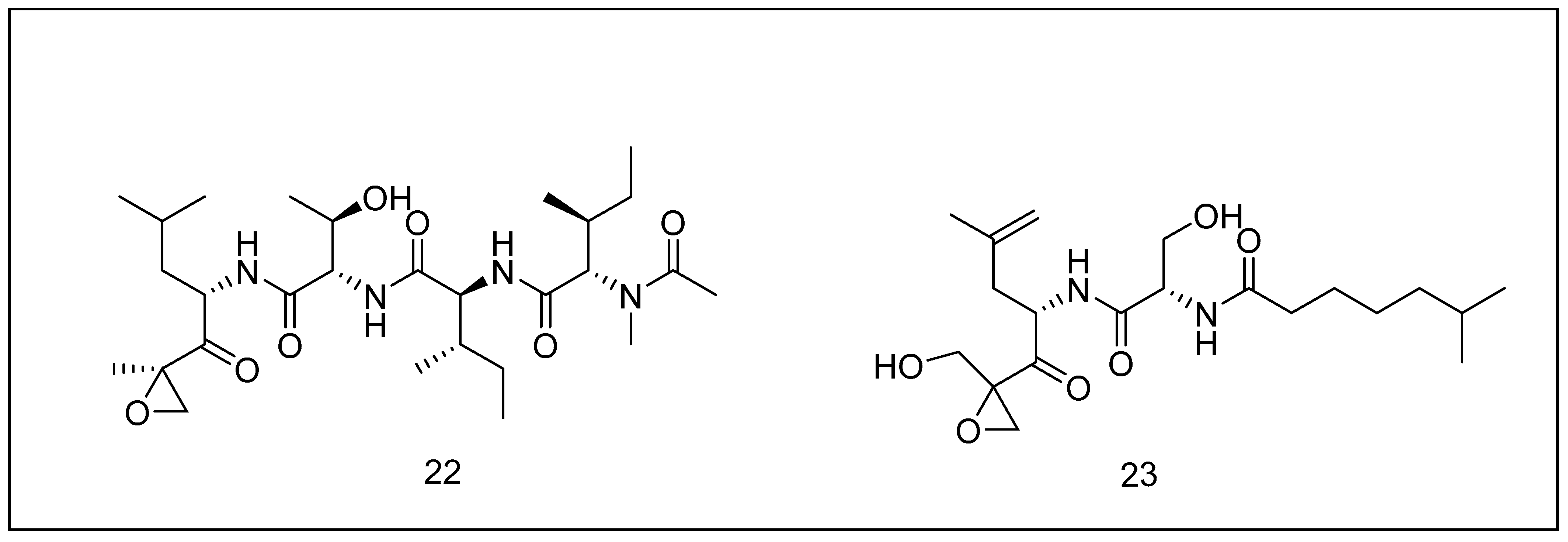
2.3.3. Epoxomicin
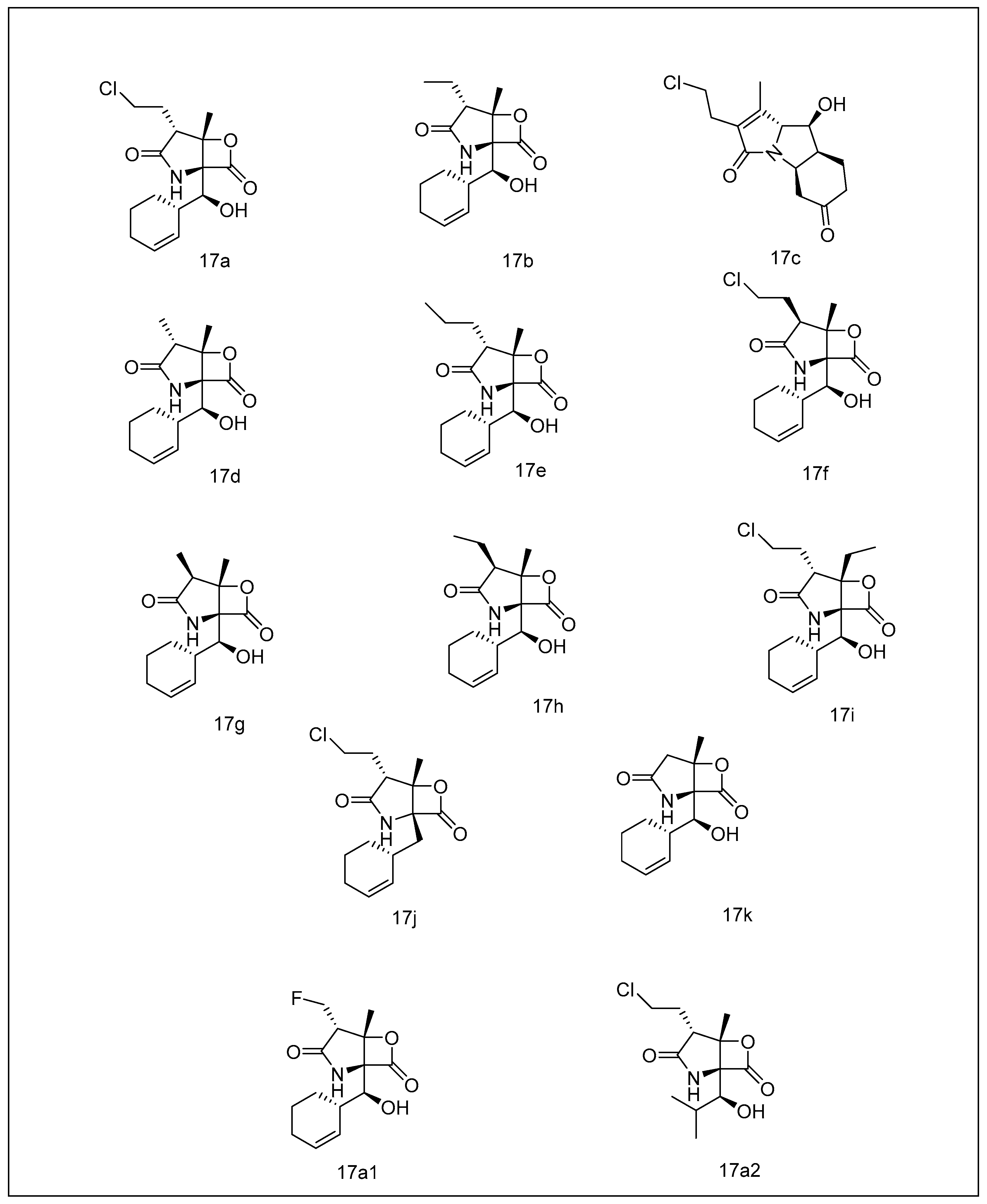
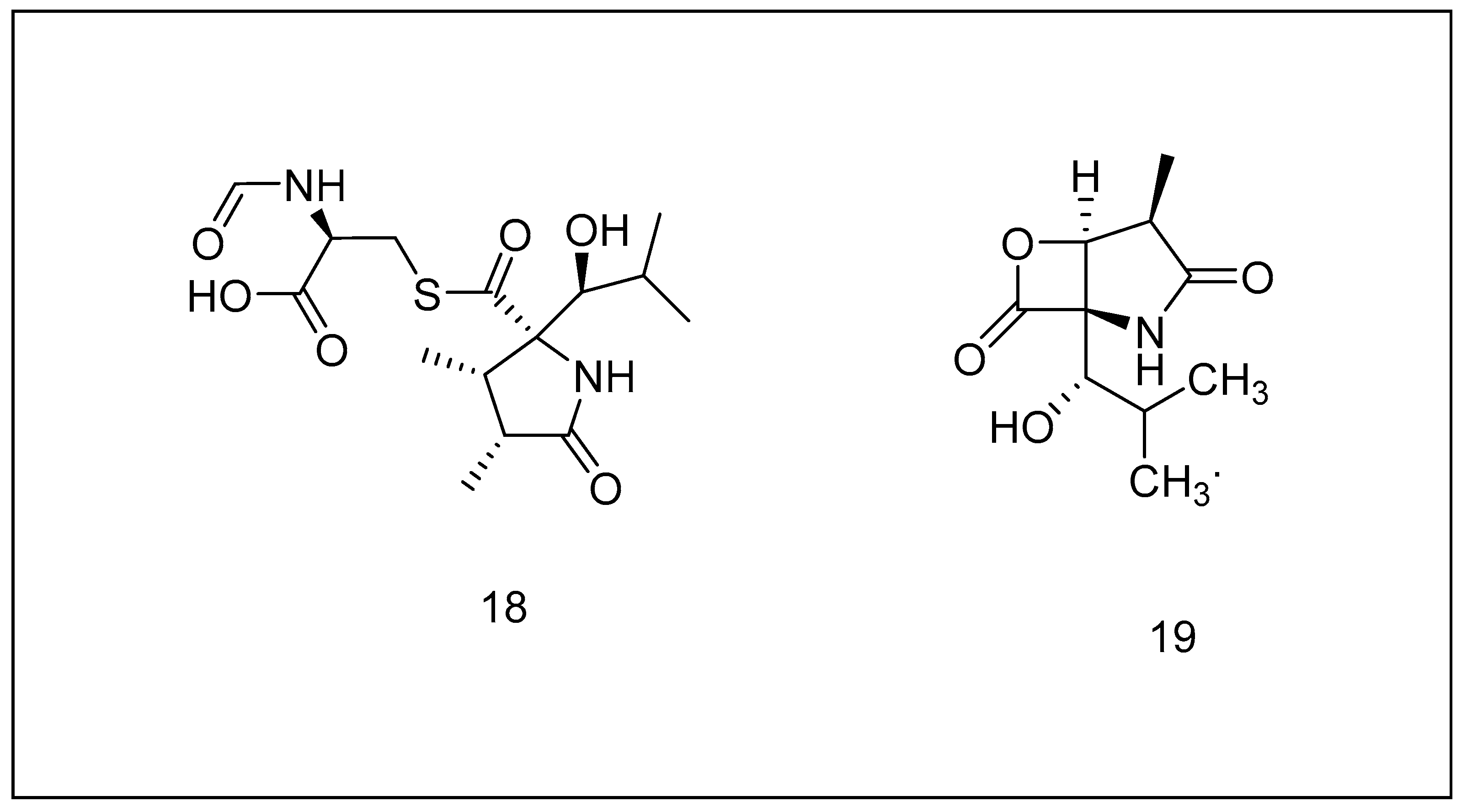
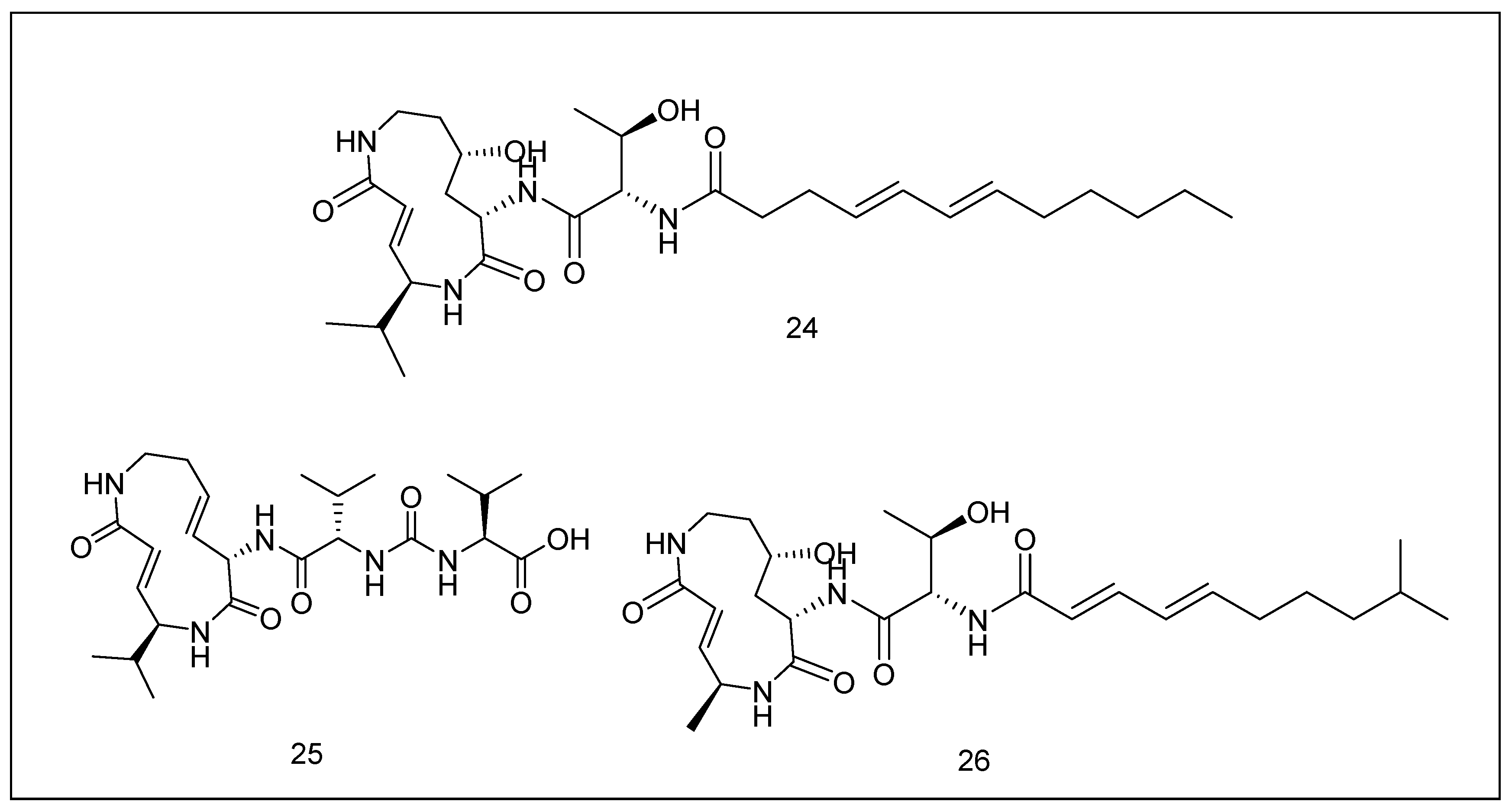
2.3.4. Syrbactins
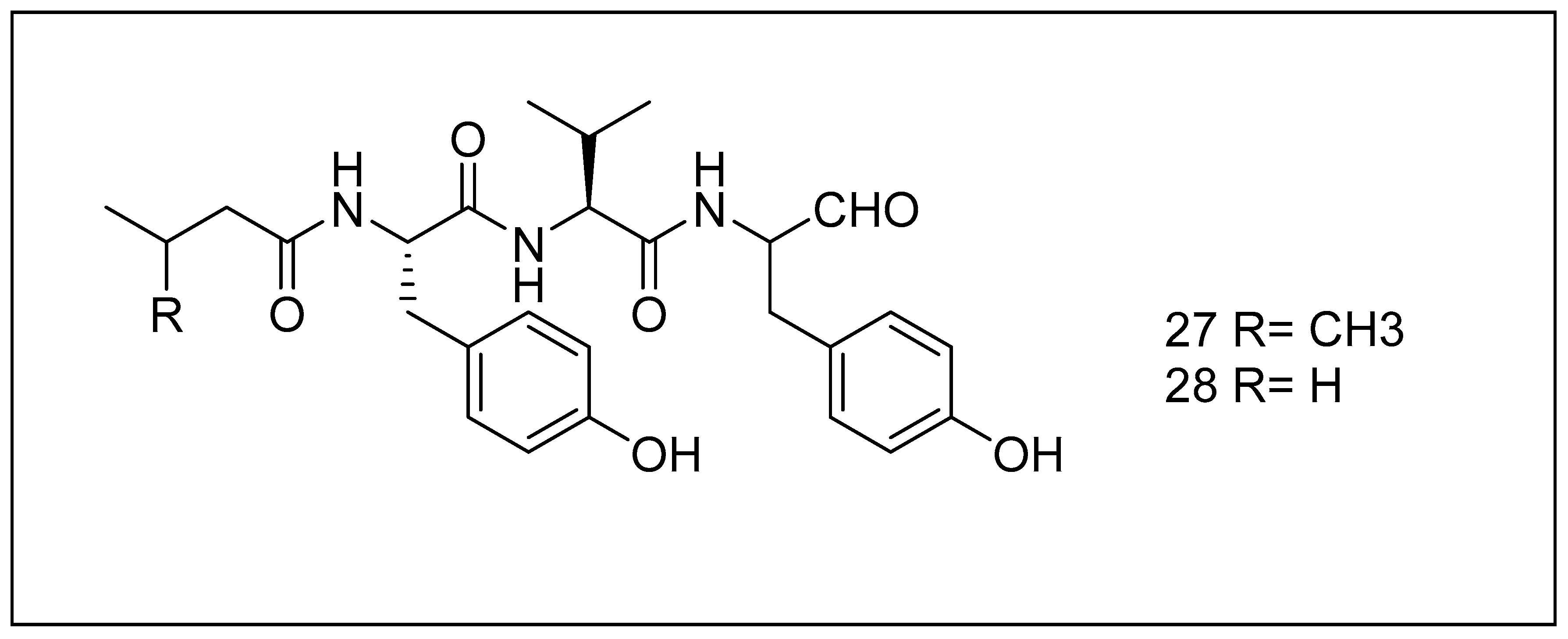
2.3.5. Tyropeptins
3. Compounds Inhibiting the Proteasome in Clinical Trials
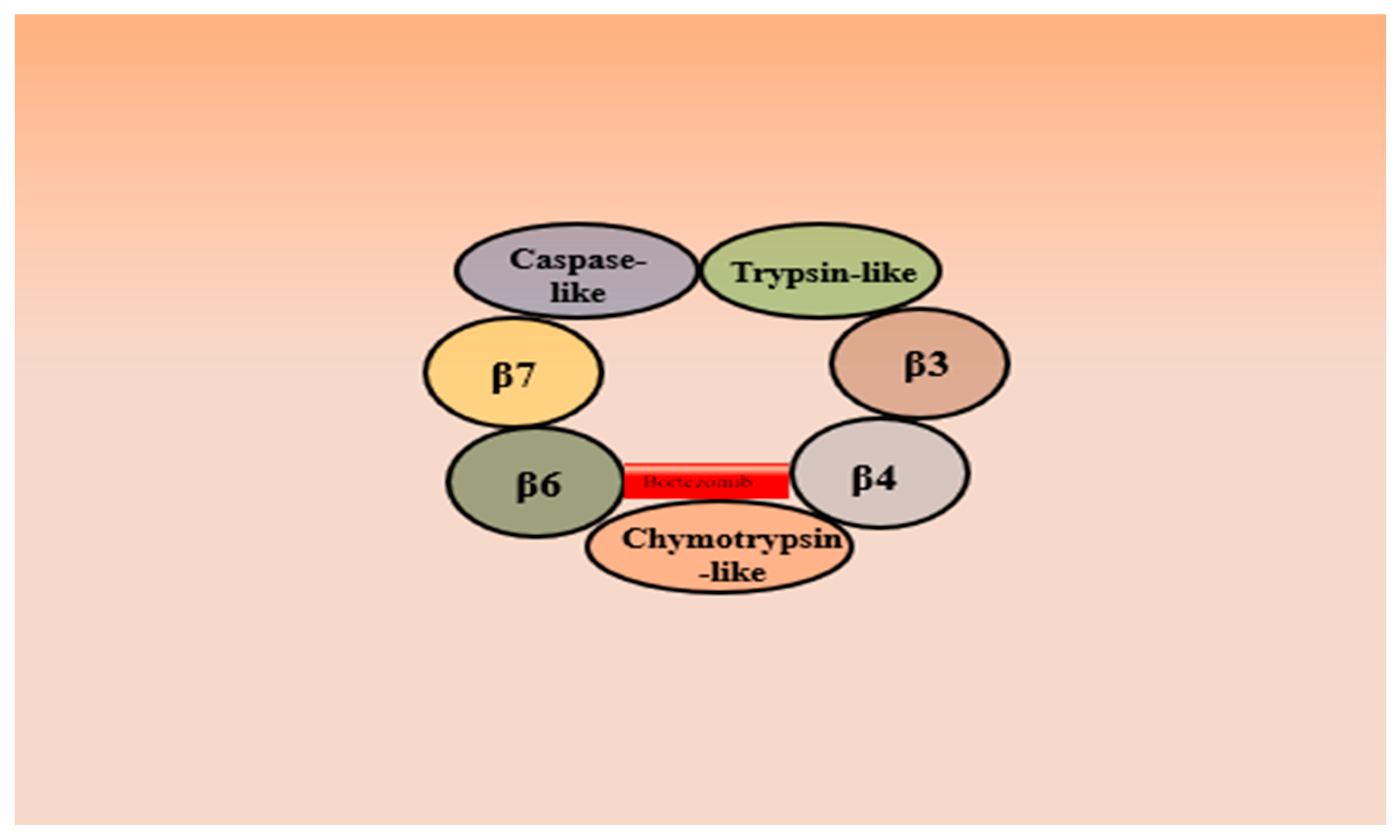
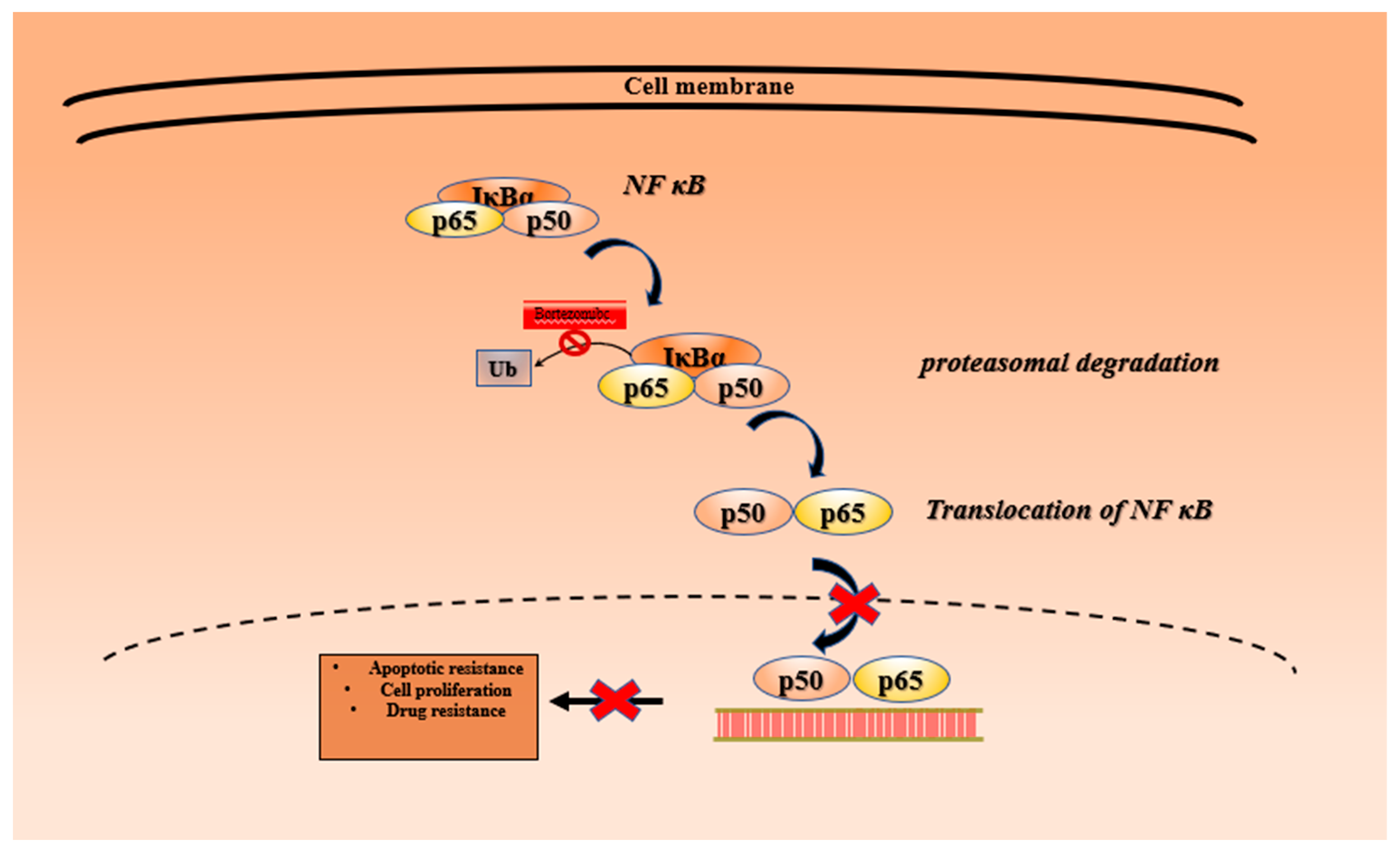
3.1. Bortezomib
3.2. Carfilzomib
3.3. Ixazomib
3.4. Marizomib (Salinosporamide A)
3.5. Oprozomib
3.6. Delanzomib
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- da Silva, D.C.; Andrade, P.B.; Ribeiro, V.; Valentao, P.; Pereira, D.M. Recent Patents on Proteasome Inhibitors of Natural Origin. Recent Pat. Anti-Cancer Drug Discov. 2017, 12, 4–15. [Google Scholar]
- Mathur, G.; Nain, S.; Sharma, P.K. Cancer: An overview. Acad. J. Cancer Res. 2015, 8, 1–9. [Google Scholar]
- Bastola, P.; Oien, D.B.; Cooley, M.; Chien, J. Emerging Cancer Therapeutic Targets in Protein Homeostasis. AAPS J. 2018, 20, 94. [Google Scholar] [PubMed]
- Park, J.E.; Miller, Z.; Jun, Y.; Lee, W.; Kim, K.B. Next-generation proteasome inhibitors for cancer therapy. Transl. Res. J. Lab. Clin. Med. 2018, 198, 1–16. [Google Scholar]
- Lecker, S.H.; Goldberg, A.L.; Mitch, W.E. Protein Degradation by the Ubiquitin–Proteasome Pathway in Normal and Disease States. J. Am. Soc. Nephrol. 2006, 17, 1807. [Google Scholar] [PubMed] [Green Version]
- Schmidt, M.; Finley, D. Regulation of proteasome activity in health and disease. Biochim. Biophys. Acta 2014, 1843, 13–25. [Google Scholar]
- Manasanch, E.E.; Orlowski, R.Z. Proteasome inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 2017, 14, 417–433. [Google Scholar]
- Roos-Mattjus, P.; Sistonen, L. The ubiquitin-proteasome pathway. Ann. Med. 2004, 36, 285–295. [Google Scholar]
- Almond, J.B.; Cohen, G.M. The proteasome: A novel target for cancer chemotherapy. Leukemia 2002, 16, 433–443. [Google Scholar]
- Tanaka, K. The proteasome: Overview of structure and functions. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2009, 85, 12–36. [Google Scholar] [PubMed] [Green Version]
- Moreau, P.; Richardson, P.G.; Cavo, M.; Orlowski, R.Z.; San Miguel, J.F.; Palumbo, A.; Harousseau, J.L. Proteasome inhibitors in multiple myeloma: 10 years later. Blood 2012, 120, 947–959. [Google Scholar] [PubMed] [Green Version]
- Chen, X.; Dou, Q.P.; Liu, J.; Tang, D. Targeting Ubiquitin-Proteasome System With Copper Complexes for Cancer Therapy. Front. Mol. Biosci. 2021, 8, 649151. [Google Scholar] [PubMed]
- Mani, A.; Gelmann, E.P. The ubiquitin-proteasome pathway and its role in cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2005, 23, 4776–4789. [Google Scholar]
- Vriend, J.; Nachtigal, M.W. Ubiquitin Proteasome Pathway Transcriptome in Epithelial Ovarian Cancer. Cancers 2021, 13, 2659. [Google Scholar] [PubMed]
- Chen, D.; Frezza, M.; Schmitt, S.; Kanwar, J.; Dou, Q.P. Bortezomib as the first proteasome inhibitor anticancer drug: Current status and future perspectives. Curr. Cancer Drug Targets 2011, 11, 239–253. [Google Scholar]
- Yarza, R.; Bover, M.; Agulló-Ortuño, M.T.; Iglesias-Docampo, L.C. Current approach and novel perspectives in nasopharyngeal carcinoma: The role of targeting proteasome dysregulation as a molecular landmark in nasopharyngeal cancer. J. Exp. Clin. Cancer Res. 2021, 40, 202. [Google Scholar]
- Morozov, A.V.; Karpov, V.L. Proteasomes and Several Aspects of Their Heterogeneity Relevant to Cancer. Front. Oncol. 2019, 9, 761. [Google Scholar]
- Hong, J. Role of natural product diversity in chemical biology. Curr. Opin. Chem. Biol. 2011, 15, 350–354. [Google Scholar]
- Butler, M.S. Natural products to drugs: Natural product-derived compounds in clinical trials. Nat. Prod. Rep. 2008, 25, 475–516. [Google Scholar]
- Cragg, G.M.; Newman, D.J. Natural products: A continuing source of novel drug leads. Biochim. Biophys. Acta (BBA) Gen. Subj. 2013, 1830, 3670–3695. [Google Scholar]
- Mohi-Ud-Din, R.; Mir, R.H.; Wani, T.U.; Shah, A.J.; Banday, N.; Pottoo, F.H.J.C.C.; Screening, H.T. Berberine in the Treatment of Neurodegenerative Diseases and Nanotechnology Enabled Targeted Delivery. Comb. Chem. High Throughput Screen. 2021, 25, 616–633. [Google Scholar]
- Thomford, N.E.; Senthebane, D.A.; Rowe, A.; Munro, D.; Seele, P.; Maroyi, A.; Dzobo, K. Natural products for drug discovery in the 21st century: Innovations for novel drug discovery. Int. J. Mol. Sci. 2018, 19, 1578. [Google Scholar]
- Dzobo, K. The role of natural products as sources of therapeutic agents for innovative drug discovery. Compr. Pharmacol. 2022, 2022, 408–422. [Google Scholar] [CrossRef]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; Supuran, C.T. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar]
- Ahmad, G.; Hassan, R.; Dhiman, N.; Ali, A.J.C.C.; Screening, H.T. Anti-inflammatory assessment of 3-Acetylmyricadiol in LPS-Stimulated Raw 264.7 Macrophages. Comb. Chem. High Throughput Screen. 2021, 25, 204–210. [Google Scholar]
- Yuan, H.; Ma, Q.; Ye, L.; Piao, G. The traditional medicine and modern medicine from natural products. Molecules 2016, 21, 559. [Google Scholar] [PubMed] [Green Version]
- Süntar, I. Importance of ethnopharmacological studies in drug discovery: Role of medicinal plants. Phytochem. Rev. 2020, 19, 1199–1209. [Google Scholar]
- Mir, R.H.; Godavari, G.; Siddiqui, N.A.; Ahmad, B.; Mothana, R.A.; Ullah, R.; Almarfadi, O.M.; Jachak, S.M.; Masoodi, M.H.J.D.D. Development; Therapy, Design, Synthesis, Molecular Modelling, and Biological Evaluation of Oleanolic Acid-Arylidene Derivatives as Potential Anti-Inflammatory Agents. Drug Des. Dev. Ther. 2021, 15, 385. [Google Scholar]
- Majolo, F.; Delwing, L.K.; Marmitt, D.J.; Bustamante-Filho, I.C.; Goettert, M.I. Medicinal plants and bioactive natural compounds for cancer treatment: Important advances for drug discovery. Phytochem. Lett. 2019, 31, 196–207. [Google Scholar]
- Pecere, T.; Gazzola, M.V.; Mucignat, C.; Parolin, C.; Dalla Vecchia, F.; Cavaggioni, A.; Basso, G.; Diaspro, A.; Salvato, B.; Carli, M.J.C.R. Aloe-emodin is a new type of anticancer agent with selective activity against neuroectodermal tumors. Cancer Res. 2000, 60, 2800–2804. [Google Scholar] [PubMed]
- Monisha, B.A.; Kumar, N.; Tiku, A. Emodin and its role in chronic diseases. Anti-Inflamm. Nutraceuticals Chronic Dis. 2016, 928, 47–73. [Google Scholar]
- Hsu, S.-C.; Chung, J.-G.J.B. Anticancer potential of emodin. BioMedicine 2012, 2, 108–116. [Google Scholar] [PubMed]
- Liu, W.; Feng, Q.; Li, Y.; Ye, L.; Hu, M.; Liu, Z.J.T. Coupling of UDP-glucuronosyltransferases and multidrug resistance-associated proteins is responsible for the intestinal disposition and poor bioavailability of emodin. Toxicol. Appl. Pharmacol. 2012, 265, 316–324. [Google Scholar] [CrossRef]
- Xing, J.Y.; Song, G.P.; Deng, J.P.; Jiang, L.Z.; Xiong, P.; Yang, B.J.; Liu, S.S. Antitumor Effects and Mechanism of Novel Emodin Rhamnoside Derivatives against Human Cancer Cells. PLoS ONE 2015, 10, e0144781. [Google Scholar]
- He, Y.; Huang, J.; Wang, P.; Shen, X.; Li, S.; Yang, L.; Liu, W.; Suksamrarn, A.; Zhang, G.; Wang, F.J.O. Emodin potentiates the antiproliferative effect of interferon α/β by activation of JAK/STAT pathway signaling through inhibition of the 26S proteasome. Oncotarget 2016, 7, 4664. [Google Scholar]
- Calderwood, S.K.; Gong, J. Heat shock proteins promote cancer: It’s a protection racket. Trends Biochem. Sci. 2016, 41, 311–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Y.-Y.; Zheng, L.-S.; Zhang, X.; Chen, L.-K.; Singh, S.; Wang, F.; Zhang, J.-Y.; Liang, Y.-J.; Dai, C.-L.; Gu, L.-Q. Blockade of Her2/neu binding to Hsp90 by emodin azide methyl anthraquinone derivative induces proteasomal degradation of Her2/neu. Mol. Pharm. 2011, 8, 1687–1697. [Google Scholar] [PubMed]
- Gililland, J.M.; Anderson, L.A.; Erickson, J.; Pelt, C.E.; Peters, C.L. Mean 5-year clinical and radiographic outcomes of cementless total hip arthroplasty in patients under the age of 30. BioMed Res. Int. 2013, 2013, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Abaza, M.-S.; Al-Attiyah, R.A.; Bhardwaj, R.; Abbadi, G.; Koyippally, M.; Afzal, M.J.P. Syringic acid from Tamarix aucheriana possesses antimitogenic and chemo-sensitizing activities in human colorectal cancer cells. Pharm. Biol. 2013, 51, 1110–1124. [Google Scholar] [CrossRef] [Green Version]
- Kampa, M.; Alexaki, V.-I.; Notas, G.; Nifli, A.-P.; Nistikaki, A.; Hatzoglou, A.; Bakogeorgou, E.; Kouimtzoglou, E.; Blekas, G.; Boskou, D.J. Antiproliferative and apoptotic effects of selective phenolic acids on T47D human breast cancer cells: Potential mechanisms of action. Breast Cancer Res. 2004, 6, R63–R74. [Google Scholar]
- Ha, S.J.; Lee, J.; Park, J.; Kim, Y.H.; Lee, N.H.; Kim, Y.E.; Song, K.-M.; Chang, P.-S.; Jeong, C.-H.; Jung, S.K. Syringic acid prevents skin carcinogenesis via regulation of NoX and EGFR signaling. Biochem. Pharmacol. 2018, 154, 435–445. [Google Scholar] [CrossRef]
- Carlsson, J.; Davidsson, S.; Helenius, G.; Karlsson, M.; Lubovac, Z.; Andrén, O.; Olsson, B.; Klinga-Levan, K.J. A miRNA expression signature that separates between normal and malignant prostate tissues. Cancer Cell Int. 2011, 11, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mir, R.H.; Mir, P.A.; Shah, A.J.; Banday, N.; Sabreen, S.; Maqbool, M.; Jan, R.; Shafi, N.; Masoodi, M.H. Curcumin as a privileged scaffold molecule for various biological targets in drug development. Stud. Nat. Prod. Chem. 2022, 73, 405–434. [Google Scholar]
- Shin, J.W.; Chun, K.-S.; Kim, D.-H.; Kim, S.-J.; Kim, S.H.; Cho, N.-C.; Na, H.-K.; Surh, Y.-J. Curcumin induces stabilization of Nrf2 protein through Keap1 cysteine modification. Biochem. Pharmacol. 2020, 173, 113820. [Google Scholar] [CrossRef]
- Huang, B.-R.; Tsai, C.-H.; Chen, C.-C.; Way, T.-D.; Kao, J.-Y.; Liu, Y.-S.; Lin, H.-Y.; Lai, S.-W.; Lu, D.-Y. Curcumin promotes connexin 43 degradation and temozolomide-induced apoptosis in glioblastoma cells. Am. J. Chin. Med. 2019, 47, 657–674. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Tao, J.; Hei, H.; Li, F.; Wang, Y.; Peng, W.; Zhang, X.J. Up-regulatory effects of curcumin on large conductance Ca2+-activated K+ channels. PLoS ONE 2015, 10, e0144800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, D.; Zhang, Z.; Chen, L.; Huang, W.; Zhang, F.; Wang, L.; Wang, Y.; Cao, P.; Zheng, S.J. Curcumin blunts epithelial-mesenchymal transition of hepatocytes to alleviate hepatic fibrosis through regulating oxidative stress and autophagy. Redox Biol. 2020, 36, 101600. [Google Scholar] [CrossRef]
- Liu, L.; Yang, J.; Ji, W.; Wang, C.J.B. Curcumin Inhibits Proliferation of Epstein–Barr Virus-Associated Human Nasopharyngeal Carcinoma Cells by Inhibiting EBV Nuclear Antigen 1 Expression. BioMed Res. Int. 2019, 2019, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Seo, S.U.; Kim, T.H.; Kim, D.E.; Min, K.-J.; Kwon, T.K. NOX4-mediated ROS production induces apoptotic cell death via down-regulation of c-FLIP and Mcl-1 expression in combined treatment with thioridazine and curcumin. Redox Biol. 2017, 13, 608–622. [Google Scholar] [CrossRef]
- Chaudhary, N.; Ueno-Shuto, K.; Ono, T.; Ohira, Y.; Watanabe, K.; Nasu, A.; Fujikawa, H.; Nakashima, R.; Takahashi, N.; Suico, M.A.J.B.; et al. Curcumin down-regulates toll-like receptor-2 gene expression and function in human cystic fibrosis bronchial epithelial cells. Biol. Pharm. Bull. 2019, 42, 489–495. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Wu, R.; Chen, W.; Liu, Y.; Liao, X.; Zeng, B.; Guo, G.; Lou, F.; Xiang, Y.; Wang, Y.J. Curcumin prevents obesity by targeting TRAF4-induced ubiquitylation in m6A-dependent manner. EMBO Rep. 2021, 22, e52146. [Google Scholar] [CrossRef] [PubMed]
- Obaidi, I.; Cassidy, H.; Ibanez Gaspar, V.; McCaul, J.; Higgins, M.; Halász, M.; Reynolds, A.L.; Kennedy, B.N.; McMorrow, T.J.B. Curcumin sensitizes kidney cancer cells to TRAIL-induced apoptosis via ROS mediated activation of JNK-CHOP pathway and upregulation of DR4. Biology 2020, 9, 92. [Google Scholar] [CrossRef]
- Buratta, S.; Chiaradia, E.; Tognoloni, A.; Gambelunghe, A.; Meschini, C.; Palmieri, L.; Muzi, G.; Urbanelli, L.; Emiliani, C.; Tancini, B.J. Effect of Curcumin on Protein Damage Induced by Rotenone in Dopaminergic PC12 Cells. Int. J. Mol. Sci. 2020, 21, 2761. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Ji, C.; Mayfield, J.E.; Goel, A.; Xiao, J.; Dixon, J.E.; Guo, X.J. Ancient drug curcumin impedes 26S proteasome activity by direct inhibition of dual-specificity tyrosine-regulated kinase 2. Proc. Natl. Acad. Sci. USA 2018, 115, 8155–8160. [Google Scholar] [CrossRef] [Green Version]
- Obata, K.; Kojima, T.; Masaki, T.; Okabayashi, T.; Yokota, S.; Hirakawa, S.; Nomura, K.; Takasawa, A.; Murata, M.; Tanaka, S.J. Curcumin prevents replication of respiratory syncytial virus and the epithelial responses to it in human nasal epithelial cells. PLoS ONE 2013, 8, e70225. [Google Scholar] [CrossRef] [PubMed]
- Khan, T.K.; You, Y.; Nelson, T.J.; Kundu, S.; Pramanik, S.K.; Das, J. Modulation of proteasome activity by curcumin and didemethylcurcumin. J. Biomol. Struct. Dyn. 2021, 40, 8332–8339. [Google Scholar] [CrossRef]
- Liu, L.; Fu, Y.; Zheng, Y.; Ma, M.; Wang, C. Curcumin inhibits proteasome activity in triple-negative breast cancer cells through regulating p300/miR-142–3p/PSMB5 axis. Phytomedicine 2020, 78, 153312. [Google Scholar] [CrossRef] [PubMed]
- Cardaci, T.D.; Machek, S.B.; Wilburn, D.T.; Hwang, P.S.; Willoughby, D.S. Ubiquitin proteasome system activity is suppressed by curcumin following exercise-induced muscle damage in human skeletal muscle. J. Am. Coll. Nutr. 2021, 40, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Milacic, V.; Banerjee, S.; Landis-Piwowar, K.R.; Sarkar, F.H.; Majumdar, A.P.; Dou, Q.P. Curcumin inhibits the proteasome activity in human colon cancer cells in vitro and in vivo. Cancer Res. 2008, 68, 7283–7292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Carvalho, J.E.R.; Verwoert, M.T.; Vogels, I.M.; Schipper-Krom, S.; Van Noorden, C.J.; Reits, E.A.; Klaassen, I.; Schlingemann, R.O. Modulation of the proteasome pathway by nano-curcumin and curcumin in retinal pigment epithelial cells. Ophthalmic Res. 2018, 59, 98–109. [Google Scholar] [CrossRef]
- Wan, S.B.; Yang, H.; Zhou, Z.; Cui, Q.C.; Chen, D.; Kanwar, J.; Mohammad, I.; Dou, O.P.; Chan, T.H. Evaluation of curcumin acetates and amino acid conjugates as proteasome inhibitors. Int. J. Mol. Med. 2010, 26, 447–455. [Google Scholar] [PubMed] [Green Version]
- Yue, X.; Zuo, Y.; Ke, H.; Luo, J.; Lou, L.; Qin, W.; Wang, Y.; Liu, Z.; Chen, D.; Sun, H.J. Identification of 4-arylidene curcumin analogues as novel proteasome inhibitors for potential anticancer agents targeting 19S regulatory particle associated deubiquitinase. Biochem. Pharmacol. 2017, 137, 29–50. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; DeSano, J.; Tang, W.; Meng, X.; Meng, Y.; Burstein, E.; Lawrence, T.S.; Xu, L. Natural proteasome inhibitor celastrol suppresses androgen-independent prostate cancer progression by modulating apoptotic proteins and NF-kappaB. PLoS ONE 2010, 5, e14153. [Google Scholar] [CrossRef] [Green Version]
- Pang, X.; Yi, Z.; Zhang, J.; Lu, B.; Sung, B.; Qu, W.; Aggarwal, B.B.; Liu, M. Celastrol suppresses angiogenesis-mediated tumor growth through inhibition of AKT/mammalian target of rapamycin pathway. Cancer Res. 2010, 70, 1951–1959. [Google Scholar]
- Raja, S.M.; Clubb, R.J.; Ortega-Cava, C.; Williams, S.H.; Bailey, T.A.; Duan, L.; Zhao, X.; Reddi, A.L.; Nyong, A.M.; Natarajan, A. Anticancer activity of Celastrol in combination with ErbB2-targeted therapeutics for treatment of ErbB2-overexpressing breast cancers. Cancer Biol. Ther. 2011, 11, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Chen, D.; Cui, Q.C.; Yuan, X.; Dou, Q.P. Celastrol, a triterpene extracted from the Chinese “Thunder of God Vine,” is a potent proteasome inhibitor and suppresses human prostate cancer growth in nude mice. Cancer Res. 2006, 66, 4758–4765. [Google Scholar] [CrossRef] [Green Version]
- Soave, C.L.; Guerin, T.; Liu, J.; Dou, Q.P. Targeting the ubiquitin-proteasome system for cancer treatment: Discovering novel inhibitors from nature and drug repurposing. Cancer Metastasis Rev. 2017, 36, 717–736. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, K.; Malla, P.; Lawrence, H.R.; Chen, Z.; Kumar-Sinha, C.; Malik, R.; Shukla, S.; Kim, J.; Coppola, D.; Lawrence, N.J. ACK1/TNK2 regulates histone H4 Tyr88-phosphorylation and AR gene expression in castration-resistant prostate cancer. Cancer Cell 2017, 31, 790–803.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Rose, A.E.; Doudican, N.; Osman, I.; Orlow, S.J. Celastrol synergistically enhances temozolomide cytotoxicity in melanoma cells. Mol. Cancer Res. 2009, 7, 1946–1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Landis-Piwowar, K.R.; Lu, D.; Yuan, P.; Li, L.; Reddy, G.P.V.; Yuan, X.; Dou, Q.P. Pristimerin induces apoptosis by targeting the proteasome in prostate cancer cells. J. Cell. Biochem. 2008, 103, 234–244. [Google Scholar] [PubMed]
- Lu, L.; Kanwar, J.; Schmitt, S.; Cui, Q.C.; Zhang, C.; Zhao, C.; Dou, Q.P. Inhibition of tumor cellular proteasome activity by triptolide extracted from the Chinese medicinal plant ‘thunder god vine’. Anticancer Res. 2011, 31, 1–10. [Google Scholar] [PubMed]
- Yang, H.; Zhou, P.; Huang, H.; Chen, D.; Ma, N.; Cui, Q.C.; Shen, S.; Dong, W.; Zhang, X.; Lian, W. Shikonin exerts antitumor activity via proteasome inhibition and cell death induction in vitro and in vivo. Int. J. Cancer 2009, 124, 2450–2459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirjalili, M.H.; Moyano, E.; Bonfill, M.; Cusido, R.M.; Palazón, J. Steroidal lactones from Withania somnifera, an ancient plant for novel medicine. Molecules 2009, 14, 2373–2393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Shi, G.; Dou, Q.P. The tumor proteasome is a primary target for the natural anticancer compound Withaferin A isolated from “Indian winter cherry”. Mol. Pharmacol. 2007, 71, 426–437. [Google Scholar] [CrossRef]
- Kashyap, D.; Mondal, R.; Tuli, H.S.; Kumar, G.; Sharma, A.K. Molecular targets of gambogic acid in cancer: Recent trends and advancements. Tumor Biol. 2016, 37, 12915–12925. [Google Scholar] [CrossRef]
- Zhou, Z.; Wan, J. Phase I human tolerability trial of gambogic acid. Chin. J. New Drugs 2007, 16, 679–682. [Google Scholar]
- Li, X.; Liu, S.; Huang, H.; Liu, N.; Zhao, C.; Liao, S.; Yang, C.; Liu, Y.; Zhao, C.; Li, S. Gambogic acid is a tissue-specific proteasome inhibitor in vitro and in vivo. Cell Rep. 2013, 3, 211–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, X.; Chen, X.; Li, X.; Lan, X.; Zhao, C.; Liu, S.; Huang, H.; Liu, N.; Liao, S.; Song, W. Gambogic acid induces apoptosis in imatinib-resistant chronic myeloid leukemia cells via inducing proteasome inhibition and caspase-dependent Bcr-Abl downregulation. Clin. Cancer Res. 2014, 20, 151–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreani, C.; Bartolacci, C.; Wijnant, K.; Crinelli, R.; Bianchi, M.; Magnani, M.; Hysi, A.; Iezzi, M.; Amici, A.; Marchini, C. Resveratrol fuels HER2 and ERα-positive breast cancer behaving as proteasome inhibitor. Aging 2017, 9, 508. [Google Scholar] [CrossRef] [Green Version]
- Mir, R.H.; Banday, N.; Sabreen, S.; Shah, A.J.; Jan, R.; Wani, T.U.; Farooq, S.; Bhat, Z.A. Resveratrol: A potential drug candidate with multispectrum therapeutic application. Stud. Nat. Prod. Chem. 2022, 73, 99–137. [Google Scholar]
- Kwon, K.J.; Kim, J.N.; Kim, M.K.; Lee, J.; Ignarro, L.J.; Kim, H.J.; Shin, C.Y.; Han, S.H. Melatonin synergistically increases resveratrol-induced heme oxygenase-1 expression through the inhibition of ubiquitin-dependent proteasome pathway: A possible role in neuroprotection. J. Pineal Res. 2011, 50, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Golonko, A.; Pienkowski, T.; Swislocka, R.; Lazny, R.; Roszko, M.; Lewandowski, W. Another look at phenolic compounds in cancer therapy the effect of polyphenols on ubiquitin-proteasome system. Eur. J. Med. Chem. 2019, 167, 291–311. [Google Scholar] [CrossRef] [PubMed]
- Bradley, J. TNF-mediated inflammatory disease. J. Pathol. A J. Pathol. Soc. Great Br. Irel. 2008, 214, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Silswal, N.; Reddy, N.; Qureshi, N. Resveratrol Modulates Cytokine Expression in LPS-induced Human Monocytes: Role of Proteasome Subunits. FASEB J. 2016, 30, 597.6. [Google Scholar]
- Sato, A.; Okada, M.; Shibuya, K.; Watanabe, E.; Seino, S.; Suzuki, K.; Narita, Y.; Shibui, S.; Kayama, T.; Kitanaka, C. Resveratrol promotes proteasome-dependent degradation of Nanog via p53 activation and induces differentiation of glioma stem cells. Stem Cell Res. 2013, 11, 601–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashemzaei, M.; Delarami Far, A.; Yari, A.; Heravi, R.E.; Tabrizian, K.; Taghdisi, S.M.; Sadegh, S.E.; Tsarouhas, K.; Kouretas, D.; Tzanakakis, G. Anticancer and apoptosis-inducing effects of quercetin in vitro and in vivo. Oncol. Rep. 2017, 38, 819–828. [Google Scholar] [PubMed] [Green Version]
- Dosenko, V.; Nagibin, V.; Tumanovskaya, L.; Zagorii, V.Y.; Moibenko, A. Effect of quercetin on the activity of purified 20S and 26S proteasomes and proteasomal activity in isolated cardiomyocytes. Biochem. (Mosc.) Suppl. Ser. B Biomed. Chem. 2007, 1, 40–44. [Google Scholar] [CrossRef]
- Klappan, A.K.; Hones, S.; Mylonas, I.; Brüning, A. Proteasome inhibition by quercetin triggers macroautophagy and blocks mTOR activity. Histochem. Cell Biol. 2012, 137, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Daniel, K.G.; Chen, M.S.; Kuhn, D.J.; Landis-Piwowar, K.R.; Dou, Q.P. Dietary flavonoids as proteasome inhibitors and apoptosis inducers in human leukemia cells. Biochem. Pharmacol. 2005, 69, 1421–1432. [Google Scholar] [PubMed]
- Chen, D.; Chen, M.S.; Cui, Q.C.; Yang, H.; Dou, Q.P. Structure-proteasome-inhibitory activity relationships of dietary flavonoids in human cancer cells. Front Biosci. 2007, 12, 1935–1945. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Zhang, C.; Qing, Y.; Cheng, Y.; Jiang, X.; Li, M.; Yang, Z.; Wang, D. Genistein induces apoptosis by stabilizing intracellular p53 protein through an APE1-mediated pathway. Free Radic. Biol. Med. 2015, 86, 209–218. [Google Scholar] [PubMed]
- Zhang, Z.; Wang, C.-Z.; Du, G.-J.; Qi, L.-W.; Calway, T.; He, T.-C.; Du, W.; Yuan, C.-S. Genistein induces G2/M cell cycle arrest and apoptosis via ATM/p53-dependent pathway in human colon cancer cells. Int. J. Oncol. 2013, 43, 289–296. [Google Scholar] [PubMed] [Green Version]
- Wu, T.-C.; Lin, Y.-C.; Chen, H.-L.; Huang, P.-R.; Liu, S.-Y.; Yeh, S.-L. The enhancing effect of genistein on apoptosis induced by trichostatin A in lung cancer cells with wild type p53 genes is associated with upregulation of histone acetyltransferase. Toxicol. Appl. Pharmacol. 2016, 292, 94–102. [Google Scholar]
- Kazi, A.; Daniel, K.G.; Smith, D.M.; Kumar, N.B.; Dou, Q.P. Inhibition of the proteasome activity, a novel mechanism associated with the tumor cell apoptosis-inducing ability of genistein. Biochem. Pharmacol. 2003, 66, 965–976. [Google Scholar]
- Zhou, N.; Yan, Y.; Li, W.; Wang, Y.; Zheng, L.; Han, S.; Yan, Y.; Li, Y. Genistein inhibition of topoisomerase IIα expression participated by Sp1 and Sp3 in HeLa cell. Int. J. Mol. Sci. 2009, 10, 3255–3268. [Google Scholar]
- Azarova, A.M.; Lin, R.-K.; Tsai, Y.-C.; Liu, L.F.; Lin, C.-P.; Lyu, Y.L. Genistein induces topoisomerase IIbeta-and proteasome-mediated DNA sequence rearrangements: Implications in infant leukemia. Biochem. Biophys. Res. Commun. 2010, 399, 66–71. [Google Scholar] [PubMed] [Green Version]
- Nitiss, J.L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. [Google Scholar] [PubMed] [Green Version]
- Sharma, V.; Joseph, C.; Ghosh, S.; Agarwal, A.; Mishra, M.K.; Sen, E. Kaempferol induces apoptosis in glioblastoma cells through oxidative stress. Mol. Cancer Ther. 2007, 6, 2544–2553. [Google Scholar] [PubMed] [Green Version]
- Shields, M. Pharmacognosy: Fundamentals, Applications and Strategies; Elsevier: Atlanta, GA, USA, 2017. [Google Scholar]
- Kim, S.-H.; Choi, K.-C. Anti-cancer effect and underlying mechanism(s) of kaempferol, a phytoestrogen, on the regulation of apoptosis in diverse cancer cell models. Toxicol. Res. 2013, 29, 229–234. [Google Scholar]
- Han, B.; Yu, Y.-Q.; Yang, Q.-L.; Shen, C.-Y.; Wang, X.-J. Kaempferol induces autophagic cell death of hepatocellular carcinoma cells via activating AMPK signaling. Oncotarget 2017, 8, 86227. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Zhou, Y.; Coughlan, K.A.; Ding, Y.; Wang, S.; Wu, Y.; Song, P.; Zou, M.-H. AMPKα1 deficiency promotes cellular proliferation and DNA damage via p21 reduction in mouse embryonic fibroblasts. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2015, 1853, 65–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegelin, M.D.; Reuss, D.E.; Habel, A.; Herold-Mende, C.; Von Deimling, A. The flavonoid kaempferol sensitizes human glioma cells to TRAIL-mediated apoptosis by proteasomal degradation of survivin. Mol. Cancer Ther. 2008, 7, 3566–3574. [Google Scholar] [CrossRef] [Green Version]
- Qiu, W.; Lin, J.; Zhu, Y.; Zhang, J.; Zeng, L.; Su, M.; Tian, Y. Kaempferol modulates DNA methylation and downregulates DNMT3B in bladder cancer. Cell. Physiol. Biochem. 2017, 41, 1325–1335. [Google Scholar] [PubMed]
- Seely, D.; Mills, E.J.; Wu, P.; Verma, S.; Guyatt, G. The effects of green tea consumption on incidence of breast cancer and recurrence of breast cancer: A systematic review and meta-analysis. Integr. Cancer Ther. 2005, 4, 144–155. [Google Scholar] [CrossRef]
- Arab, L.; Il’yasova, D.J. The epidemiology of tea consumption and colorectal cancer incidence. J. Nutr. 2003, 133, 3310S–3318S. [Google Scholar]
- Imai, K.; Suga, K.; Nakachi, K.J. Cancer-preventive effects of drinking green tea among a Japanese population. Prev. Med. 1997, 26, 769–775. [Google Scholar] [CrossRef]
- Mir, R.H.; Masoodi, M.H. Anti-inflammatory plant polyphenolics and cellular action mechanisms. Curr. Bioact. Compd. 2020, 16, 809–817. [Google Scholar]
- Dou, Q.; Landis-Piwowar, K.; Chen, D.; Huo, C.; Wan, S.; Chan, T.J. Green tea polyphenols as a natural tumour cell proteasome inhibitor. Inflammopharmacology 2008, 16, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Nam, S.; Smith, D.M.; Dou, Q.P. Ester bond-containing tea polyphenols potently inhibit proteasome activity in vitro and in vivo. J. Biol. Chem. 2001, 276, 13322–13330. [Google Scholar] [PubMed] [Green Version]
- Thangapazham, R.L.; Singh, A.K.; Sharma, A.; Warren, J.; Gaddipati, J.P.; Maheshwari, R.K. Green tea polyphenols and its constituent epigallocatechin gallate inhibits proliferation of human breast cancer cells in vitro and in vivo. Cancer Lett. 2007, 245, 232–241. [Google Scholar] [CrossRef]
- Smith, D.M.; Wang, Z.; Kazi, A.; Li, L.-H.; Chan, T.-H.; Dou, Q.P. Synthetic analogs of green tea polyphenols as proteasome inhibitors. Mol. Med. 2002, 8, 382–392. [Google Scholar] [CrossRef] [Green Version]
- Kazi, A.; Wang, Z.; Kumar, N.; Falsetti, S.C.; Chan, T.-H.; Dou, Q.P. Structure-activity relationships of synthetic analogs of (-)-epigallocatechin-3-gallate as proteasome inhibitors. Anticancer Res. 2004, 24, 943–954. [Google Scholar]
- Chen, Z.P.; Schell, J.B.; Ho, C.-T.; Chen, K.Y. Green tea epigallocatechin gallate shows a pronounced growth inhibitory effect on cancerous cells but not on their normal counterparts. Cancer Lett. 1998, 129, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, D.; Lam, W.H.; Kazi, A.; Daniel, K.G.; Song, S.; Chow, L.; Chan, T.H.; Dou, Q.P. Synthetic peracetate tea polyphenols as potent proteasome inhibitors and apoptosis inducers in human cancer cells. Front. Biosci. Landmark 2005, 10, 1010–1023. [Google Scholar]
- Quan, Y.; Li, L.; Dong, L.; Wang, S.; Jiang, X.; Zhang, T.; Jin, P.; Fan, J.; Mao, S.; Fan, X.; et al. Epigallocatechin-3-gallate (EGCG) inhibits aggregation of pulmonary fibrosis associated mutant surfactant protein A2 via a proteasomal degradation pathway. Int. J. Biochem. Cell Biol. 2019, 116, 105612. [Google Scholar] [CrossRef]
- Jordan, J.D.; He, J.C.; Eungdamrong, N.J.; Gomes, I.; Ali, W.; Nguyen, T.; Bivona, T.G.; Philips, M.R.; Devi, L.A.; Iyengar, R.J. Cannabinoid receptor-induced neurite outgrowth is mediated by Rap1 activation through Gαo/i-triggered proteasomal degradation of Rap1GAPII. J. Biol. Chem. 2005, 280, 11413–11421. [Google Scholar] [CrossRef] [Green Version]
- Nam, B.; Rho, J.K.; Shin, D.-M.; Son, J.J. Gallic acid induces apoptosis in EGFR-mutant non-small cell lung cancers by accelerating EGFR turnover. Bioorg. Med. Chem. Lett. 2016, 26, 4571–4575. [Google Scholar] [CrossRef]
- Zhang, L.; Wei, Y.; Zhang, J.J. Novel mechanisms of anticancer activities of green tea component epigallocatechin-3-gallate. Anti-Cancer Agents Med. Chem. 2014, 14, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Xiang, L.-P.; Wang, A.; Ye, J.-H.; Zheng, X.-Q.; Polito, C.A.; Lu, J.-L.; Li, Q.-S.; Liang, Y.-R. Suppressive effects of tea catechins on breast cancer. Nutrients 2016, 8, 458. [Google Scholar] [CrossRef] [PubMed]
- Ju, J.; Lu, G.; Lambert, J.D.; Yang, C.S. Inhibition of carcinogenesis by tea constituents. Semin. Cancer Biol. 2007, 17, 395–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davalli, P.; Rizzi, F.; Caldara, G.F.; Davoli, S.; Corti, A.; Silva, A.; Astancolle, S.; Vitale, M.; Bettuzzi, S.; Arcari, M.J. Chronic administration of green tea extract to TRAMP mice induces the collapse of Golgi apparatus in prostate secretory cells and results in alterations of protein post-translational processing. Int. J. Oncol. 2011, 39, 1521–1527. [Google Scholar] [PubMed] [Green Version]
- Lam, W.H.; Kazi, A.; Kuhn, D.J.; Chow, L.M.; Chan, A.S.; Dou, Q.P.; Chan, T.H. A potential prodrug for a green tea polyphenol proteasome inhibitor: Evaluation of the peracetate ester of (−)-epigallocatechin gallate [(−)-EGCG]. Bioorg. Med. Chem. 2004, 12, 5587–5593. [Google Scholar] [CrossRef] [PubMed]
- Nabavi, S.F.; Atanasov, A.G.; Khan, H.; Barreca, D.; Trombetta, D.; Testai, L.; Sureda, A.; Tejada, S.; Vacca, R.A.; Pittalà, V.; et al. Targeting ubiquitin-proteasome pathway by natural, in particular polyphenols, anticancer agents: Lessons learned from clinical trials. Cancer Lett. 2018, 434, 101–113. [Google Scholar] [CrossRef]
- Kuhn, D.J.; Burns, A.C.; Kazi, A.; Dou, Q.P. Direct inhibition of the ubiquitin–proteasome pathway by ester bond-containing green tea polyphenols is associated with increased expression of sterol regulatory element-binding protein 2 and LDL receptor. Mol. Cell Biol. Lipids 2004, 1682, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kanwar, J.; Taskeen, M.; Mohammad, I.; Huo, C.; Chan, T.H.; Dou, Q.P. Recent advances on tea polyphenols. Front. Biosci. 2012, 4, 111. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.S.; Kang, S.U.; Park, J.K.; Kim, Y.E.; Kim, Y.S.; Baek, S.J.; Lee, S.-H.; Kim, C.-H. Anti-cancer effect of (-)-epigallocatechin-3-gallate (EGCG) in head and neck cancer through repression of transactivation and enhanced degradation of β-catenin. Phytomedicine 2016, 23, 1344–1355. [Google Scholar] [CrossRef]
- Meador, B.; Mirza, K.; Tian, M.; Skelding, M.; Reaves, L.; Edens, N.; Tisdale, M.; Pereira, S.J. The Green Tea Polyphenol Epigallocatechin-3-Gallate (EGCg) Attenuates Skeletal Muscle Atrophy in a Rat Model of Sarcopenia. J. Frailty Aging 2015, 4, 209–215. [Google Scholar] [CrossRef]
- Zhang, J.; Lei, Z.; Huang, Z.; Zhang, X.; Zhou, Y.; Luo, Z.; Zeng, W.; Su, J.; Peng, C.; Chen, X.J.O. Epigallocatechin-3-gallate (EGCG) suppresses melanoma cell growth and metastasis by targeting TRAF6 activity. Oncotarget 2016, 7, 79557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, L.; Li, C.; Xu, Y.; Wang, L.; Liu, J.; Wang, D.; Hong, C.; Jiang, Z.; Ma, Y.; Chen, Q.J. Epigallocatechin gallate promotes p53 accumulation and activity via the inhibition of MDM2-mediated p53 ubiquitination in human lung cancer cells. Oncol. Rep. 2013, 29, 1983–1990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, A.R.; Kale, A.J.; Fenley, A.T.; Byrum, T.; Debonsi, H.M.; Gilson, M.K.; Valeriote, F.A.; Moore, B.S.; Gerwick, W.H. The Carmaphycins, new proteasome inhibitors exhibiting an α, β-epoxyketone warhead from a marine cyanobacterium. ChemBioChem 2012, 13, 810. [Google Scholar] [CrossRef] [Green Version]
- Trivella, D.B.; Pereira, A.R.; Stein, M.L.; Kasai, Y.; Byrum, T.; Valeriote, F.A.; Tantillo, D.J.; Groll, M.; Gerwick, W.H.; Moore, B.S. Enzyme inhibition by hydroamination: Design and mechanism of a hybrid carmaphycin-syringolin enone proteasome inhibitor. Chem. Biol. 2014, 21, 782–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, L.T.; Phyo, M.Y. Marine cyanobacteria: A source of lead compounds and their clinically-relevant molecular targets. Molecules 2020, 25, 2197. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Kobayashi, J.I.; Ohizumi, Y.; Hirata, Y.J. Isolation and structure of aaptamine a novel heteroaromatic substance possessing α-blocking activity from the sea sponge Aaptos aaptos. Tetrahedron Lett. 1982, 23, 5555–5558. [Google Scholar] [CrossRef]
- Shaari, K.; Ling, K.C.; Mat Rashid, Z.; Jean, T.P.; Abas, F.; Raof, S.M.; Zainal, Z.; Lajis, N.H.; Mohamad, H.; Ali, A.M. Cytotoxic aaptamines from Malaysian Aaptos aaptos. Mar. Drugs 2009, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, S.; Yamanokuchi, R.; Yoshitomi, M.; Sato, K.; Ikeda, T.; Rotinsulu, H.; Mangindaan, R.E.; de Voogd, N.J.; van Soest, R.W.; Yokosawa, H.J. Aaptamine, an alkaloid from the sponge Aaptos suberitoides, functions as a proteasome inhibitor. Bioorg. Med. Chem. Lett. 2010, 20, 3341–3343. [Google Scholar] [CrossRef]
- Nadar, V.M.; Manivannan, S.; Chinnaiyan, R.; Govarthanan, M.; Ponnuchamy, K. Review on marine sponge alkaloid, aaptamine: A potential antibacterial and anticancer drug. Chem. Biol. Drug Des. 2021, 99, 103–110. [Google Scholar] [CrossRef]
- Potts, B.C.; Albitar, M.X.; Anderson, K.C.; Baritaki, S.; Berkers, C.; Bonavida, B.; Chandra, J.; Chauhan, D.; Cusack, J.C.; Fenical, W.J. Marizomib, a proteasome inhibitor for all seasons: Preclinical profile and a framework for clinical trials. Curr. Cancer Drug Targets 2011, 11, 254–284. [Google Scholar] [CrossRef] [Green Version]
- Feling, R.H.; Buchanan, G.O.; Mincer, T.J.; Kauffman, C.A.; Jensen, P.R.; Fenical, W.J. Salinosporamide A: A highly cytotoxic proteasome inhibitor from a novel microbial source, a marine bacterium of the new genus Salinospora. Angew. Chem. Int. Ed. 2003, 42, 355–357. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Gao, Q. Review on Patents for Ubiquitin-Proteasome Inhibitor as Medical Advance in Major Human Diseases. Recent Pat. Biomed. Eng. 2009, 2, 180–192. [Google Scholar] [CrossRef]
- Corey, E.J. Analogs of Salinosporamide, A. United States Patent US 7,511,156, 31 March 2009. [Google Scholar]
- Chauhan, D.; Catley, L.; Li, G.; Podar, K.; Hideshima, T.; Velankar, M.; Mitsiades, C.; Mitsiades, N.; Yasui, H.; Letai, A.J. A novel orally active proteasome inhibitor induces apoptosis in multiple myeloma cells with mechanisms distinct from Bortezomib. Cancer Cell 2005, 8, 407–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manam, R.R.; McArthur, K.A.; Chao, T.-H.; Weiss, J.; Ali, J.A.; Palombella, V.J.; Groll, M.; Lloyd, G.K.; Palladino, M.A.; Neuteboom, S.T. Leaving groups prolong the duration of 20S proteasome inhibition and enhance the potency of salinosporamides. J. Med. Chem. 2008, 51, 6711–6724. [Google Scholar] [CrossRef]
- Groll, M.; McArthur, K.A.; Macherla, V.R.; Manam, R.R.; Potts, B.C. Snapshots of the fluorosalinosporamide/20S complex offer mechanistic insights for fine tuning proteasome inhibition. J. Med. Chem. 2009, 52, 5420–5428. [Google Scholar] [CrossRef]
- Reed, K.A.; Manam, R.R.; Mitchell, S.S.; Xu, J.; Teisan, S.; Chao, T.-H.; Deyanat-Yazdi, G.; Neuteboom, S.T.; Lam, K.S.; Potts, B.C. Salinosporamides D− J from the marine actinomycete Salinispora tropica, bromosalinosporamide, and thioester derivatives are potent inhibitors of the 20S proteasome. J. Nat. Prod. 2007, 70, 269–276. [Google Scholar] [CrossRef]
- Nett, M.; Gulder, T.A.; Kale, A.J.; Hughes, C.C.; Moore, B.S. Function-oriented biosynthesis of β-lactone proteasome inhibitors in Salinispora tropica. J. Med. Chem. 2009, 52, 6163–6167. [Google Scholar] [CrossRef] [Green Version]
- Macherla, V.R.; Mitchell, S.S.; Manam, R.R.; Reed, K.A.; Chao, T.-H.; Nicholson, B.; Deyanat-Yazdi, G.; Mai, B.; Jensen, P.R.; Fenical, W.F. Structure−activity relationship studies of salinosporamide A (NPI-0052), a novel marine derived proteasome Inhibitor. J. Med. Chem. 2005, 48, 3684–3687. [Google Scholar] [CrossRef]
- Gulder, T.A.; Moore, B.S. Salinosporamide natural products: Potent 20 S proteasome inhibitors as promising cancer chemotherapeutics. Angew. Chem. Int. Ed. 2010, 49, 9346–9367. [Google Scholar] [CrossRef]
- Ahn, K.S.; Sethi, G.; Chao, T.-H.; Neuteboom, S.T.; Chaturvedi, M.M.; Palladino, M.A.; Younes, A.; Aggarwal, B.B. The Journal of the American Society of Hematology, Salinosporamide A (NPI-0052) potentiates apoptosis, suppresses osteoclastogenesis, and inhibits invasion through down-modulation of NF-κB–regulated gene products. J. Am. Soc. Hematol. 2007, 110, 2286–2295. [Google Scholar]
- Baritaki, S.; Yeung, K.; Palladino, M.; Berenson, J.; Bonavida, B.J. Pivotal roles of snail inhibition and RKIP induction by the proteasome inhibitor NPI-0052 in tumor cell chemoimmunosensitization. Cancer Res. 2009, 69, 8376–8385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baritaki, S.; Chapman, A.; Yeung, K.; Spandidos, D.; Palladino, M.; Bonavida, B. Inhibition of epithelial to mesenchymal transition in metastatic prostate cancer cells by the novel proteasome inhibitor, NPI-0052: Pivotal roles of Snail repression and RKIP induction. Oncogene 2009, 28, 3573–3585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, C.P.; Ban, K.; Dujka, M.E.; McConkey, D.J.; Munsell, M.; Palladino, M.; Chandra, J. The Journal of the American Society of Hematology, NPI-0052, a novel proteasome inhibitor, induces caspase-8 and ROS-dependent apoptosis alone and in combination with HDAC inhibitors in leukemia cells. J. Am. Soc. Hematol. 2007, 110, 267–277. [Google Scholar]
- Millward, M.; Price, T.; Townsend, A.; Sweeney, C.; Spencer, A.; Sukumaran, S.; Longenecker, A.; Lee, L.; Lay, A.; Sharma, G. Phase 1 clinical trial of the novel proteasome inhibitor marizomib with the histone deacetylase inhibitor vorinostat in patients with melanoma, pancreatic and lung cancer based on in vitro assessments of the combination. Investig. New Drugs 2012, 30, 2303–2317. [Google Scholar] [CrossRef]
- Harrison, S.J.; Mainwaring, P.; Price, T.; Millward, M.J.; Padrik, P.; Underhill, C.R.; Cannell, P.K.; Reich, S.D.; Trikha, M.; Spencer, A.J. Phase I clinical trial of marizomib (NPI-0052) in patients with advanced malignancies including multiple myeloma: Study NPI-0052-102 final results. Clin. Cancer Res. 2016, 22, 4559–4566. [Google Scholar] [CrossRef] [Green Version]
- Levin, N.; Spencer, A.; Harrison, S.J.; Chauhan, D.; Burrows, F.J.; Anderson, K.C.; Reich, S.D.; Richardson, P.G.; Trikha, M.J. Marizomib irreversibly inhibits proteasome to overcome compensatory hyperactivation in multiple myeloma and solid tumour patients. Br. J. Haematol. 2016, 174, 711–720. [Google Scholar] [PubMed]
- Di, K.; Lloyd, G.K.; Abraham, V.; MacLaren, A.; Burrows, F.J.; Desjardins, A.; Trikha, M.; Bota, D. Marizomib activity as a single agent in malignant gliomas: Ability to cross the blood-brain barrier. Neuro-Oncology 2016, 18, 840–848. [Google Scholar] [CrossRef] [Green Version]
- Potts, B.C.; Lam, K.S. Generating a generation of proteasome inhibitors: From microbial fermentation to total synthesis of salinosporamide a (marizomib) and other salinosporamides. Mar. Drugs 2010, 8, 835–880. [Google Scholar] [CrossRef] [Green Version]
- Fenteany, G.; Standaert, R.F.; Lane, W.S.; Choi, S.; Corey, E.J.; Schreiber, S.L. Inhibition of proteasome activities and subunit-specific amino-terminal threonine modification by lactacystin. Science 1995, 268, 726–731. [Google Scholar] [CrossRef] [PubMed]
- Fenteany, G.; Schreiber, S. Lactacystin, proteasome function, and cell fate. J. Biol. Chem. 1998, 273, 8545–8548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aikawa, S.-I.; Matsuzawa, F.; Satoh, Y.; Kadota, Y.; Doi, H.; Itoh, K. Proteomics, Prediction of the mechanism of action of omuralide (clasto-lactacystin β-lactone) on human cathepsin A based on a structural model of the yeast proteasome β5/PRE2-subunit/omuralide complex. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2006, 1764, 1372–1380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Liu, H.; Zu, X.; Liu, Y.; Chen, L.; Zhu, X.; Zhang, L.; Zhou, Z.; Xiao, G.; Wang, W. The ubiquitin-proteasome system is essential for the productive entry of Japanese encephalitis virus. Virology 2016, 498, 116–127. [Google Scholar] [CrossRef]
- Li, X.; Yang, D.; Li, L.; Peng, C.; Chen, S.; Le, W. Proteasome inhibitor lactacystin disturbs the intracellular calcium homeostasis of dopamine neurons in ventral mesencephalic cultures. Neurochem. Int. 2007, 50, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Xing, L.-F.; Wang, D.-T.; Yang, Y.; Pan, S.-Y. Effect of HDAC-6 on PD cell induced by lactacystin. Asian Pac. J. Trop. Med. 2015, 8, 855–859. [Google Scholar] [CrossRef] [Green Version]
- Soucy, F.; Grenier, L.; Behnke, M.L.; Destree, A.T.; McCormack, T.A.; Adams, J.; Plamondon, L.J. A novel and efficient synthesis of a highly active analogue of clasto-lactacystin β-lactone. J. Am. Chem. Soc. 1999, 121, 9967–9976. [Google Scholar] [CrossRef]
- Verma, R.; Peters, N.R.; D’Onofrio, M.; Tochtrop, G.P.; Sakamoto, K.M.; Varadan, R.; Zhang, M.; Coffino, P.; Fushman, D.; Deshaies, R.J. Ubistatins inhibit proteasome-dependent degradation by binding the ubiquitin chain. Science 2004, 306, 117–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellows, D.S.; Tyers, M.J. Chemical genetics hits ”reality”. Science 2004, 306, 67–68. [Google Scholar] [PubMed]
- Hanada, M.; Sugawara, K.; Kaneta, K.; Toda, S.; Nishiyama, Y.; Tomita, K.; Yamamoto, H.; Konishi, M.; Oki, T. Epoxomicin, a new antitumor agent of microbial origin. J. Antibiot. 1992, 45, 1746–1752. [Google Scholar]
- Sugawara, K.; Hatori, M.; Nishiyama, Y.; Tomita, K.; Kamei, H.; Konishi, M.; Oki, T. eponemycin, a new antibiotic active against b16 melanoma i. production, isolation, structure and biological activity. J. Antibiot. 1990, 43, 8–18. [Google Scholar]
- Di Giovanni, C.; Ettari, R.; Sarno, S.; Rotondo, A.; Bitto, A.; Squadrito, F.; Altavilla, D.; Schirmeister, T.; Novellino, E.; Grasso, S. Identification of noncovalent proteasome inhibitors with high selectivity for chymotrypsin-like activity by a multistep structure-based virtual screening. Eur. J. Med. Chem. 2016, 121, 578–591. [Google Scholar] [CrossRef]
- Ziogas, D.C.; Terpos, E.; Kastritis, E.; Dimopoulos, M. Carfilzomib for treating myeloma. Expert Opin. Orphan Drugs 2016, 4, 989–999. [Google Scholar]
- Archer, C.R.; Koomoa, D.-L.T.; Mitsunaga, E.M.; Clerc, J.; Shimizu, M.; Kaiser, M.; Schellenberg, B.; Dudler, R.; Bachmann, A. Syrbactin class proteasome inhibitor-induced apoptosis and autophagy occurs in association with p53 accumulation and Akt/PKB activation in neuroblastoma. Biochem. Pharmacol. 2010, 80, 170–178. [Google Scholar] [PubMed]
- Groll, M.; Schellenberg, B.; Bachmann, A.S.; Archer, C.R.; Huber, R.; Powell, T.K.; Lindow, S.; Kaiser, M.; Dudler, R. A plant pathogen virulence factor inhibits the eukaryotic proteasome by a novel mechanism. Nature 2008, 452, 755–758. [Google Scholar] [CrossRef]
- Ramel, C.; Tobler, M.; Meyer, M.; Bigler, L.; Ebert, M.-O.; Schellenberg, B.; Dudler, R.J. Biosynthesis of the proteasome inhibitor syringolin A: The ureido group joining two amino acids originates from bicarbonate. BMC Biochem. 2009, 10, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachmann, K.A.; Lewis, J.D. Predicting inhibitory drug—Drug interactions and evaluating drug interaction reports using inhibition constants. Ann. Pharmacother. 2005, 39, 1064–1072. [Google Scholar] [CrossRef]
- Coleman, C.; Rocetes, J.; Park, D.; Wallick, C.; Warn-Cramer, B.; Michel, K.; Dudler, R.; Bachmann, A. Syringolin A, a new plant elicitor from the phytopathogenic bacterium Pseudomonas syringae pv. syringae, inhibits the proliferation of neuroblastoma and ovarian cancer cells and induces apoptosis. Cell Prolif. 2006, 39, 599–609. [Google Scholar]
- Bachmann, A.S.; Dudler, R.; Groll, M. Pharmaceutical Compositions for the Treatment of Conditions Responsive to Proteasome Inhibition. United States Patent US 8,597,904, 3 December 2013. [Google Scholar]
- Pirrung, M. Synthesis of Syrbactin Proteasome Inhibitors. United States Patent US 9,221,772, 29 December 2015. [Google Scholar]
- Momose, I.; Sekizawa, R.; Hashizume, H.; Kinoshita, N.; Homma, Y.; Hamada, M.; Iinuma, H.; Takeuchi, T. Tyropeptins A and B, new proteasome inhibitors produced by Kitasatospora sp. MK993-dF2. J. Antibiot. 2001, 54, 997–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, T.; Momose, I.; Abe, M.; Abe, H.; Sawa, R.; Umezawa, Y.; Ikeda, D.; Takahashi, Y.; Akamatsu, Y. Synthesis of boronic acid derivatives of tyropeptin: Proteasome inhibitors. Bioorganic Med. Chem. Lett. 2009, 19, 2343–2345. [Google Scholar] [CrossRef]
- Momose, I.; Sekizawa, R.; Iinuma, H.; Takeuchi, T. Inhibition of proteasome activity by tyropeptin A in PC12 cells. Biosci. Biotechnol. Biochem. 2002, 66, 2256–2258. [Google Scholar] [CrossRef] [Green Version]
- Momose, I.; Abe, H.; Watanabe, T.; Ohba, S.I.; Yamazaki, K.; Dan, S.; Yamori, T.; Masuda, T.; Nomoto, A.J. Antitumor effects of tyropeptin-boronic acid derivatives: New proteasome inhibitors. Cancer Sci. 2014, 105, 1609–1615. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Hideshima, T.; Anderson, K.C. Bortezomib (PS-341): A novel, first-in-class proteasome inhibitor for the treatment of multiple myeloma and other cancers. Cancer Control J. Moffitt Cancer Cent. 2003, 10, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Raedler, L. Velcade (Bortezomib) Receives 2 New FDA Indications: For Retreatment of Patients with Multiple Myeloma and for First-Line Treatment of Patients with Mantle-Cell Lymphoma. Am. Health Drug Benefits 2015, 8, 135–140. [Google Scholar] [PubMed]
- Boccadoro, M.; Morgan, G.; Cavenagh, J. Preclinical evaluation of the proteasome inhibitor bortezomib in cancer therapy. Cancer Cell Int. 2005, 5, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, M.-H.; Bagain, L.; Chen, Z.; Karpova, T.; Yang, X.; Silvin, C.; Voss, T.C.; McNally, J.G.; Van Waes, C.; Hager, G.L. Dynamic effect of bortezomib on nuclear factor-kappaB activity and gene expression in tumor cells. Mol. Pharm. 2008, 74, 1215–1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nawrocki, S.T.; Carew, J.S.; Dunner, K., Jr.; Boise, L.H.; Chiao, P.J.; Huang, P.; Abbruzzese, J.L.; McConkey, D.J. Bortezomib inhibits PKR-like endoplasmic reticulum (ER) kinase and induces apoptosis via ER stress in human pancreatic cancer cells. Cancer Res. 2005, 65, 11510–11519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bui, H.T.T.; Le, N.H.; Le, Q.A.; Kim, S.E.; Lee, S.; Kang, D. Synergistic apoptosis of human gastric cancer cells by bortezomib and TRAIL. Int. J. Med. Sci. 2019, 16, 1412–1423. [Google Scholar]
- Yuan, B.Z.; Chapman, J.; Reynolds, S.H. Proteasome inhibitors induce apoptosis in human lung cancer cells through a positive feedback mechanism and the subsequent Mcl-1 protein cleavage. Oncogene 2009, 28, 3775–3786. [Google Scholar] [CrossRef] [Green Version]
- Brüning, A.; Burger, P.; Vogel, M.; Rahmeh, M.; Friese, K.; Lenhard, M.; Burges, A. Bortezomib treatment of ovarian cancer cells mediates endoplasmic reticulum stress, cell cycle arrest, and apoptosis. Investig. New Drugs 2009, 27, 543–551. [Google Scholar]
- Teicher, B.A.; Tomaszewski, J.E. Proteasome inhibitors. Biochem. Pharmacol. 2015, 96, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Jayaweera, S.P.E.; Wanigasinghe Kanakanamge, S.P.; Rajalingam, D.; Silva, G.N. Carfilzomib: A Promising Proteasome Inhibitor for the Treatment of Relapsed and Refractory Multiple Myeloma. Front. Oncol. 2021, 11, e740796. [Google Scholar] [CrossRef]
- Park, J.; Cho, J.; Song, E.J. Ubiquitin-proteasome system (UPS) as a target for anticancer treatment. Arch. Pharmacal. Res. 2020, 43, 1144–1161. [Google Scholar] [CrossRef]
- Khan, M.L.; Stewart, A.K. Carfilzomib: A novel second-generation proteasome inhibitor. Future Oncol. 2011, 7, 607–612. [Google Scholar] [PubMed] [Green Version]
- Xie, J.; Wan, N.; Liang, Z.; Zhang, T.; Jiang, J. Ixazomib—The first oral proteasome inhibitor. Leuk. Lymphoma 2019, 60, 610–618. [Google Scholar] [CrossRef]
- Wang, J.; Shi, Y.; Jiang, D. β-lactone derivatives and their anticancer activities: A short review. Curr. Top. Med. Chem. 2021, 21, 1645–1656. [Google Scholar] [CrossRef] [PubMed]
- Seyed, M.A.; Ayesha, S. Marine-derived pipeline anticancer natural products: A review of their pharmacotherapeutic potential and molecular mechanisms. Future J. Pharm. Sci. 2021, 7, 203. [Google Scholar] [CrossRef]
- Chauhan, D.; Singh, A.; Brahmandam, M.; Podar, K.; Hideshima, T.; Richardson, P.; Munshi, N.; Palladino, M.A.; Anderson, K.C. Combination of proteasome inhibitors bortezomib and NPI-0052 trigger in vivo synergistic cytotoxicity in multiple myeloma. Blood 2008, 111, 1654–1664. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, S.; Krupnik, Y.; Keating, M.; Chandra, J.; Palladino, M.; McConkey, D. The proteasome inhibitor NPI-0052 is a more effective inducer of apoptosis than bortezomib in lymphocytes from patients with chronic lymphocytic leukemia. Mol. Cancer Ther. 2006, 5, 1836–1843. [Google Scholar] [CrossRef] [Green Version]
- Cusack, J.C., Jr.; Liu, R.; Xia, L.; Chao, T.H.; Pien, C.; Niu, W.; Palombella, V.J.; Neuteboom, S.T.; Palladino, M.A. NPI-0052 enhances tumoricidal response to conventional cancer therapy in a colon cancer model. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006, 12, 6758–6764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badros, A.; Singh, Z.; Dhakal, B.; Kwok, Y.; MacLaren, A.; Richardson, P.; Trikha, M.; Hari, P. Marizomib for central nervous system-multiple myeloma. Br. J. Haematol. 2017, 177, 221–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Accardi, F.; Toscani, D.; Bolzoni, M.; Dalla Palma, B.; Aversa, F.; Giuliani, N. Mechanism of Action of Bortezomib and the New Proteasome Inhibitors on Myeloma Cells and the Bone Microenvironment: Impact on Myeloma-Induced Alterations of Bone Remodeling. BioMed Res. Int. 2015, 2015, 172458. [Google Scholar] [CrossRef] [PubMed] [Green Version]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Agent | Active Moiety | Therapeutic (UPS) Target | Binding Kinetics (Towards Proteasome) |
|---|---|---|---|
| Bortezomib | Boronate | CT-L > T-L | Reversible |
| Carfilzomib | Epoxyketone | CT-L | Irreversible |
| Ixazomib | Boronate | CT-L | Reversible |
| Marizomib | β-Lactone | CT-L,T-L > C-L | Irreversible |
| Oprozomib | Epoxyketone | CT-L | Irreversible |
| Delanzomib | Boronate | CT-L, C-L | Reversible |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mir, R.H.; Mir, P.A.; Uppal, J.; Chawla, A.; Patel, M.; Bardakci, F.; Adnan, M.; Mohi-ud-din, R. Evolution of Natural Product Scaffolds as Potential Proteasome Inhibitors in Developing Cancer Therapeutics. Metabolites 2023, 13, 509. https://doi.org/10.3390/metabo13040509
Mir RH, Mir PA, Uppal J, Chawla A, Patel M, Bardakci F, Adnan M, Mohi-ud-din R. Evolution of Natural Product Scaffolds as Potential Proteasome Inhibitors in Developing Cancer Therapeutics. Metabolites. 2023; 13(4):509. https://doi.org/10.3390/metabo13040509
Chicago/Turabian StyleMir, Reyaz Hassan, Prince Ahad Mir, Jasreen Uppal, Apporva Chawla, Mitesh Patel, Fevzi Bardakci, Mohd Adnan, and Roohi Mohi-ud-din. 2023. "Evolution of Natural Product Scaffolds as Potential Proteasome Inhibitors in Developing Cancer Therapeutics" Metabolites 13, no. 4: 509. https://doi.org/10.3390/metabo13040509
APA StyleMir, R. H., Mir, P. A., Uppal, J., Chawla, A., Patel, M., Bardakci, F., Adnan, M., & Mohi-ud-din, R. (2023). Evolution of Natural Product Scaffolds as Potential Proteasome Inhibitors in Developing Cancer Therapeutics. Metabolites, 13(4), 509. https://doi.org/10.3390/metabo13040509