
Natural Product Discovery Using Planes of Principal Component Analysis in R (PoPCAR)

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
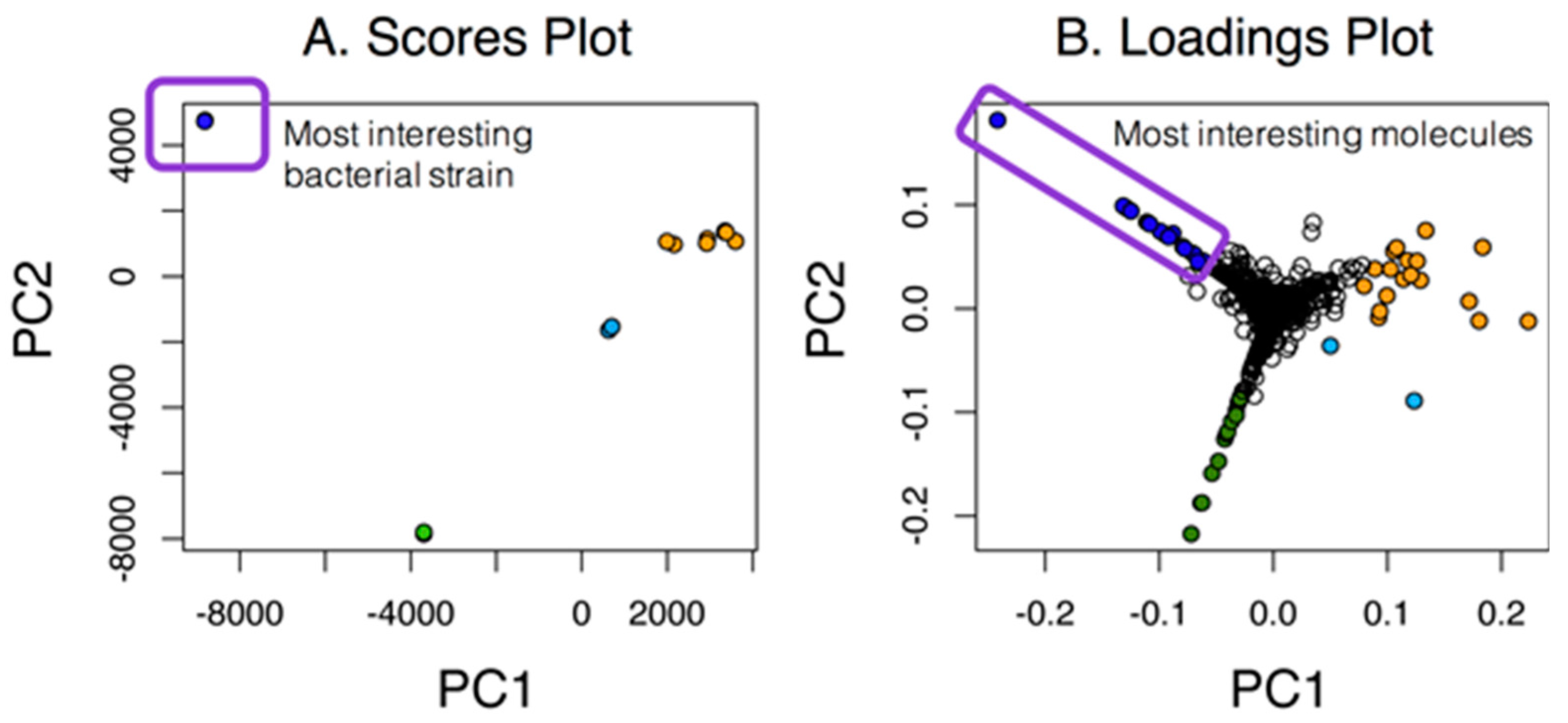
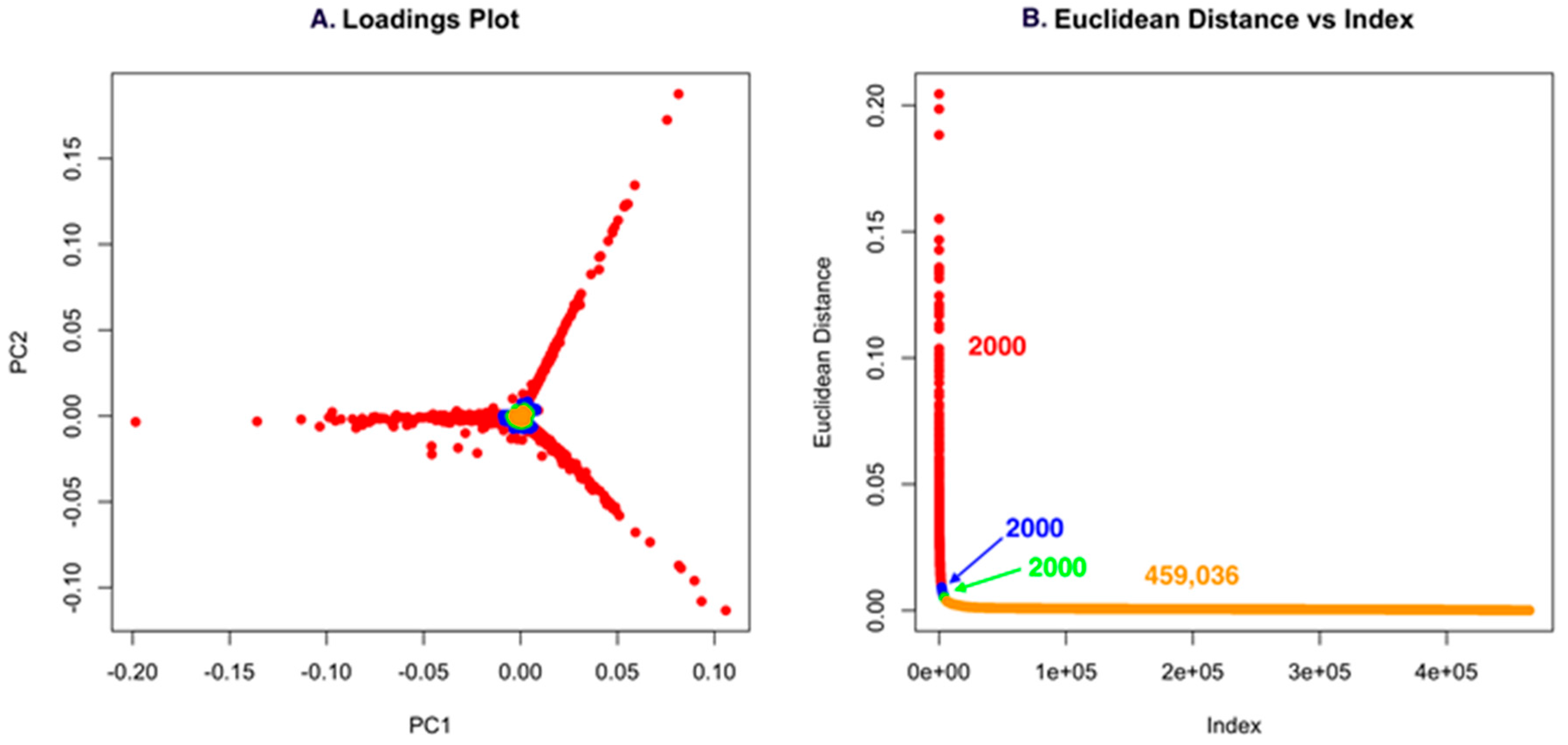
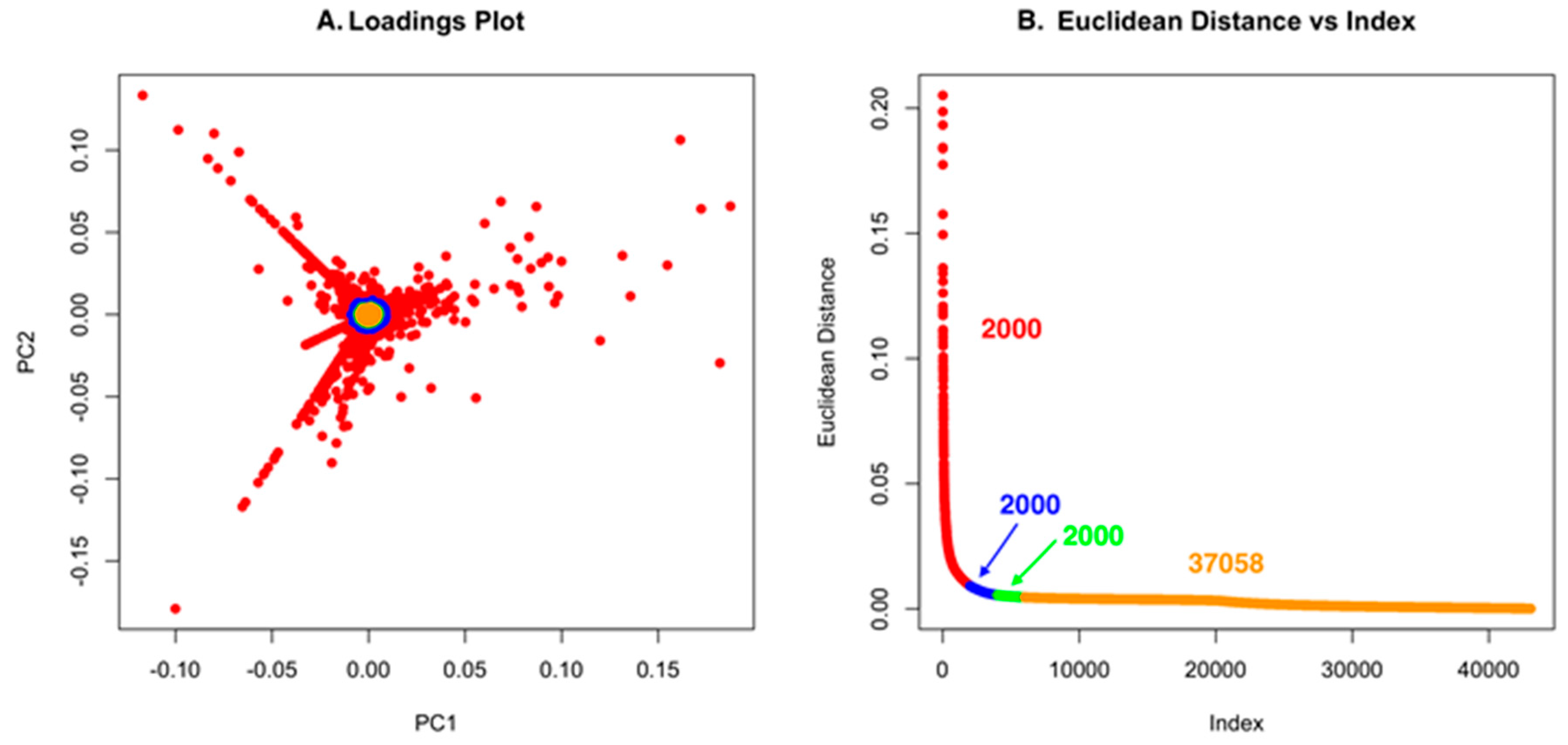
2.1. Untargeted Metabolomics and Feature Analysis
2.2. Dependence of the Analysis on the Group
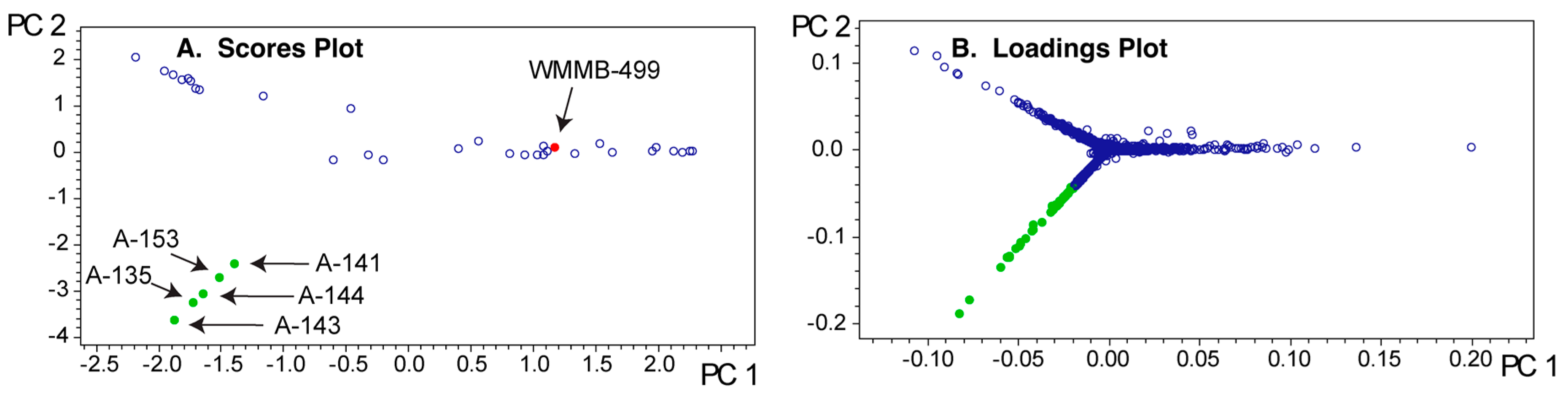
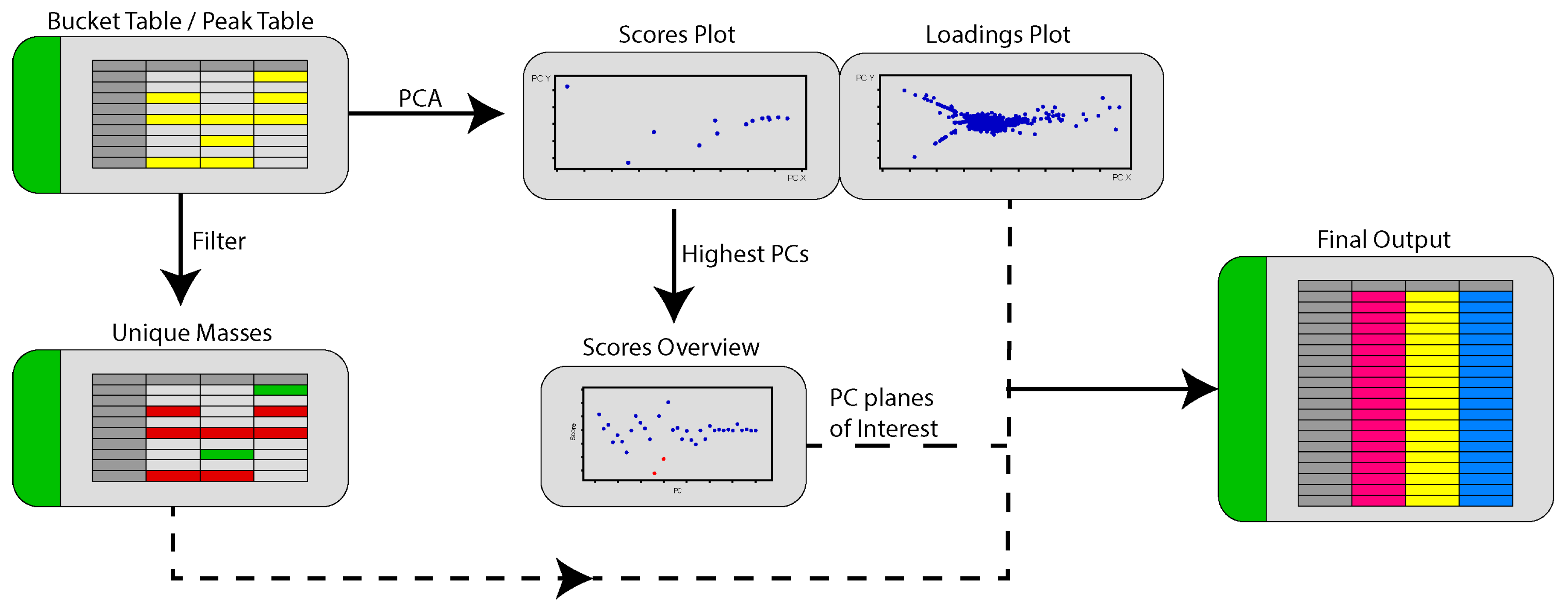
2.3. PoPCAR
3. Discussion
4. Materials and Methods
4.1. Bacterial Cultivation
4.2. Agar-based Media for LC-MS Profiling
4.3. Sample Preparation for UHPLC/HRESI-TOF-MS
4.4. UHPLC/HRMS Analysis.
4.5. Data Processing and PCA
4.6. PoPCAR
Supplementary Material
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wyche, T.P.; Ruzzini, A.C.; Beemelmanns, C.; Kim, K.H.; Klassen, J.L.; Cao, S.; Poulsen, M.; Bugni, T.S.; Currie, C.R.; Clardy, J. Linear Peptides Are the Major Products of a Biosynthetic Pathway That Encodes for Cyclic Depsipeptides. Org. Lett. 2017, 19, 1772–1775. [Google Scholar] [CrossRef] [PubMed]
- Beemelmanns, C.; Ramadhar, T.R.; Kim, K.H.; Klassen, J.L.; Cao, S.; Wyche, T.P.; Hou, Y.; Poulsen, M.; Bugni, T.S.; Currie, C.R.; et al. Macrotermycins A-D, Glycosylated Macrolactams from a Termite-Associated Amycolatopsis sp. M39. Org. Lett. 2017, 19, 1000–1003. [Google Scholar] [CrossRef] [PubMed]
- Van Arnam, E.B.; Ruzzini, A.C.; Sit, C.S.; Horn, H.; Pinto-Tomas, A.A.; Currie, C.R.; Clardy, J. Selvamicin, an atypical antifungal polyene from two alternative genomic contexts. Proc. Natl. Acad. Sci. USA 2016, 113, 12940–12945. [Google Scholar] [CrossRef] [PubMed]
- Derewacz, D.K.; McNees, C.R.; Scalmani, G.; Covington, C.L.; Shanmugam, G.; Marnett, L.J.; Polavarapu, P.L.; Bachmann, B.O. Structure and Stereochemical Determination of Hypogeamicins from a Cave-Derived Actinomycete. J. Nat. Prod. 2014, 77, 1759–1763. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Elshahawi, S.I.; Cai, W.; Zhang, Y.; Ponomareva, L.V.; Chen, X.; Copley, G.C.; Hower, J.C.; Zhan, C.G.; Parkin, S.; et al. Bi- and Tetracyclic Spirotetronates from the Coal Mine Fire Isolate Streptomyces sp. LC-6-2. J. Nat. Prod. 2017, 80, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, Y.; Ponomareva, L.V.; Qiu, Q.; Woodcock, R.; Elshahawi, S.I.; Chen, X.; Zhou, Z.; Hatcher, B.E.; Hower, J.C.; et al. Mccrearamycins A-D, Geldanamycin-Derived Cyclopentenone Macrolactams from an Eastern Kentucky Abandoned Coal Mine Microbe. Angew. Chem. Int. Ed. Engl. 2017, 56, 2994–2998. [Google Scholar] [CrossRef] [PubMed]
- Shaaban, K.A.; Saunders, M.A.; Zhang, Y.; Tran, T.; Elshahawi, S.I.; Ponomareva, L.V.; Wang, X.; Zhang, J.; Copley, G.C.; Sunkara, M.; et al. Spoxazomicin D and Oxachelin C, Potent Neuroprotective Carboxamides from the Appalachian Coal Fire-Associated Isolate Streptomyces sp. RM-14-6. J. Nat. Prod. 2017, 80, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Leutou, A.S.; Yang, I.; Kang, H.; Seo, E.K.; Nam, S.J.; Fenical, W. Nocarimidazoles A and B from a Marine-Derived Actinomycete of the Genus Nocardiopsis. J. Nat. Prod. 2015, 78, 2846–2849. [Google Scholar] [CrossRef] [PubMed]
- Nam, S.J.; Kauffman, C.A.; Jensen, P.R.; Moore, C.E.; Rheingold, A.L.; Fenical, W. Actinobenzoquinoline and Actinophenanthrolines A-C, Unprecedented Alkaloids from a Marine Actinobacterium. Org. Lett. 2015, 17, 3240–3243. [Google Scholar] [CrossRef] [PubMed]
- Edlund, A.; Loesgen, S.; Fenical, W.; Jensen, P.R. Geographic distribution of secondary metabolite genes in the marine actinomycete Salinispora arenicola. Appl. Environ. Microbiol. 2011, 77, 5916–5925. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Franco, C.M.; Zhang, W. Sponge-associated actinobacterial diversity: Validation of the methods of actinobacterial DNA extraction and optimization of 16S rRNA gene amplification. Appl. Microbiol. Biotechnol. 2015, 99, 8731–8740. [Google Scholar] [CrossRef] [PubMed]
- Abdelmohsen, U.R.; Yang, C.; Horn, H.; Hajjar, D.; Ravasi, T.; Hentschel, U. Actinomycetes from Red Sea sponges: Sources for chemical and phylogenetic diversity. Mar. Drugs 2014, 12, 2771–2789. [Google Scholar] [CrossRef] [PubMed]
- Abdelmohsen, U.R.; Bayer, K.; Hentschel, U. Diversity, abundance and natural products of marine sponge-associated actinomycetes. Nat. Prod. Rep. 2014, 31, 381–399. [Google Scholar] [CrossRef] [PubMed]
- Wyche, T.P.; Piotrowski, J.S.; Hou, Y.; Braun, D.; Deshpande, R.; McIlwain, S.; Ong, I.M.; Myers, C.L.; Guzei, I.A.; Westler, W.M.; et al. Forazoline A: Marine-derived polyketide with antifungal in vivo efficacy. Angew. Chem. Int. Ed. Engl. 2014, 53, 11583–11586. [Google Scholar] [CrossRef] [PubMed]
- Krug, D.; Zurek, G.; Revermann, O.; Vos, M.; Velicer, G.J.; Muller, R. Discovering the hidden secondary metabolome of Myxococcus xanthus: A study of intraspecific diversity. Appl. Environ. Microbiol. 2008, 74, 3058–3068. [Google Scholar] [CrossRef] [PubMed]
- Krug, D.; Zurek, G.; Schneider, B.; Garcia, R.; Muller, R. Efficient mining of myxobacterial metabolite profiles enabled by liquid chromatography-electrospray ionisation-time-of-flight mass spectrometry and compound-based principal component analysis. Anal. Chim. Acta 2008, 624, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Braun, D.R.; Michel, C.R.; Klassen, J.L.; Adnani, N.; Wyche, T.P.; Bugni, T.S. Microbial Strain Prioritization Using Metabolomics Tools for the Discovery of Natural Products. Anal. Chem. 2012, 84, 4277–4283. [Google Scholar] [CrossRef] [PubMed]
- Robertson, V.; Haltli, B.; McCauley, E.P.; Overy, D.P.; Kerr, R.G. Highly Variable Bacterial Communities Associated with the Octocoral Antillogorgia elisabethae. Microorganisms 2016, 4, 23. [Google Scholar] [CrossRef] [PubMed]
- Forner, D.; Berrue, F.; Correa, H.; Duncan, K.; Kerr, R.G. Chemical dereplication of marine actinomycetes by liquid chromatography-high resolution mass spectrometry profiling and statistical analysis. Anal. Chim. Acta 2013, 805, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Covington, B.C.; McLean, J.A.; Bachmann, B.O. Comparative mass spectrometry-based metabolomics strategies for the investigation of microbial secondary metabolites. Nat. Prod. Rep. 2017, 34, 6–24. [Google Scholar] [CrossRef] [PubMed]
- Derewacz, D.K.; Covington, B.C.; McLean, J.A.; Bachmann, B.O. Mapping Microbial Response Metabolomes for Induced Natural Product Discovery. ACS Chem. Biol. 2015, 10, 1998–2006. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, C.R.; Covington, B.C.; Derewacz, D.K.; McNees, C.R.; Wikswo, J.P.; McLean, J.A.; Bachmann, B.O. Structuring Microbial Metabolic Responses to Multiplexed Stimuli via Self-Organizing Metabolomics Maps. Chem. Biol. 2015, 22, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, C.R.; Sherrod, S.D.; Marasco, C.C.; Bachmann, B.O.; Schramm-Sapyta, N.; Wikswo, J.P.; McLean, J.A. Phenotypic mapping of metabolic profiles using self-organizing maps of high-dimensional mass spectrometry data. Anal. Chem. 2014, 86, 6563–6571. [Google Scholar] [CrossRef] [PubMed]
- Betancur, L.A.; Naranjo-Gaybor, S.J.; Vinchira-Villarraga, D.M.; Moreno-Sarmiento, N.C.; Maldonado, L.A.; Suarez-Moreno, Z.R.; Acosta-Gonzalez, A.; Padilla-Gonzalez, G.F.; Puyana, M.; Castellanos, L.; et al. Marine Actinobacteria as a source of compounds for phytopathogen control: An integrative metabolic-profiling/bioactivity and taxonomical approach. PLoS ONE 2017, 12, e0170148. [Google Scholar] [CrossRef] [PubMed]
- Carr, G.; Poulsen, M.; Klassen, J.L.; Hou, Y.; Wyche, T.P.; Bugni, T.S.; Currie, C.R.; Clardy, J. Microtermolides A and B from termite-associated Streptomyces sp. and structural revision of vinylamycin. Org. Lett. 2012, 14, 2822–2825. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Tianero, M.D.; Kwan, J.C.; Wyche, T.P.; Michel, C.R.; Ellis, G.A.; Vazquez-Rivera, E.; Braun, D.R.; Rose, W.E.; Schmidt, E.W.; et al. Structure and biosynthesis of the antibiotic bottromycin D. Org. Lett. 2012, 14, 5050–5053. [Google Scholar] [CrossRef] [PubMed]
- Doroghazi, J.R.; Albright, J.C.; Goering, A.W.; Ju, K.S.; Haines, R.R.; Tchalukov, K.A.; Labeda, D.P.; Kelleher, N.L.; Metcalf, W.W. A roadmap for natural product discovery based on large-scale genomics and metabolomics. Nat. Chem. Biol. 2014, 10, 963–968. [Google Scholar] [CrossRef] [PubMed]
- Jensen, P.R.; Moore, B.S.; Fenical, W. The marine actinomycete genus Salinispora: A model organism for secondary metabolite discovery. Nat. Prod. Rep. 2015, 32, 738–751. [Google Scholar] [CrossRef] [PubMed]
- Ziemert, N.; Lechner, A.; Wietz, M.; Millan-Aguinaga, N.; Chavarria, K.L.; Jensen, P.R. Diversity and evolution of secondary metabolism in the marine actinomycete genus Salinispora. Proc. Natl. Acad. Sci. USA 2014, 111, E1130–E1139. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef] [PubMed]
- Wyche, T.P.; Standiford, M.; Hou, Y.; Braun, D.; Johnson, D.A.; Johnson, J.A.; Bugni, T.S. Activation of the nuclear factor E2-related factor 2 pathway by novel natural products halomadurones A-D and a synthetic analogue. Mar. Drugs 2013, 11, 5089–5099. [Google Scholar] [CrossRef] [PubMed]
- Reasoner, D.J.; Geldreich, E.E. A new medium for the enumeration and subculture of bacteria from potable water. Appl. Environ. Microbiol. 1985, 49, 1–7. [Google Scholar] [PubMed]
- Shirling, E.B.; Gottlieb, D. Methods for characterization of Streptomyces species. Int. J. Syst. Evol. Microbiol. 1966, 16, 313–340. [Google Scholar] [CrossRef]
- Zhang, H.; Lee, Y.K.; Zhang, W.; Lee, H.K. Culturable actinobacteria from the marine sponge Hymeniacidon perleve: Isolation and phylogenetic diversity by 16S rRNA gene-RFLP analysis. Antonie Van Leeuwenhoek 2006, 90, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Gauze, G.F.; Preobrazhenskaya, J.P.; Kudrina, E.E.; Blinov, N.O.; Ryabova, I.D.; Sveshnikova, M.A. Problems in the Classification of Antagonistic Actinomycetes; Medgiz, State Publishing House of Medical Literature: Moscow, Russia, 1957. [Google Scholar]
- Harrison, P.J.; Waters, R.E.; Taylor, F.J.R. A Broad-Spectrum Artificial Seawater Medium for Coastal and Open Ocean Phytoplankton. J. Phycol. 1980, 16, 28–35. [Google Scholar] [CrossRef]
- Maldonado, L.A.; Fragoso-Yanez, D.; Perez-Garcia, A.; Rosellon-Druker, J.; Quintana, E.T. Actinobacterial diversity from marine sediments collected in Mexico. Antonie Van Leeuwenhoek 2009, 95, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, K.F.; Mansson, M.; Rank, C.; Frisvad, J.C.; Larsen, T.O. Dereplication of microbial natural products by LC-DAD-TOFMS. J. Nat. Prod. 2011, 74, 2338–2348. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, R.A.; Hoefsloot, H.C.; Westerhuis, J.A.; Smilde, A.K.; van der Werf, M.J. Centering, scaling, and transformations: Improving the biological information content of metabolomics data. BMC Genom. 2006, 7, 142. [Google Scholar] [CrossRef] [PubMed]
- RStudio Team. RStudio: Integrated Development for R; RStudio Inc.: Boston, MA, USA, 2016; Available online: https://www.rstudio.com/ (accessed on 29 April 2017).
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2017; Available online: https://www.R-project.org/ (accessed on 29 April 2017).
- Dowle, M.; Srinivasan, A. Data.Table: Extension of ‘Data.Frame’. R package version 1.10.4. 2017. Available online: https://CRAN.R-project.org/package=data.table (accessed on 29 April 2017).
- Wickham, H. Stringr: Simple, Consistent Wrappers for Common String Operations. R Package version 1.2.0. 2017. Available online: https://CRAN.R-project.org/package=stringr (accessed on 29 April 2017).
- Dragulescu, A.A. xlsx: Read, Write, Format Excel 2007 and Excel 97/2000/XP/2003 Files. R package version 0.5.7. 2014. Available online: https://CRAN.R-project.org/package=xlsx (accessed on 29 April 2017).








© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chanana, S.; Thomas, C.S.; Braun, D.R.; Hou, Y.; Wyche, T.P.; Bugni, T.S. Natural Product Discovery Using Planes of Principal Component Analysis in R (PoPCAR). Metabolites 2017, 7, 34. https://doi.org/10.3390/metabo7030034
Chanana S, Thomas CS, Braun DR, Hou Y, Wyche TP, Bugni TS. Natural Product Discovery Using Planes of Principal Component Analysis in R (PoPCAR). Metabolites. 2017; 7(3):34. https://doi.org/10.3390/metabo7030034
Chicago/Turabian StyleChanana, Shaurya, Chris S. Thomas, Doug R. Braun, Yanpeng Hou, Thomas P. Wyche, and Tim S. Bugni. 2017. "Natural Product Discovery Using Planes of Principal Component Analysis in R (PoPCAR)" Metabolites 7, no. 3: 34. https://doi.org/10.3390/metabo7030034
APA StyleChanana, S., Thomas, C. S., Braun, D. R., Hou, Y., Wyche, T. P., & Bugni, T. S. (2017). Natural Product Discovery Using Planes of Principal Component Analysis in R (PoPCAR). Metabolites, 7(3), 34. https://doi.org/10.3390/metabo7030034