A Lipidomic Analysis of Docosahexaenoic Acid (22:6, ω3) Mediated Attenuation of Western Diet Induced Nonalcoholic Steatohepatitis in Male Ldlr -/- Mice

 ,
,
Abstract
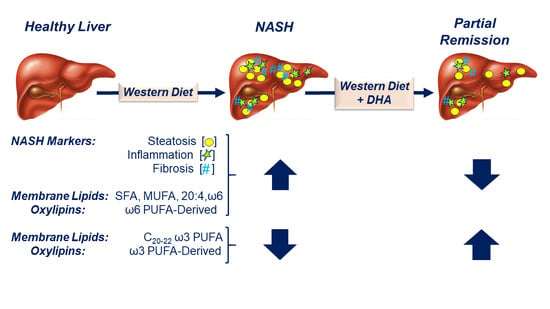
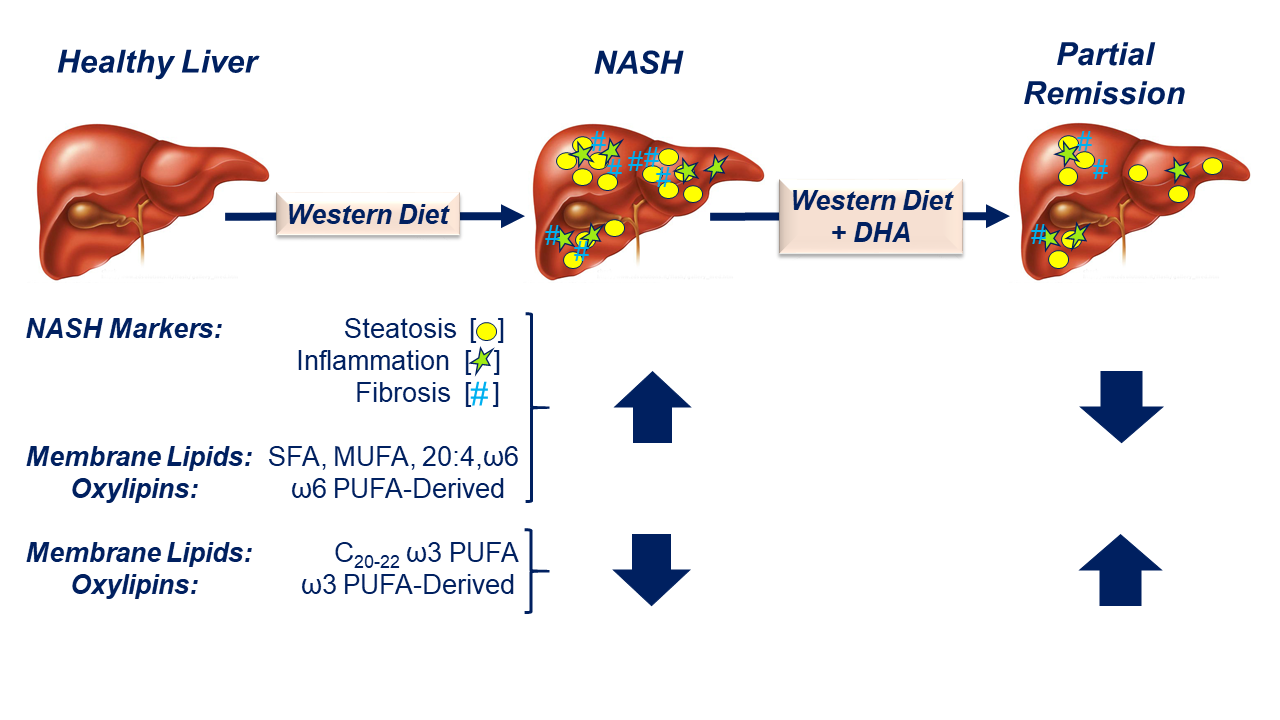
:
1. Introduction
2. Results
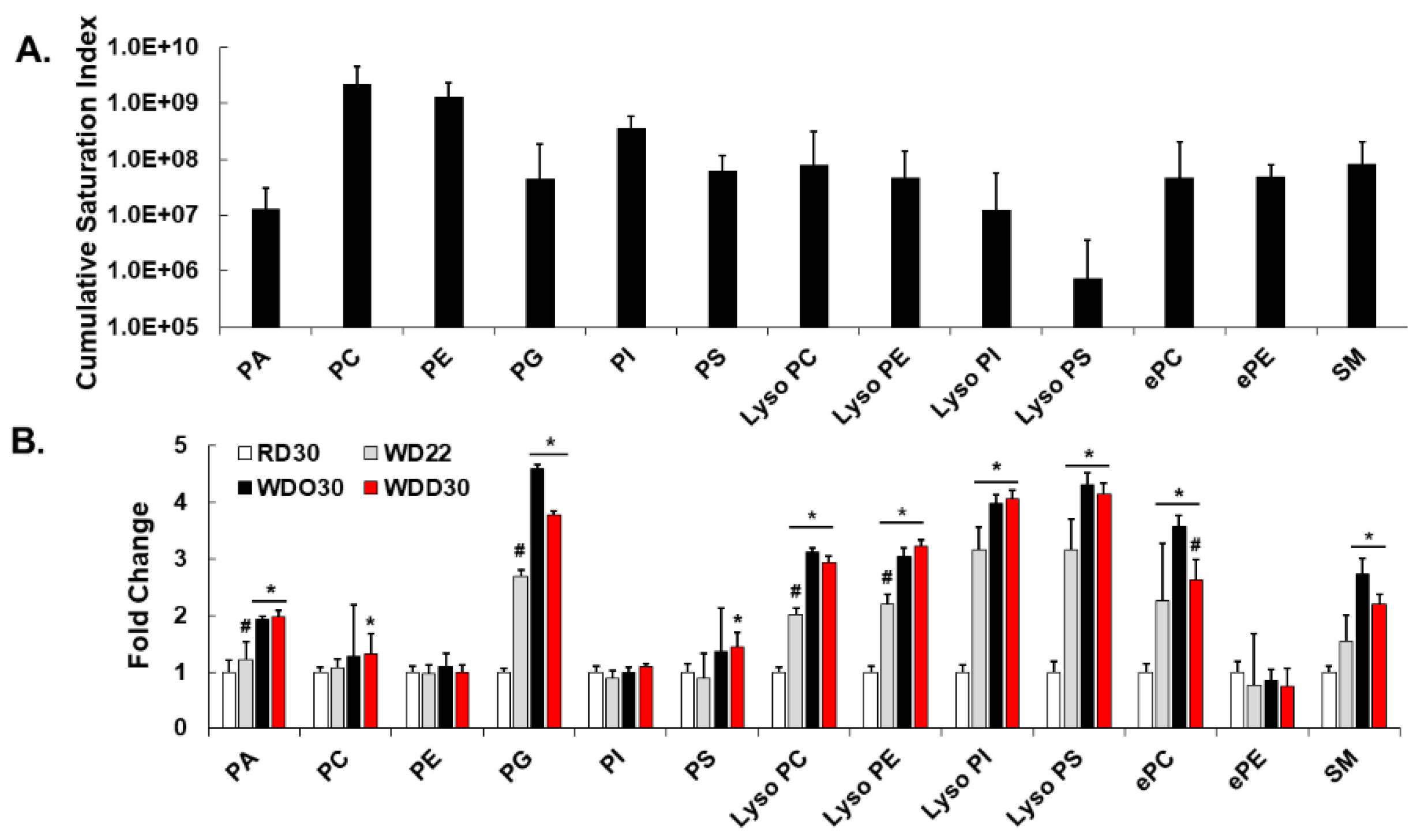
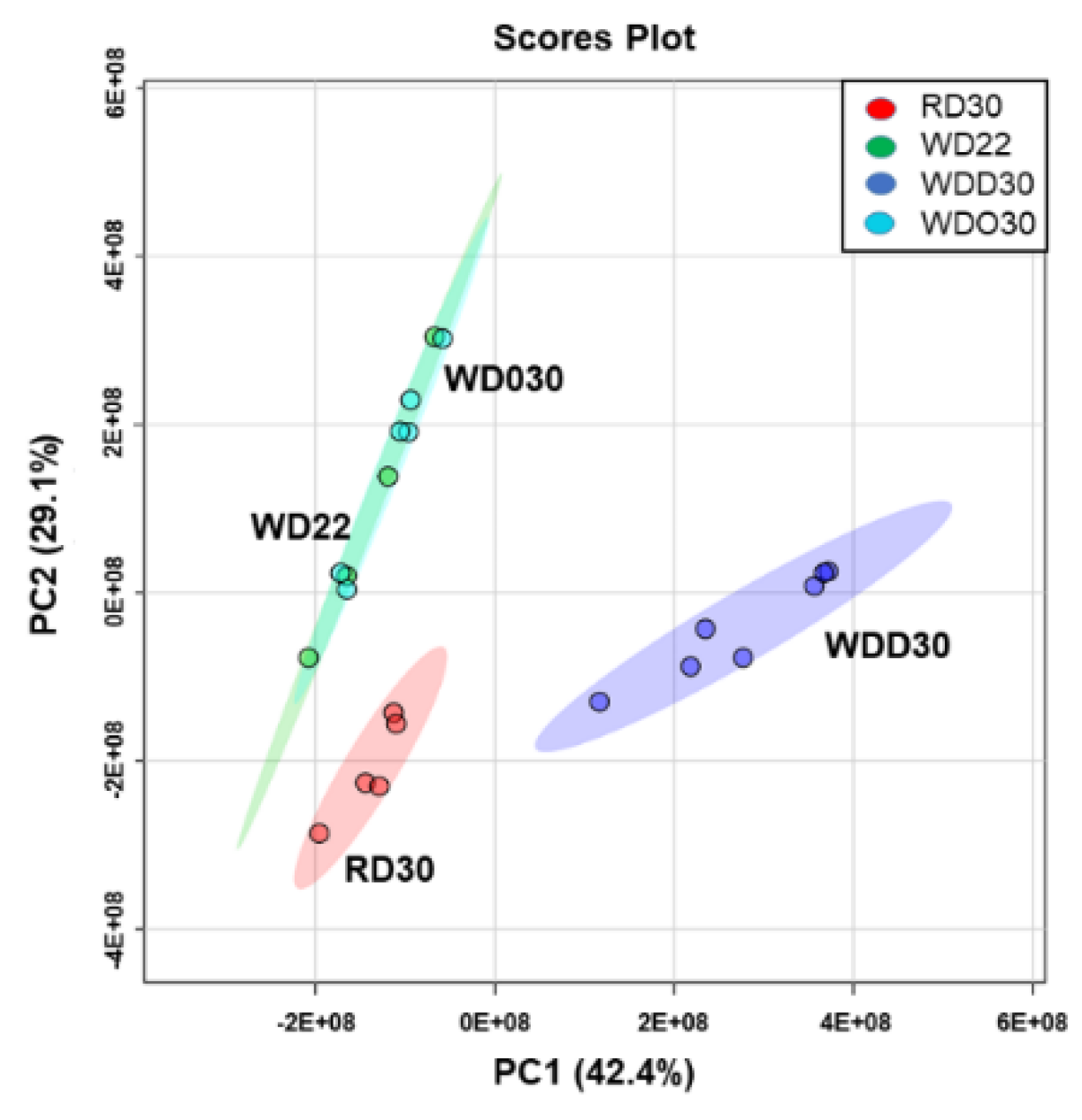
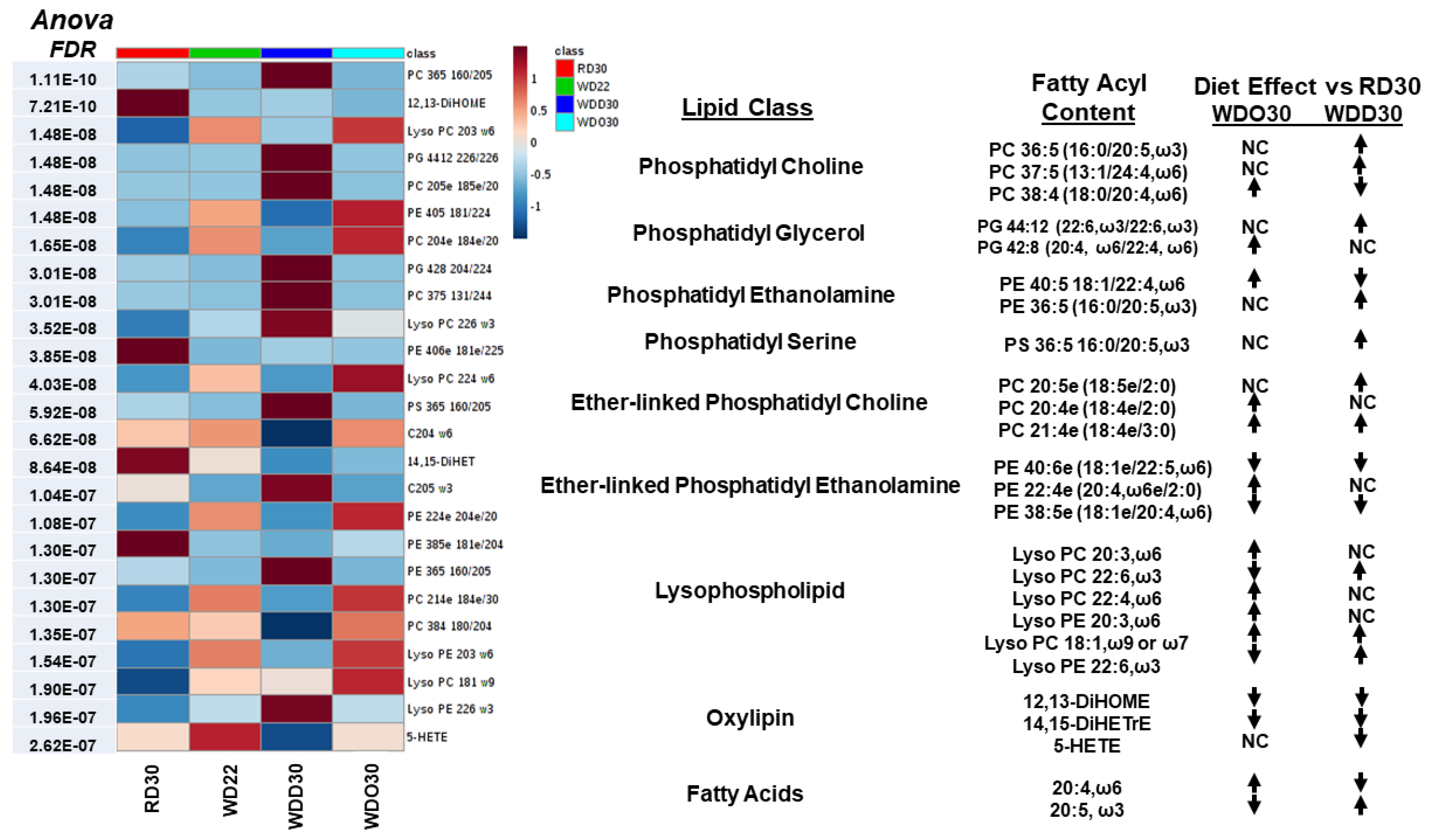
2.1. Impact of the Western Diet (WD) on Membrane Lipids
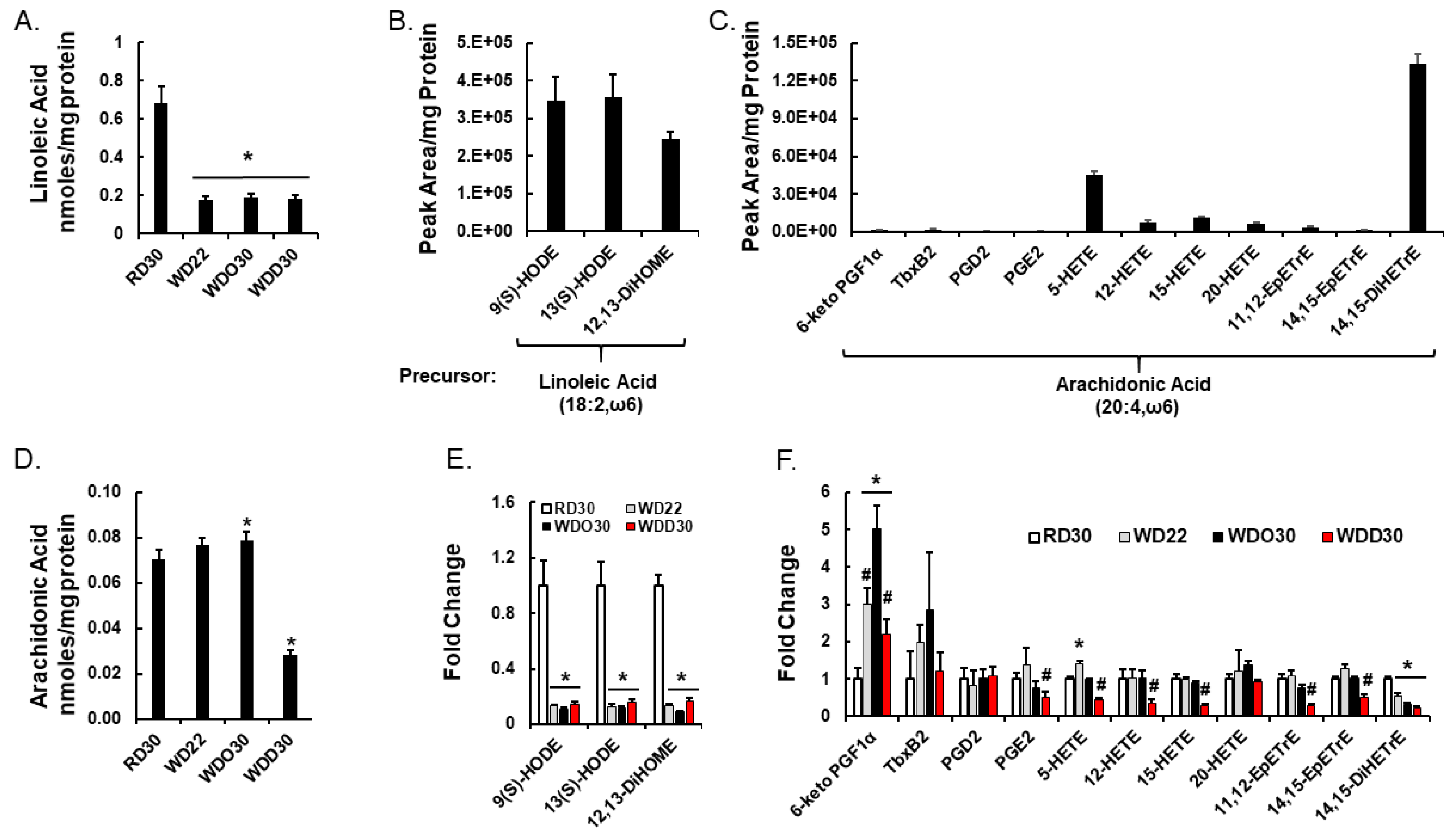
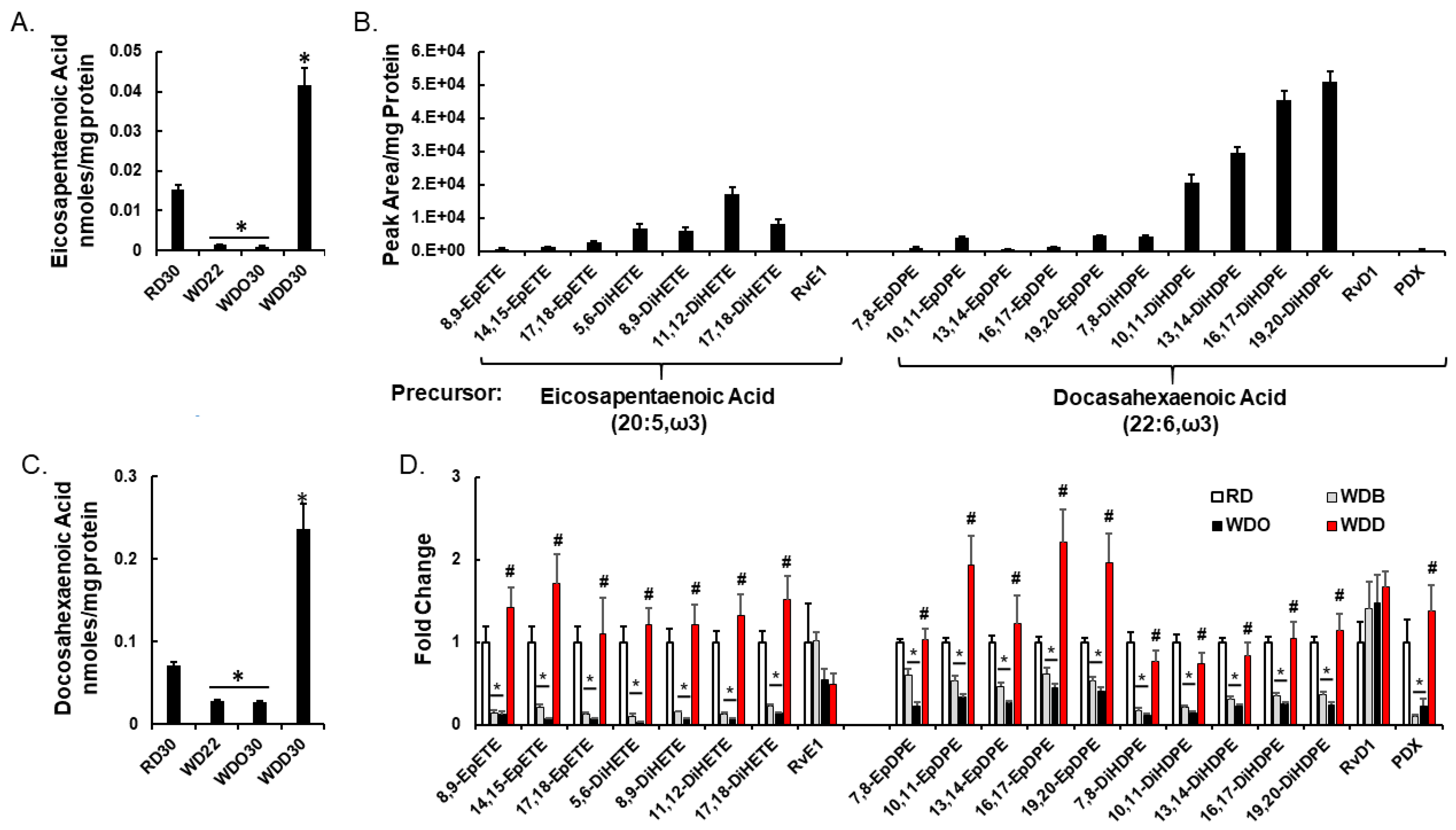
2.2. Diet Effects on Hepatic Non-Esterified Oxylipins
2.3. Associations between Hepatic Lipids and NASH Markers of Inflammation and Fibrosis
2.3.1. Inflammation
2.3.2. Fibrosis
3. Discussion
4. Materials and Methods
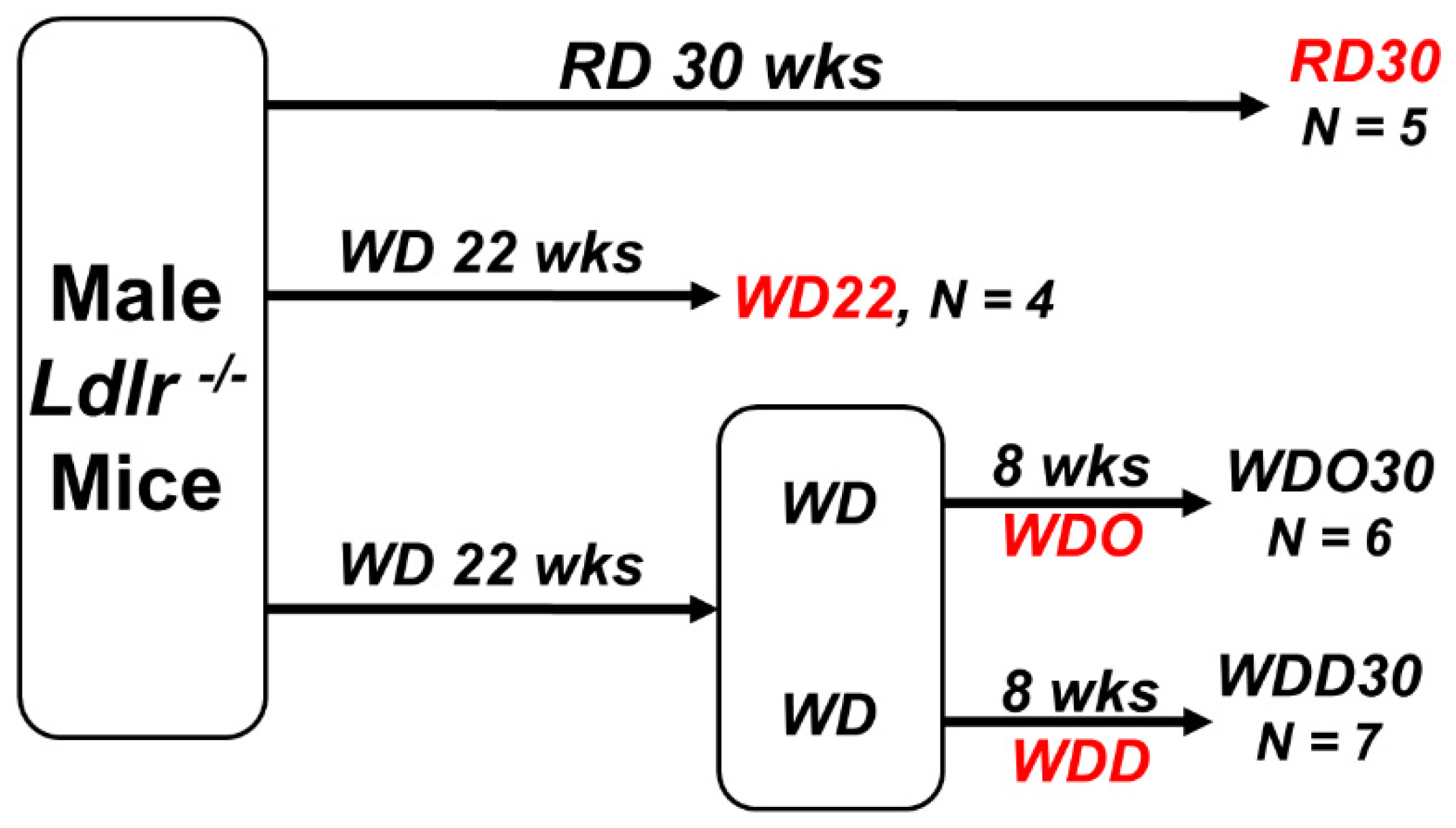
4.1. Study Design for DHA-Mediated NASH Remission in Male Ldlr -/- Mice
4.2. RNA Extraction and qRTPCR
4.3. Sample Preparation for Lipidomic Analysis
4.4. Sample Preparation for Oxylipins Analysis
4.5. Chromatographic and Mass Spectrometry Conditions for Lipids and Oxylipins Analysis
4.5.1. Untargeted Lipidomics
4.5.2. Targeted Oxylipidomics
4.6. Data Processing
4.6.1. Untargeted Lipidomics
4.6.2. Targeted Analysis of Oxylipins
4.6.3. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bellentani, S.; Scaglioni, F.; Marino, M.; Bedogni, G. Epidemiology of non-alcoholic fatty liver disease. Dig. Dis. 2010, 28, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Golabi, P.; de Avila, L.; Paik, J.M.; Srishord, M.; Fukui, N.; Qiu, Y.; Burns, L.; Afendy, A.; Nader, F. The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: A systematic review and meta-analysis. J. Hepatol. 2019, 4, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Angulo, P.; Lindor, K.D. Non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2002, 17, S186–S190. [Google Scholar] [CrossRef] [PubMed]
- Neuschwander-Tetri, B.A.; Caldwell, S.H. Nonalcoholic steatohepatitis: Summary of an AASLD Single Topic Conference. Hepatology 2003, 37, 1202–1219. [Google Scholar] [CrossRef]
- Farrell, G.C.; Larter, C.Z. Nonalcoholic fatty liver disease: From steatosis to cirrhosis. Hepatology 2006, 43, S99–S112. [Google Scholar] [CrossRef]
- Cohen, J.C.; Horton, J.D.; Hobbs, H.H. Human fatty liver disease: Old questions and new insights. Science 2011, 332, 1519–1523. [Google Scholar] [CrossRef]
- Lauby-Secretan, B.; Scoccianti, C.; Loomis, D.; Grosse, Y.; Bianchini, F.; Straif, K. Body fatness and cancer-viewpoint of the IARC working group. N. Engl. J. Med. 2016, 375, 794–798. [Google Scholar] [CrossRef]
- Amarapurkar, D.; Kamani, P.; Patel, N.; Gupte, P.; Kumar, P.; Agal, S.; Baijal, R.; Lala, S.; Chaudhary, D.; Deshpande, A. Prevalence of non-alcoholic fatty liver disease: Population based study. Ann. Hepatol. 2007, 6, 161–163. [Google Scholar] [CrossRef]
- Center for Disease Control and Prevention. Adult Obesity Facts. 2015. Available online: http://www.cdc.gov/obesity/data/adult.html (accessed on 21 September 2019).
- Center for Disease Control and Prevention. Childhood Obesity Facts. 2015. Available online: http://www.cdc.gov/obesity/data/childhood.html (accessed on 21 September 2019).
- Spooner, M.H.; Jump, D.B. Omega-3 Fatty Acids and Nonalcoholic Fatty Liver Disease in Adults and Children: Where Do We stand? Current Opinion in Clinical Nutrition and Metabolic Care; Wolters Kluwer: Alphen aan den Rijn, The Netherlands, 2019. [Google Scholar]
- Cussons, A.J.; Watts, G.F.; Mori, T.A.; Stuckey, B.G. Omega-3 fatty acid supplementaion decreases liver fat content in polycystic ovary syndrome: A randomized controlled trial employing proton magnetic resonance spectroscopy. J. Clin. Endocrinol. Metabol. 2009, 94, 3842–3848. [Google Scholar] [CrossRef]
- Soderberg, C.; Stal, P.; Askling, J.; Glaumann, H.; Lindberg, G.; Marmur, J.; Hultcrantz, R. Decreased survival of subjects with elevated liver function tests during a 28-year follow-up. Hepatology 2010, 51, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Ekstedt, M.; Franzen, L.E.; Mathiesen, U.L.; Thorelius, L.; Holmqvist, M.; Bodemar, G.; Kechagias, S. Long-term follow-up of patients with NAFLD and elevated liver enzymes. Hepatology 2006, 44, 865–873. [Google Scholar] [CrossRef] [PubMed]
- Adams, L.A.; Lymp, J.F.; St Sauver, J.; Sanderson, S.O.; Lindor, K.D.; Feldstein, A.; Angulo, P. The natural history of nonalcoholic fatty liver disease: A population-based cohort study. Gastroenterology 2005, 129, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Diehl, A.M.; Brunt, E.M.; Cusi, K.; Charlton, M.; Sanyal, A.J. The diagnosis and management of non-alcoholic fatty liver disease: Practice guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Am. J. Gastroenterol. 2012, 107, 811–826. [Google Scholar] [CrossRef]
- Alberti, K.G.; Zimmet, P.; Shaw, J. The metabolic syndrome—A new worldwide definition. Lancet 2005, 366, 1059–1062. [Google Scholar] [CrossRef]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- LaBrecque, D.; Abbas, Z.; Anania, F.; Ferenci, P.; Gahafoor Kahn, A.; Goh, K.-L.; Hamid, S.S.; Isakov, V.; Lizarzabal, M.; Mojica Pernaranda, M.; et al. Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis; World Gastroentrology Organization Global Guidelines: Milwaukee, WI, USA, 2012; pp. 1–29. [Google Scholar]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef]
- Schuppan, D.; Kim, Y.O. Evolving therapies for liver fibrosis. J. Clin. Invest. 2013, 123, 1887–1901. [Google Scholar] [CrossRef] [Green Version]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef]
- Angulo, P. Nonalcoholic fatty liver disease. N. Engl. J. Med. 2002, 346, 1221–1231. [Google Scholar] [CrossRef]
- Friedman, S.L. Mechanisms of hepatic fibrogenesis. Gastroenterology 2008, 134, 1655–1669. [Google Scholar] [CrossRef] [PubMed]
- Koyama, Y.; Brenner, D.A. New therapies for hepatic fibrosis. Clin. Res. Hepatol. Gastroenterol. 2015, 39, S75–S79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenbloom, J.; Mendoza, F.A.; Jimenez, S.A. Strategies for anti-fibrotic therapies. Biochim. Biophys. Acta 2013, 1832, 1088–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilar-Gomez, E.; Martenez-Perez, Y.; Calzadilla-Bertot, L.; Torres-Gonzalez, A.; Gra-Oramas, B.; Gonzalez-Fabian, L.; Friedman, S.L.; Diago, M.; Romero-Gomez, M. Weight loss through lifestyle modification significantly reduces features of nonalcoholic steatohepatitis. Gastroenterology 2015, 149, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.L.; de Silva, H.J.; Leung, N.W.; Lim, S.G.; Farrell, G.C. How should we manage patients with non-alcoholic fatty liver disease in 2007. J. Gastroenterol. Hepatol. 2007, 22, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Leslie, M. The liver’s weight problem. Science 2015, 6243, 18–20. [Google Scholar]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Diehl, A.M.; Brunt, E.M.; Cusi, K.; Charlton, M.; Sanyal, A.J. The diagnosis and management of non-alcoholic fatty liver disease: Practice guideline by the American Gastroenterological Association, American Association for the Study of Liver Diseases, and American College of Gastroenterology. Gastroenterology 2012, 142, 1592–1609. [Google Scholar] [CrossRef]
- Lee, Y.A.; Wallace, M.C.; Friedman, S.L. Pathology of liver fibrosis: A translational success story. Gut 2015, 64, 830–841. [Google Scholar] [CrossRef]
- Kappler, R. From crawl to sprint: The race to treat NASH. Life Sci. VC 2015. Available online: https://lifescivc.com/2015/05/from-crawl-to-sprint-the-race-to-treat-nash/ (accessed on 21 September 2019).
- Musso, G.; Cassader, M.; Rosina, F.; Gambino, R. Impact of current treatments on liver disease, glucose metabolism and cardiovascular risk in non-alcoholic fatty liver disease (NAFLD): A systematic review and meta-analysis of randomised trials. Diabetologia 2012, 55, 885–904. [Google Scholar] [CrossRef]
- Petit, J.M.; Guiu, B.; Duvillard, L.; Jooste, V.; Brindisi, M.C.; Athias, A.; Bouillet, B.; Habchi, M.; Cottet, V.; Gambert, P.; et al. Increased erythrocytes n-3 and n-6 polyunsaturated fatty acids is significantly associated with a lower prevalence of steatosis in patients with type 2 diabetes. Clin. Nutr. 2012, 31, 520–525. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.S.; Xu, A.; Huang, T.; Yu, X.; Li, D. Low docosahexaenoic acid content in plasma phospholipids is associated with increased non-alcoholic fatty liver disease in China. Lipids 2012, 47, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Parker, H.M.; Johnson, N.A.; Burdon, C.A.; Cohn, J.S.; O’Connor, H.T.; George, J. Omega-3 supplementation and non-alcoholic fatty liver disease: A systematic review and meta-analysis. J. Hepatol. 2012, 56, 944–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Minno, M.N.; Russolillo, A.; Lupoli, R.; Ambrosino, P.; Di Minno, A.; Tarantino, G. Omega-3 fatty acids for the treatment of non-alcoholic fatty liver disease. World J. Gastroenterol. 2012, 18, 5839–5847. [Google Scholar] [CrossRef]
- Nobili, V.; Bedogni, G.; Alisi, A.; Pietrobattista, A.; Rise, P.; Galli, C.; Agostoni, C. Docosahexaenoic acid supplementation decreases liver fat content in children with non-alcoholic fatty liver disease: Double-blind randomised controlled clinical trial. Arch. Dis. Child. 2011, 96, 350–353. [Google Scholar] [CrossRef]
- Shapiro, H.; Tehilla, M.; Attal-Singer, J.; Bruck, R.; Luzzatti, R.; Singer, P. The therapeutic potential of long-chain omega-3 fatty acids in nonalcoholic fatty liver disease. Clin. Nutr. 2011, 30, 6–19. [Google Scholar] [CrossRef]
- Glass, L.M.; Dickson, R.C.; Anderson, J.C.; Sufiawinata, A.A.; Putra, J.; Berk, B.S.; Toor, A. Total body weight loss of ≥10% is associated with improved hepatic fibrosis in patients with nonalcoholic steatohepatitis. Dig. Dis. Sci. 2015, 60, 1024–1030. [Google Scholar] [CrossRef]
- Sofi, F.; Casini, A. Mediterranean diet and non-alcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 7339–7346. [Google Scholar] [CrossRef]
- Marchesini, G.; Mazzotti, A. NAFLD incidence and remission: Only a matter of weight gain and weight loss. J. Hepatol. 2015, 62, 15–17. [Google Scholar] [CrossRef]
- Panera, N.; Barbaro, B.; Della Corte, C.; Mosca, A.; Nobili, V.; Alisi, A. A review of the pathogenic and therapeutic role of nutrition in pediatric nonalcoholic fatty liver disease. Nutr. Res. 2018, 58, 1–16. [Google Scholar] [CrossRef]
- Sumida, Y.; Yoneda, M. Current and future pharmacological therapies for NAFLD/NASH. J. Gastroenterol. 2018, 53, 362–376. [Google Scholar] [CrossRef] [PubMed]
- Albhaisi, S.; Sanyal, A. Recent advances in understanding and managing non-alcoholic fatty liver disease. F1000Research 2018, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Jump, D.B.; Lytle, K.A.; Depner, C.M.; Tripathy, S. Omega-3 polyunsaturated fatty acids as a treatment strategy for nonalcoholic fatty liver disease. Pharmacol. Ther. 2018, 181, 108–125. [Google Scholar] [CrossRef] [PubMed]
- Banini, B.A.; Sanyal, A.J. Current and future pharmacologic treatment of nonalcoholic steatohepatitis. Curr. Opin. Gastroenterol. 2017, 33, 134–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCollough, A.J. Epidemiology of the metabolic syndrome in the USA. J. Dig. Dis. 2011, 12, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Depner, C.M.; Philbrick, K.A.; Jump, D.B. Docosahexaenoic acid attenuates hepatic inflammation, oxidative stress, and fibrosis without decreasing hepatosteatosis in a Ldlr(-/-) mouse model of western diet-induced nonalcoholic steatohepatitis. J. Nutr. 2013, 143, 315–323. [Google Scholar] [CrossRef]
- Lytle, K.A.; Jump, D.B. Is western diet-induced nonalcoholic steatohepatitis in Ldlr-/- mice reversible. PLoS ONE 2016, 11, e0146942. [Google Scholar] [CrossRef]
- Lytle, K.A.; Wong, C.P.; Jump, D.B. Docosahexaenoic acid blocks progression of western diet-induced nonalcoholic steatohepatitis in obese Ldlr-/- mice. PLoS ONE 2017, 12, e0173376. [Google Scholar] [CrossRef]
- Simopoulos, A.P. n-3 fatty acid-enriched eggs, lipids, and Western diet: Time for change. Nutrition 1993, 9, 561–562. [Google Scholar]
- Cordain, L.; Eaton, S.B.; Sebastian, A.; Mann, N.; Lindeberg, S.; Watkins, B.A.; O’Keefe, J.H.; Brand-Miller, J. Orgins and evolution of the western diet: Health implications for the 21st century. Am. J. Clin. Nutr. 2005, 81, 341–354. [Google Scholar] [CrossRef]
- Allard, J.B.; Aghdassi, E.; Mohammed, S.; Raman, M.; Avand, G.; Arendt, B.M.; Jalali, P.; Kandasamy, T.; Prayitno, N.; Sherman, M.; et al. Nutrition assessment and hepatic fatty acid composition in non-alcoholic fatty liver disease (NAFLD): A cross-sectional study. J. Hepatol. 2007, 48, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Arendt, B.M.; Comelli, E.M.; Ma, D.W.; Lou, W.; Teterina, A.; Kim, T.; Fung, D.K.H.; McGilvray, I.; Fischer, S.E.; Allard, J.P. Altered hepatic gene expression in non-alcoholic fatty liver disease is associated with lower n-3 and n-6 polyunsaturated fatty acids. Hepatology 2015, 61, 1565–1578. [Google Scholar] [CrossRef] [PubMed]
- Spahis, S.; Alverez, F.; Dubois, J.; Ahmed, N.; Peretti, N.; Levy, E. Plasma fatty acid composition in French-Canadian children with non-alsocholic fatty liver disease: Effect of n-3 PUFA supplementation. Prostaglandins Leukot. Essent. Fat. Acids 2015, 99, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Jump, D.B. The biochemistry of n-3 polyunsaturated fatty acids. J. Biol. Chem. 2002, 277, 8755–8758. [Google Scholar] [CrossRef] [PubMed]
- Jump, D.B.; Tripathy, S.; Depner, C.M. Fatty Acid-regulated transcription factors in the liver. Ann. Rev. Nutr. 2013, 33, 249–269. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Jaramillo, M.; Spooner, M.H.; Loehr, C.V.; Wong, C.P.; Zhang, W.; Jump, D.B. Lipidomic and transcriptomic analysis of western diet-induced nonalcoholic steatohepatitis (NASH) in female Ldlr-/- mice. PLoS ONE 2019, 14, E0214387. [Google Scholar] [CrossRef]
- Gabbs, M.; Leng, S.; Devassy, J.G.; Monirujjaman, M.; Aukema, H.M. Advances in our understanding of oxylipins derived from dietary PUFAs. Adv. Nutr. 2015, 6, 513–540. [Google Scholar] [CrossRef]
- Jump, D.B.; Depner, C.M.; Tripathy, S. Omega-3 fatty acid supplementation and cardiovascular disease. J. Lipid Res. 2012, 53, 2525–2545. [Google Scholar] [CrossRef] [Green Version]
- Scorletti, E.; Bhatia, L.; McCormick, K.G.; Clough, G.F.; Nash, K.; Hodson, L.; Moyses, H.E.; Calder, P.C.; Byrne, C.D.; The Welcome Study Investigators. Effects of purified eicosapentaenoic and docosahexaenoic acids in non-alcoholic fatty liver disease: Results from the *WELCOME study. Hepatology 2014, 60, 1211–1221. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Abdelmalek, M.F.; Suzuki, A.; Cummings, O.W.; Chojkier, M. No significant effects of ethyl-eicosapentaenoic acid on histologic features of nonalcoholic steatohepatitis in a Phase 2 trial. Gastroenterology 2014, 147, 377–384. [Google Scholar] [CrossRef]
- Barter, P.; Ginsberg, H.N. Effectiveness of combined statin plus omega-3 fatty acid therapy for mixed dyslipidemia. Am. J. Cardiol. 2008, 102, 1040–1045. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef] [PubMed]
- Mater, M.K.; Thelen, A.P.; Jump, D.B. Arachidonic acid and PGE2 regulation of hepatic lipogenic gene expression. J. Lipid Res. 1999, 40, 1045–1052. [Google Scholar]
- Lopane, C.; Agosti, P.; Gigante, I.; Sabba, C.; Mazzocca, A. Implications of the lysophosphatidic acid signaling axis in liver cancer. Biochim. Biophys. Acta 2017, 1868, 277–282. [Google Scholar] [CrossRef]
- Ishii, I.; Fukushima, N.; Ye, X.; Chun, J. Lysophospholipid receptors: Signaling and biology. Ann. Rev. Biochem. 2004, 73, 321–354. [Google Scholar] [CrossRef]
- Gardell, S.E.; Dubin, A.E.; Chun, J. Emerging medicinal roles for lysophospholipid signaling. Trends Mol. Med. 2006, 12, 65–75. [Google Scholar] [CrossRef]
- Frasch, S.C.; Bratton, D.L. Emerging roles for lysophosphatidylserine in resolution of inflammation. Prog. Lipid Res. 2012, 51, 199–207. [Google Scholar] [CrossRef] [Green Version]
- Tsukahara, T.; Matsuda, Y.; Haniu, H. Lysophospholipid-Related Diseases and PPARgamma Signaling Pathway. Int. J. Mol. Sci. 2017, 18, 2730. [Google Scholar] [CrossRef]
- D’Souza, K.; Paramel, G.V.; Kienesberger, P.C. Lysophosphatidic Acid Signaling in Obesity and Insulin Resistance. Nutrients 2018, 10, 399. [Google Scholar] [CrossRef]
- Brown, A.J.; Jupe, S.; Briscoe, C.P. A family of fatty acid binding receptors. DNA Cell Biol. 2005, 24, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Funk, C.D. Prostaglandins and leukotrienes: Advances in eicosanoid biology. Science 2001, 294, 1871–1875. [Google Scholar] [CrossRef] [PubMed]
- Kostenis, E. A glance at G-protein-coupled receptors for lipid mediators: A growing receptor family with remarkably diverse ligands. Pharmacol. Ther. 2004, 102, 243–257. [Google Scholar] [CrossRef] [PubMed]
- Graves, J.P.; Gruzdev, A.; Bradbury, J.A.; DeGraff, L.M.; Li, H.; House, J.S.; Hoopes, S.L.; Edin, M.L.; Zeldin, D.C. Quantitative Polymerase Chain Reaction Analysis of the Mouse Cyp2j Subfamily: Tissue Distribution and Regulation. Drug Metab. Dispos. 2015, 43, 1169–1180. [Google Scholar] [CrossRef] [PubMed]
- Feldstein, A.E.; Lopez, R.; Tamimi, T.A.; Yerian, L.; Chung, Y.M.; Berk, M.; Zhang, R.; McIntyre, T.M.; Hazen, S.L. Mass spectrometric profiling of oxidized lipid products in human nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. J. Lipid Res. 2010, 51, 3046–3054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasegawa, E.; Inafuku, S.; Mulki, L.; Okunuki, Y.; Yanai, R.; Smith, K.E.; Kim, C.B.; Klokman, G.; Bielenberg, D.R.; Puli, N.; et al. Cytochrome P450 monooxygenase lipid metabolites are significant second messengers in the resolution of choroidal neovascularization. Proc. Natl. Acad. Sci. USA 2017, 114, E7545–E7553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calder, P.C. Marine omega-3 fatty acids and inflammatory processes: Effects, mechanisms and clinical relevance. Biochim. Biophys. Acta 2015, 1851, 469–484. [Google Scholar] [CrossRef] [PubMed]
- Palladini, G.; Di Pasqua, L.G.; Berardo, C.; Siciliano, V.; Richelmi, P.; Mannucci, B.; Croce, A.C.; Rizzo, V.; Perlini, S.; Vairetti, M.; et al. Fatty Acid Desaturase Involvement in Non-Alcoholic Fatty Liver Disease Rat Models: Oxidative Stress Versus Metalloproteinases. Nutrients 2019, 11, 799. [Google Scholar] [CrossRef]
- Stankovic, M.N.; Mladenovic, D.; Ninkovic, M.; Ethuricic, I.; Sobajic, S.; Jorgacevic, B.; de Luka, S.; Vukicevic, R.J.; Radosavljević, T.S. The effects of alpha-lipoic acid on liver oxidative stress and free fatty acid composition in methionine-choline deficient diet-induced NAFLD. J. Med. Food. 2014, 17, 254–261. [Google Scholar] [CrossRef]
- Lopez-Vicario, C.; Alcaraz-Quiles, J.; Garcia-Alonso, V.; Rius, B.; Hwang, S.H.; Titos, E.; Clària, J. Inhibition of soluble epoxide hydrolase modulates inflammation and autophagy in obese adipose tissue and liver: Role for omega-3 epoxides. Proc. Natl. Acad. Sci. USA 2015, 112, 536–541. [Google Scholar] [CrossRef]
- Matyash, V.; Liebisch, G.; Kurzchalia, T.V.; Shevchenko, A.; Schwudke, D. Lipid extraction by methyl-tert-butyl ether for high-throughput lipidomics. J. Lipid Res. 2008, 49, 1137–1146. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, T.L.; Newman, J.W. Establishing and performing targeted multi-residue analysis for lipid mediators and fatty acids in small clinical plasma samples. Methods Mol. Biol. 2018, 1730, 175–212. [Google Scholar] [PubMed]
- La Franko, M.R.; Hernandez-Carretero, A.; Weber, N.; Borkowski, K.; Pedersen, T.L.; Osborn, O.; Newman, J.W. Diet-induced obesity and weight loss alter bile acid concentrations and bile acid-sensitive gene expression in insulin target tissues of C57BL/6J mice. Nutr. Res. 2017, 46, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Cajka, T.; Fiehn, O. Increasing lipidomic coverage by selecting optimal mobile-phase modifiers in LC-MS of blood plasma. Metabolomics 2016, 12, 1–11. [Google Scholar] [CrossRef]
- Cajka, T.; Davis, R.; Austin, K.J.; Newman, J.W.; German, J.B.; Feihn, O.; Smilowitz, J.T. Using a lipidomics approach for nutritional phenotyping in resposne to a test meal containing gamma-linolenic acid. Metabolomics 2016, 12, 1–22. [Google Scholar] [CrossRef]
- Tsugawa, H.; Cajka, T.; Kind, T.; Ma, Y.; Higgins, B.; Ikeda, K.; Kanazawa, M.; VanderGheynst, J.; Fiehn, O.; Arita, M. MS-DIAL: Data-independent MS/MS deconvolution for comprehensive metabolome analysis. Nat. Methods. 2015, 12, 523–526. [Google Scholar] [CrossRef] [PubMed]
- Kind, T.; Liu, K.H.; Lee, D.Y.; DeFelice, B.; Meissen, J.K.; Fiehn, O. LipidBlast in silico tandem mass spectrometry database for lipid identification. Nat. Methods 2013, 10, 755–758. [Google Scholar] [CrossRef] [Green Version]
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, M.H.; Beger, R.; Daykin, C.A.; Fan, T.W.; Fiehn, O.; Goodacre, R.; Griffin, J.L.; et al. Proposed minimum reporting standards for chemical analysis Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics 2007, 3, 211–221. [Google Scholar] [CrossRef]
- Benjamini, Y.; Krieger, A.M.; Yekutieli, D. Adaptive linear step-up procedures that control the false discovery rate. Biometrika 2006, 93, 491–507. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
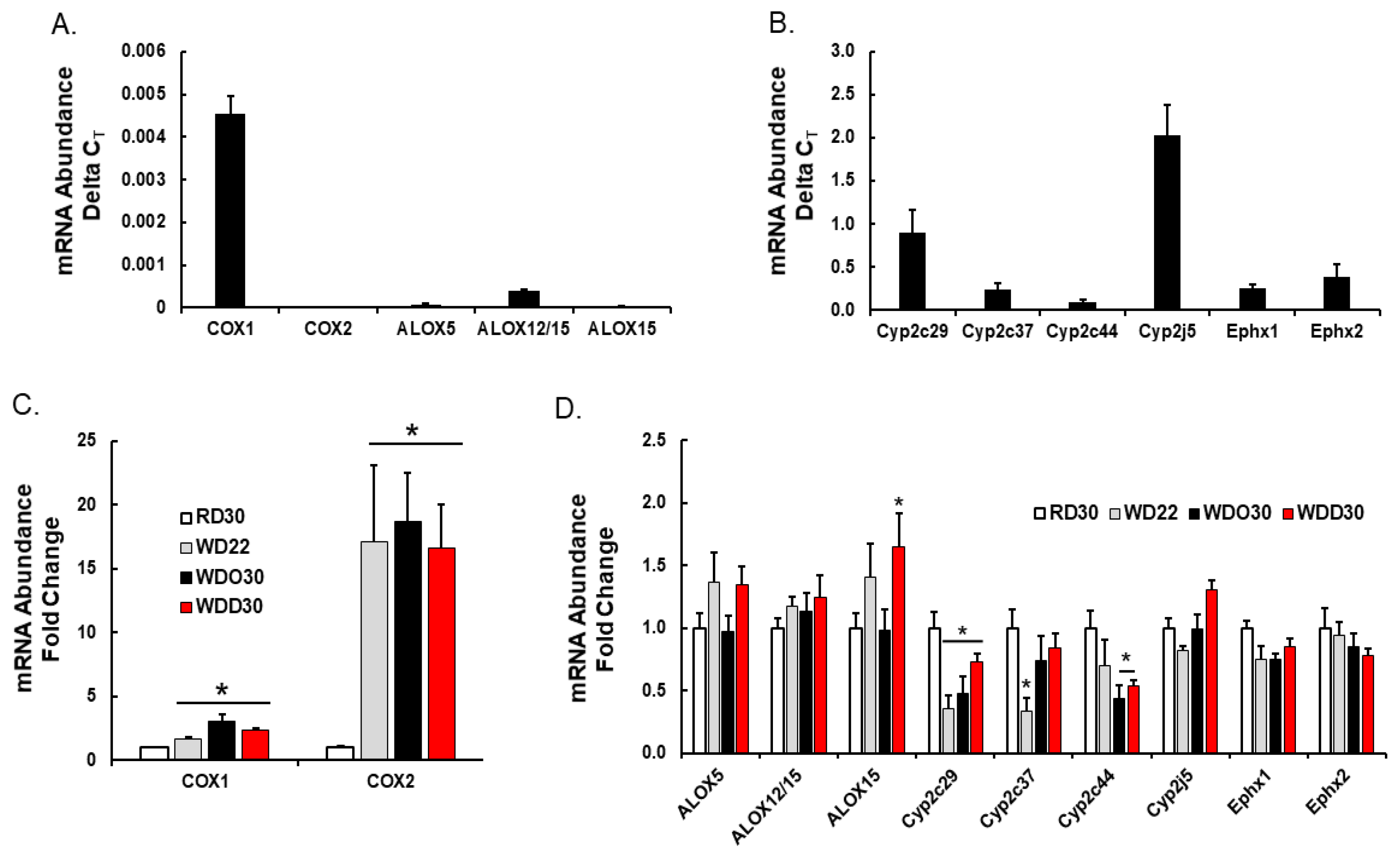{kind=link}
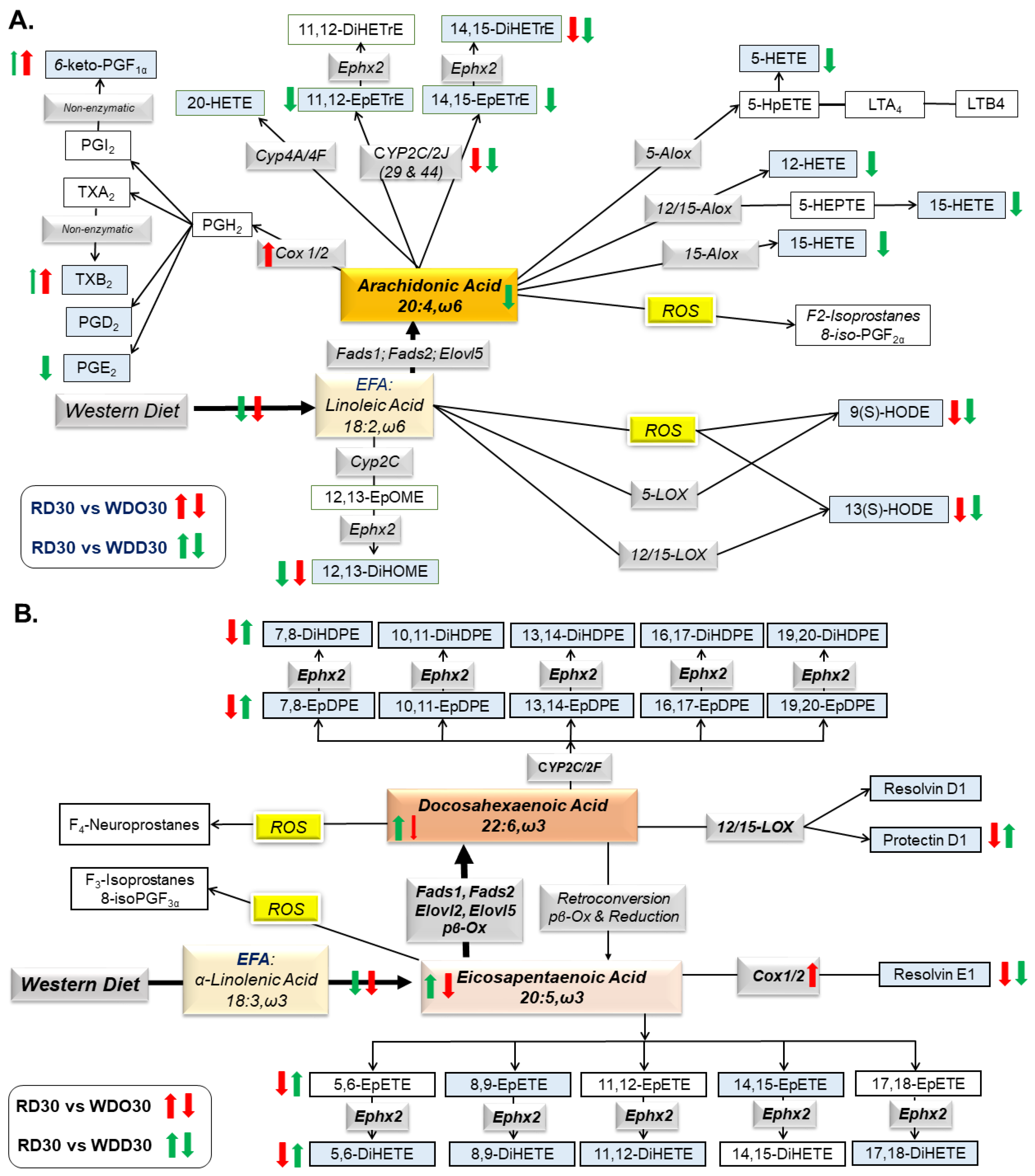{kind=link}
| Gene | Opn | Mcp1 | CD68 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Association | Lipid | CC 2 | q-Value 3 | Lipid | CC | q-Value | Lipid | CC | q-Value |
| Positive | PG 38:4 18:1/20:3,ω6 | 0.74 | 5.9 × 10−4 | Lyso PC 18:1 | 0.86 | 8.6 × 10−6 | Lyso PC 18:1 | 0.78 | 5.3 × 10−4 |
| Positive | Lyso PC 22:4,ω6 | 0.74 | 6.4 × 10−4 | PC 17:0e 14:0e/3:0 | 0.85 | 1.3 × 10−5 | SM d36:2 d14:0/22:2 | 0.78 | 5.4 × 10−4 |
| Positive | Lyso PE 22:4,ω6 | 0.74 | 6.6 × 10−4 | PC 16:0e 14:0e/2:0 | 0.84 | 2.3 × 10−5 | Lyso PC 20:3,ω6 | 0.77 | 6.4 × 10−4 |
| Positive | PG 36:4 16:0/20:4,ω6 | 0.73 | 8.4 × 10−4 | SM d35:1 d14:0/21:1 | 0.82 | 3.8 × 10−5 | PS 42:3 26:0/16:3 | 0.76 | 6.9 × 10−4 |
| Positive | PG 38:5 18:1/20:4,ω6 | 0.72 | 9.9 × 10−4 | PA 36:2 18:1/18:1 | 0.82 | 4.3 × 10−5 | PC 17:0e 14:0e/3:0 | 0.76 | 7.7 × 10−4 |
| Positive | 20:1,ω9 | 0.72 | 1.1 × 10−3 | PC 21:3e 18:3e/3:0 | 0.81 | 6.6 × 10−5 | PC 21:3e 18:3e/3:0 | 0.74 | 1.0 × 10−3 |
| Positive | PG 38:2 18:1/20:1 | 0.71 | 1.2 × 10−3 | PC 18:1e 16:1e/2:0 | 0.81 | 6.7 × 10−5 | SM d34:0 d14:0/20:0 | 0.74 | 1.1 × 10−3 |
| Positive | PG 36:2 18:1/18:1 | 0.71 | 1.3 × 10−3 | PE 20:1e 14:1e/6:0 | 0.80 | 7.8 × 10−5 | PC 18:1e 16:1e/2:0 | 0.72 | 1.4 × 10−3 |
| Positive | PG 42:9 20:3,ω6/22:6,ω3 | 0.71 | 1.3 × 10−3 | PC 19:1e 14:1e/5:0 | 0.80 | 8.6 × 10−5 | PC 16:0e 14:0e/2:0 | 0.72 | 1.4 × 10−3 |
| Positive | Lyso PE 20:3,ω6 | 0.70 | 1.4 × 10−3 | SM d34:1 d14:0/20:1 | 0.80 | 9.2 × 10−5 | PE 20:1e 14:1e/6:0 | 0.72 | 1.6 × 10−3 |
| Negative | PA 34:3 16:1,ω7/18:2,ω6 | −0.69 | 1.9 × 10−3 | 12,13-DiHOME | −0.78 | 1.3 × 10−4 | PI 36:2 18:1/18:1 | −0.77 | 6.4 × 10−4 |
| Negative | 7,8-DiHDPE | −0.62 | 6.9 × 10−3 | 18:3,ω3 | −0.76 | 2.9 × 10−4 | PE 34:2 16:0/18:2,ω6 | −0.76 | 6.9 × 10−4 |
| Negative | 13,14-DiHDPE | −0.62 | 6.9 × 10−3 | PI 36:2 18:1/18:1 | −0.76 | 2.9 × 10−4 | PI 36:4 16:0/20:4,ω6 | −0.76 | 7.7 × 10−4 |
| Negative | 7,8-EpDPE | −0.62 | 6.9 × 10−3 | PE 40:6e 18:1e/22:5,ω3 | −0.74 | 4.9 × 10−4 | PI 36:3 16:0/20:3,ω6 | −0.74 | 1.0 × 10−3 |
| Negative | PE 34:2 16:0/18:2,ω6 | −0.62 | 7.2 × 10−3 | PI 36:4 16:0/20:4.ω6 | −0.73 | 6.0 × 10−4 | PE 34:3e 16:1e/18:2,ω3 | −0.74 | 1.2 × 10−3 |
| Negative | 10,11-DiHDPE | −0.62 | 7.2 × 10−3 | PE 34:2 16:0/18:2,ω6 | −0.73 | 6.2 × 10−4 | 12,13-DiHOME | −0.73 | 1.4 × 10−3 |
| Negative | Lyso PE 18:2,ω6 | −0.62 | 7.4 × 10−3 | 18:2,ω6 | −0.73 | 6.3 × 10−4 | PE 36:2 18:0/18:2,ω6 | −0.72 | 1.4 × 10−3 |
| Negative | PI 36:3 16:0/20:3,ω6 | −0.61 | 8.9 × 10−3 | PA 34:3 16:1,ω7/18:2,ω6 | −0.72 | 6.8 × 10−4 | PE 36:2 18:1/18:1 | −0.72 | 1.5 × 10−3 |
| Negative | PC 35:2 13:1/22:1 | −0.59 | 1.1 × 10−2 | 14,15-DiHETrE | −0.72 | 6.9 × 10−4 | PE 40:6e 18:1e/22:5,ω3 | −0.71 | 1.8 × 10−3 |
| Negative | 5,6-DiHETE | −0.59 | 1.1 × 10−2 | 13(S)-HODE | −0.71 | 1.1 × 10−3 | 18:3,ω3 | −0.71 | 1.8 × 10−3 |
| GENE | Col1A2 | Timp1 | Lox | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Association | Lipid | CC 2 | q-Value 3 | Lipid | CC | q-Value | Lipid | CC | q-Value |
| Positive | 20:1,ω9 | 0.81 | 4.2 × 10−5 | 20:1,ω9 | 0.81 | 5.6 × 10−5 | Lyso PC 22:4,ω6 | 0.84 | 1.2 × 10−5 |
| Positive | 18:1,ω9 | 0.81 | 4.9 × 10−5 | 18:1,ω7 | 0.80 | 9.0 × 10−5 | PC 38:4 18:1/20:3,ω6 | 0.82 | 2.9 × 10−5 |
| Positive | 18:1,ω7 | 0.80 | 6.4 × 10−5 | 18:1,ω9 | 0.79 | 1.1 × 10−4 | Lyso PE 22:4,ω6 | 0.80 | 5.3 × 10−5 |
| Positive | Lyso PC 20:3,ω6 | 0.74 | 5.3 × 10−4 | Lyso PC 20:3,ω6 | 0.74 | 6.7 × 10−4 | PG 38:4 18:1/20:3,ω6 | 0.80 | 5.6 × 10−5 |
| Positive | Lyso PE 20:3,ω6 | 0.73 | 6.7 × 10−4 | 16:0 | 0.73 | 8.1 × 10−4 | PG 38:5 18:1/20:4,ω6 | 0.79 | 7.0 × 10−5 |
| Positive | PC 38:4 18:1/20:3,ω6 | 0.72 | 1.1 × 10−3 | Lyso PE 20:3, ω6 | 0.72 | 9.7 × 10−4 | PG 38:1 20:0/18:1 | 0.79 | 7.0 × 10−5 |
| Positive | Lyso PC 18:1,ω9 | 0.71 | 1.2 × 10−3 | PC 38:4 18:1/20:3,ω6 | 0.71 | 1.3 × 10−3 | PG 40:6 18:1/22:5,ω6 | 0.78 | 1.1 × 10−4 |
| Positive | SM d35:1 d14:0/21:1 | 0.71 | 1.2 × 10−3 | PC 21:3e 18:3e/3:0 | 0.71 | 1.3 × 10−3 | PG 38:2 18:1/20:1 | 0.78 | 1.1 × 10−4 |
| Positive | SM d36:2 d14:0/22:2 | 0.70 | 1.4 × 10−3 | Lyso PC 18:1,ω9 | 0.71 | 1.3 × 10−3 | PG 40:5 18:1/22:4,ω6 | 0.78 | 1.1 × 10−4 |
| Positive | PA 36:2 18:1/18:1 | 0.70 | 1.4 × 10−3 | SM d36:2 d14:0/22:2 | 0.69 | 2.2 × 10−3 | PG 36:2 18:1/18:1 | 0.77 | 1.5 × 10−4 |
| Negative | PA 34:3 16:1,ω7/18:2,ω6 | −0.77 | 1.9 × 10−4 | PA 34:3 16:1,ω7/18:2,ω6 | −0.75 | 4.8 × 10−4 | Lyso PE 18:2,ω6 | −0.57 | 1.3 × 10−2 |
| Negative | PE 34:2 16:0/18:2,ω6 | −0.75 | 3.7 × 10−4 | PE 34:2 16:0/18:2,ω6 | 0.73 | 7.1 × 10−4 | 8,9-DiHETE | −0.56 | 1.5 × 10−2 |
| Negative | PI 36:3 16:0/20:3,ω6 | −0.73 | 7.2 × 10−4 | PI 36:3 16:0/20:3,ω6 | −0.71 | 1.2 × 10−3 | 11,12-DiHETE | −0.56 | 1.5 × 10−2 |
| Negative | PI 36:2 18:1/18:1 | −0.72 | 8.7 × 10−4 | PI 36:2 18:1/18:1 | −0.71 | 1.3 × 10−3 | 8,9-EpETE | −0.56 | 1.5 × 10−2 |
| Negative | PC 35:2 13:1/22:1 | −0.71 | 1.1 × 10−3 | PI 36:4 16:0/20:4,ω6 | −0.69 | 1.9 × 10−3 | 14,15-EpETE | −0.56 | 1.7 × 10−2 |
| Negative | PI 36:4 16:0/20:4,ω6 | −0.71 | 1.1 × 10−3 | PC 35:2 13:1/22:1 | −0.69 | 2.0 × 10−3 | 17,18-DiHETE | −0.54 | 2.0 × 10−2 |
| Negative | PE 36:2 18:1/18:1 | −0.70 | 1.5 × 10−3 | PE 36:2 18:1/18:1 | −0.69 | 2.2 × 10−3 | 20:5,ω3 | −0.54 | 2.2 × 10−2 |
| Negative | PE 36:2 18:0/18:2,ω6 | −0.68 | 2.4 × 10−3 | PE 36:2 18:0/18:2,ω6 | −0.66 | 3.7 × 10−3 | PI 36:4 16:0/20:4,ω6 | −0.53 | 2.3 × 10−2 |
| Negative | PA 34:2 16:0/18:2,ω6 | −0.66 | 3.5 × 10−3 | 10,11-DiHDPE | −0.65 | 4.3 × 10−3 | PS 40:6 18:0/22:6,ω3 | −0.53 | 2.3 × 10−2 |
| Negative | 7,8-DiHDPE | −0.66 | 3.8 × 10−3 | PE 34:3e 16:1,ω7e/18:2,ω6 | −0.65 | 4.4 × 10−3 | 10,11-EpDPE | −0.52 | 2.6 × 10−2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Jaramillo, M.; Lytle, K.A.; Spooner, M.H.; Jump, D.B. A Lipidomic Analysis of Docosahexaenoic Acid (22:6, ω3) Mediated Attenuation of Western Diet Induced Nonalcoholic Steatohepatitis in Male Ldlr -/- Mice. Metabolites 2019, 9, 252. https://doi.org/10.3390/metabo9110252
García-Jaramillo M, Lytle KA, Spooner MH, Jump DB. A Lipidomic Analysis of Docosahexaenoic Acid (22:6, ω3) Mediated Attenuation of Western Diet Induced Nonalcoholic Steatohepatitis in Male Ldlr -/- Mice. Metabolites. 2019; 9(11):252. https://doi.org/10.3390/metabo9110252
Chicago/Turabian StyleGarcía-Jaramillo, Manuel, Kelli A. Lytle, Melinda H. Spooner, and Donald B. Jump. 2019. "A Lipidomic Analysis of Docosahexaenoic Acid (22:6, ω3) Mediated Attenuation of Western Diet Induced Nonalcoholic Steatohepatitis in Male Ldlr -/- Mice" Metabolites 9, no. 11: 252. https://doi.org/10.3390/metabo9110252
APA StyleGarcía-Jaramillo, M., Lytle, K. A., Spooner, M. H., & Jump, D. B. (2019). A Lipidomic Analysis of Docosahexaenoic Acid (22:6, ω3) Mediated Attenuation of Western Diet Induced Nonalcoholic Steatohepatitis in Male Ldlr -/- Mice. Metabolites, 9(11), 252. https://doi.org/10.3390/metabo9110252