Integration of Metabolomic and Other Omics Data in Population-Based Study Designs: An Epidemiological Perspective

 ,
,  , ,
, ,  , ,
, ,  ,
, 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
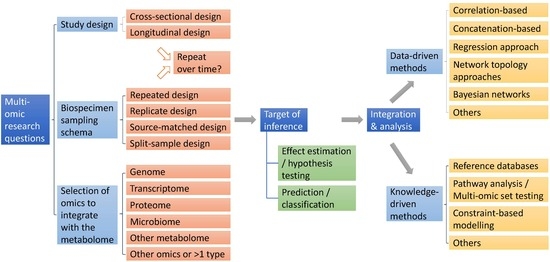
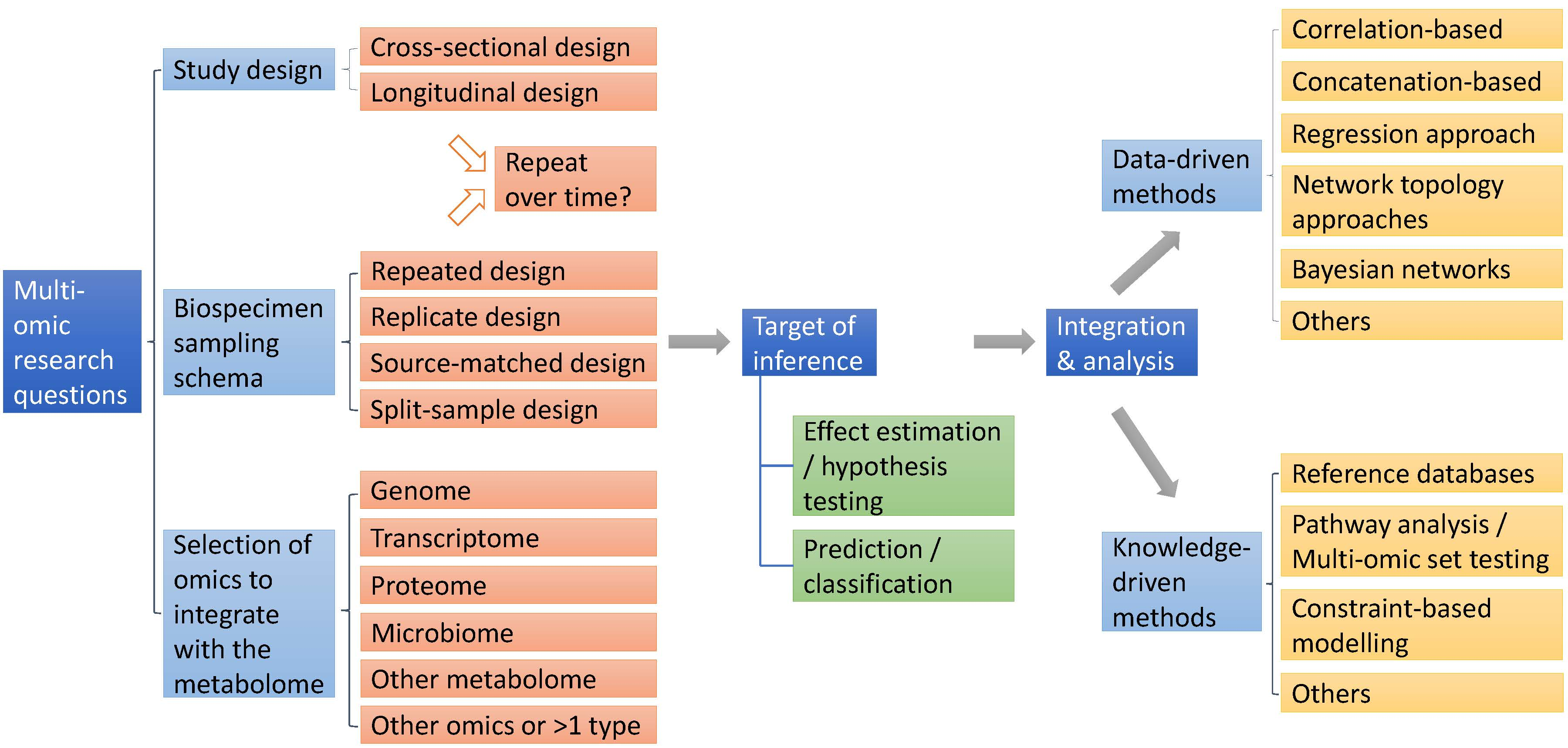
2. Study Design and Biosampling Design
2.1. Study Design
2.1.1. Cross-Sectional Study Design
2.1.2. Longitudinal Study Designs
2.2. Biospecimen Sampling Schema
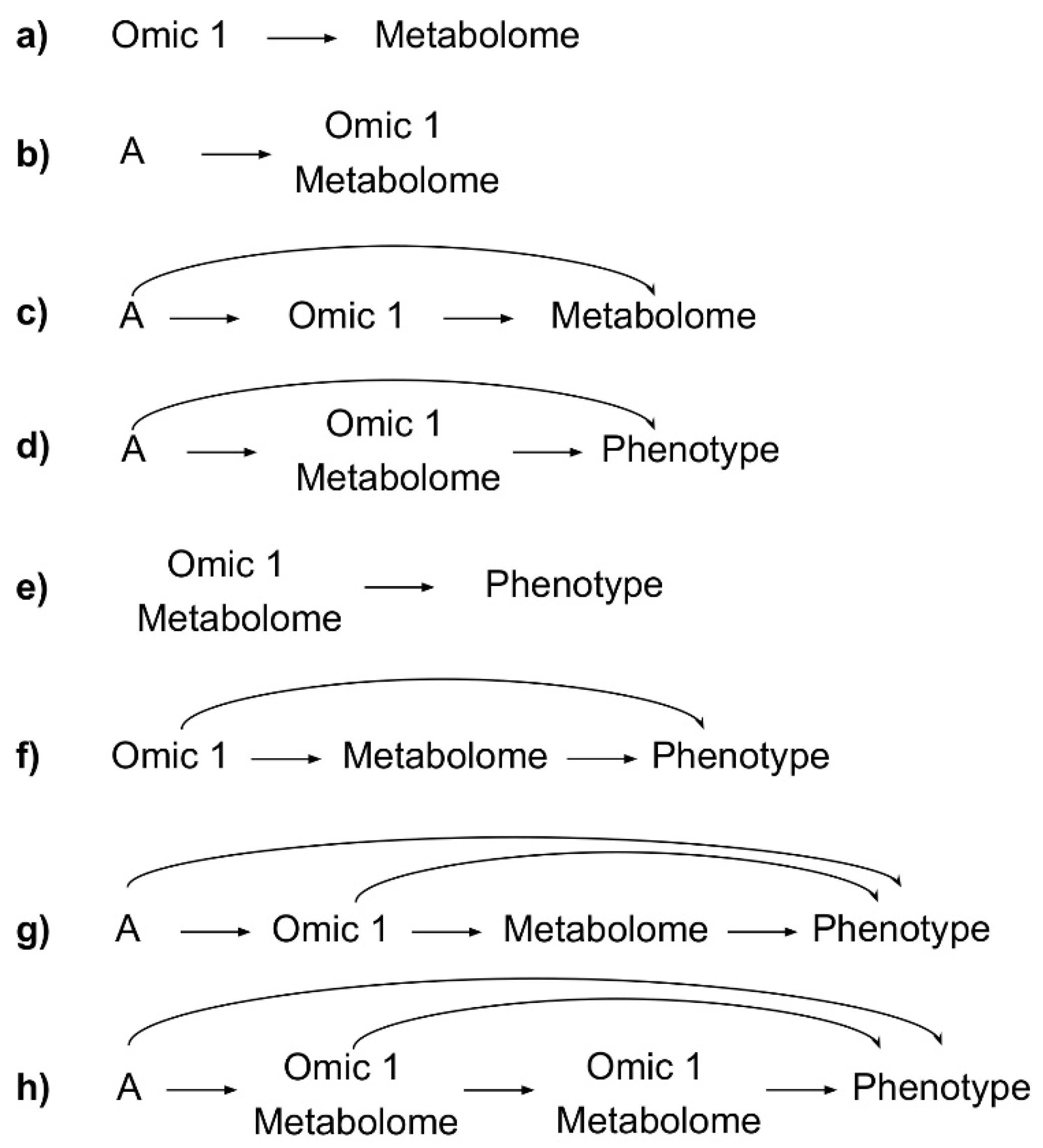
2.3. Selection of Additional Omic Type to Integrate with Metabolomics
2.3.1. Genome and Metabolome
2.3.2. Transcriptome and Metabolome
2.3.3. Proteome and Metabolome
2.3.4. Microbiome and Metabolome
2.3.5. Metabolome and Metabolome
3. Integration Paradigms and Analytic Approaches
3.1. Data-Driven Methods
3.1.1. Correlation-Based
3.1.2. Networks/Topological Structure
3.1.3. Bayesian Networks
3.1.4. Regression Approaches
3.2. Knowledge-Driven Methods
3.2.1. Reference Databases
3.2.2. Pathway-Based Analysis and Multi-Omic Set Testing
3.2.3. Constraint-Based Modeling: Flux Balance Analysis
3.3. Strategies for Type I Error Protection in Multi-Omics Analyses
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Fiehn, O. Metabolomics—The link between genotypes and phenotypes. Plant Mol. Biol. 2002, 48, 155–171. [Google Scholar] [CrossRef] [PubMed]
- Bictash, M.; Ebbels, T.M.; Chan, Q.; Loo, R.L.; Yap, I.K.; Brown, I.J.; de Iorio, M.; Daviglus, M.L.; Holmes, E.; Stamler, J.; et al. Opening up the “Black Box”: Metabolic phenotyping and metabolome-wide association studies in epidemiology. J. Clin. Epidemiol. 2010, 63, 970–979. [Google Scholar] [CrossRef] [PubMed]
- Bundy, J.G.; Davey, M.P.; Viant, M.R. Environmental metabolomics: A critical review and future perspectives. Metabolomics 2008, 5, 3–21. [Google Scholar] [CrossRef]
- Cavill, R.; Jennen, D.; Kleinjans, J.; Briede, J.J. Transcriptomic and metabolomic data integration. Brief. Bioinform. 2016, 17, 891–901. [Google Scholar] [CrossRef] [PubMed]
- Hasin, Y.; Seldin, M.; Lusis, A. Multi-omics approaches to disease. Genome Biol. 2017, 18, 83. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Snyder, M.P. Integrative omics for health and disease. Nat. Rev. Genet. 2018, 19, 299–310. [Google Scholar] [CrossRef]
- Pinu, F.R.; Beale, D.J.; Paten, A.M.; Kouremenos, K.; Swarup, S.; Schirra, H.J.; Wishart, D. Systems Biology and Multi-Omics Integration: Viewpoints from the Metabolomics Research Community. Metabolites 2019, 9, 76. [Google Scholar] [CrossRef]
- Hernán, M.A.; Robins, J.M. Causal Inference; Chapman & Hall/CRC: Boca Raton, FL, USA, 2019; forthcoming. [Google Scholar]
- VanderWeele, T.J. Explanation in Causal Inference: Methods for Mediation and Interaction; Oxford University Press: Oxford, UK, 2015. [Google Scholar]
- Leek, J.T.; Scharpf, R.B.; Bravo, H.C.; Simcha, D.; Langmead, B.; Johnson, W.E.; Geman, D.; Baggerly, K.; Irizarry, R.A. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat. Rev. Genet. 2010, 11, 733–739. [Google Scholar] [CrossRef] [Green Version]
- Kelly, R.S.; Chawes, B.L.; Blighe, K.; Virkud, Y.V.; Croteau-Chonka, D.C.; McGeachie, M.J.; Clish, C.B.; Bullock, K.; Celedon, J.C.; Weiss, S.T.; et al. An Integrative Transcriptomic and Metabolomic Study of Lung Function in Children With Asthma. Chest 2018, 154, 335–348. [Google Scholar] [CrossRef] [Green Version]
- Hallan, S.; Afkarian, M.; Zelnick, L.R.; Kestenbaum, B.; Sharma, S.; Saito, R.; Darshi, M.; Barding, G.; Raftery, D.; Ju, W.; et al. Metabolomics and Gene Expression Analysis Reveal Down-regulation of the Citric Acid (TCA) Cycle in Non-diabetic CKD Patients. EBioMedicine 2017, 26, 68–77. [Google Scholar] [CrossRef] [Green Version]
- Doerge, R.W. Multifactorial genetics: Mapping and analysis of quantitative trait loci in experimental populations. Nat. Rev. Genet. 2002, 3, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Kendziorski, C.; Wang, P. A review of statistical methods for expression quantitative trait loci mapping. Mamm. Genome 2006, 17, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Long, T.; Hicks, M.; Yu, H.C.; Biggs, W.H.; Kirkness, E.F.; Menni, C.; Zierer, J.; Small, K.S.; Mangino, M.; Messier, H.; et al. Whole-genome sequencing identifies common-to-rare variants associated with human blood metabolites. Nat. Genet. 2017, 49, 568–578. [Google Scholar] [CrossRef]
- Ried, J.S.; Shin, S.-Y.; Krumsiek, J.; Illig, T.; Theis, F.J.; Spector, T.D.; Adamski, J.; Wichmann, H.-E.; Strauch, K.; Soranzo, N. Novel genetic associations with serum level metabolites identified by phenotype set enrichment analyses. Hum. Mol. Genet. 2014, 23, 5847–5857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inouye, M.; Ripatti, S.; Kettunen, J.; Lyytikäinen, L.-P.; Oksala, N.; Laurila, P.-P.; Kangas, A.J.; Soininen, P.; Savolainen, M.J.; Viikari, J.; et al. Novel Loci for metabolic networks and multi-tissue expression studies reveal genes for atherosclerosis. PLoS Genet. 2012, 8, e1002907. [Google Scholar] [CrossRef]
- Kastenmuller, G.; Raffler, J.; Gieger, C.; Suhre, K. Genetics of human metabolism: An update. Hum. Mol. Genet. 2015, 24, R93–R101. [Google Scholar] [CrossRef] [PubMed]
- Stempler, S.; Yizhak, K.; Ruppin, E. Integrating transcriptomics with metabolic modeling predicts biomarkers and drug targets for Alzheimer’s disease. PLoS ONE 2014, 9, e105383. [Google Scholar] [CrossRef]
- Budhu, A.; Roessler, S.; Zhao, X.; Yu, Z.; Forgues, M.; Ji, J.; Karoly, E.; Qin, L.X.; Ye, Q.H.; Jia, H.L.; et al. Integrated metabolite and gene expression profiles identify lipid biomarkers associated with progression of hepatocellular carcinoma and patient outcomes. Gastroenterology 2013, 144, 1066–1075. [Google Scholar] [CrossRef]
- Zhang, G.; He, P.; Tan, H.; Budhu, A.; Gaedcke, J.; Ghadimi, B.M.; Ried, T.; Yfantis, H.G.; Lee, D.H.; Maitra, A.; et al. Integration of metabolomics and transcriptomics revealed a fatty acid network exerting growth inhibitory effects in human pancreatic cancer. Clin. Cancer Res. 2013, 19, 4983–4993. [Google Scholar] [CrossRef]
- Terunuma, A.; Putluri, N.; Mishra, P.; Mathe, E.A.; Dorsey, T.H.; Yi, M.; Wallace, T.A.; Issaq, H.J.; Zhou, M.; Killian, J.K.; et al. MYC-driven accumulation of 2-hydroxyglutarate is associated with breast cancer prognosis. J. Clin. Investig. 2014, 124, 398–412. [Google Scholar] [CrossRef]
- Su, G.; Burant, C.F.; Beecher, C.W.; Athey, B.D.; Meng, F. Integrated metabolome and transcriptome analysis of the NCI60 dataset. BMC Bioinform. 2011, 12. [Google Scholar] [CrossRef] [PubMed]
- Zelezniak, A.; Sheridan, S.; Patil, K.R. Contribution of network connectivity in determining the relationship between gene expression and metabolite concentration changes. PLoS Comput. Biol. 2014, 10, e1003572. [Google Scholar] [CrossRef] [PubMed]
- Buescher, J.M.; Driggers, E.M. Integration of omics: More than the sum of its parts. Cancer Metab. 2016, 4, 4. [Google Scholar] [CrossRef] [PubMed]
- Auslander, N.; Yizhak, K.; Weinstock, A.; Budhu, A.; Tang, W.; Wang, X.W.; Ambs, S.; Ruppin, E. A joint analysis of transcriptomic and metabolomic data uncovers enhanced enzyme-metabolite coupling in breast cancer. Sci. Rep. 2016, 6, 29662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, R.S.; Croteau-Chonka, D.C.; Dahlin, A.; Mirzakhani, H.; Wu, A.C.; Wan, E.S.; McGeachie, M.J.; Qiu, W.; Sordillo, J.E.; Al-Garawi, A.; et al. Integration of metabolomic and transcriptomic networks in pregnant women reveals biological pathways and predictive signatures associated with preeclampsia. Metabolomics 2017, 13. [Google Scholar] [CrossRef]
- Wahl, S.; Vogt, S.; Stuckler, F.; Krumsiek, J.; Bartel, J.; Kacprowski, T.; Schramm, K.; Carstensen, M.; Rathmann, W.; Roden, M.; et al. Multi-omic signature of body weight change: Results from a population-based cohort study. BMC Med. 2015, 13, 48. [Google Scholar] [CrossRef]
- Chen, R.; Mias, G.I.; Li-Pook-Than, J.; Jiang, L.; Lam, H.Y.; Chen, R.; Miriami, E.; Karczewski, K.J.; Hariharan, M.; Dewey, F.E.; et al. Personal omics profiling reveals dynamic molecular and medical phenotypes. Cell 2012, 148, 1293–1307. [Google Scholar] [CrossRef]
- Miller, M.A.; Danhorn, T.; Cruickshank-Quinn, C.I.; Leach, S.M.; Jacobson, S.; Strand, M.J.; Reisdorph, N.A.; Bowler, R.P.; Petrache, I.; Kechris, K.; et al. Gene and metabolite time-course response to cigarette smoking in mouse lung and plasma. PLoS ONE 2017, 12, e0178281. [Google Scholar] [CrossRef]
- Li, X.; Dunn, J.; Salins, D.; Zhou, G.; Zhou, W.; Schussler-Fiorenza Rose, S.M.; Perelman, D.; Colbert, E.; Runge, R.; Rego, S.; et al. Digital Health: Tracking Physiomes and Activity Using Wearable Biosensors Reveals Useful Health-Related Information. PLoS Biol. 2017, 15, e2001402. [Google Scholar] [CrossRef]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Thul, P.J.; Akesson, L.; Wiking, M.; Mahdessian, D.; Geladaki, A.; Ait Blal, H.; Alm, T.; Asplund, A.; Bjork, L.; Breckels, L.M.; et al. A subcellular map of the human proteome. Science 2017, 356. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjostedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357. [Google Scholar] [CrossRef] [PubMed]
- Schaffer, L.V.; Shortreed, M.R.; Cesnik, A.J.; Frey, B.L.; Solntsev, S.K.; Scalf, M.; Smith, L.M. Expanding Proteoform Identifications in Top-Down Proteomic Analyses by Constructing Proteoform Families. Anal. Chem. 2018, 90, 1325–1333. [Google Scholar] [CrossRef] [PubMed]
- Toby, T.K.; Fornelli, L.; Kelleher, N.L. Progress in Top-Down Proteomics and the Analysis of Proteoforms. Annu. Rev. Anal. Chem. (Palo Alto Calif.) 2016, 9, 499–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, L.M.; Kelleher, N.L.; Consortium for Top Down, P. Proteoform: A single term describing protein complexity. Nat. Methods 2013, 10, 186–187. [Google Scholar] [CrossRef] [PubMed]
- Gold, L.; Ayers, D.; Bertino, J.; Bock, C.; Bock, A.; Brody, E.N.; Carter, J.; Dalby, A.B.; Eaton, B.E.; Fitzwater, T.; et al. Aptamer-based multiplexed proteomic technology for biomarker discovery. PLoS ONE 2010, 5, e15004. [Google Scholar] [CrossRef] [PubMed]
- Nakayasu, E.S.; Nicora, C.D.; Sims, A.C.; Burnum-Johnson, K.E.; Kim, Y.M.; Kyle, J.E.; Matzke, M.M.; Shukla, A.K.; Chu, R.K.; Schepmoes, A.A.; et al. MPLEx: A Robust and Universal Protocol for Single-Sample Integrative Proteomic, Metabolomic, and Lipidomic Analyses. mSystems 2016, 1. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, D.B.; Gant-Branum, R.L.; Romer, C.E.; Farrow, M.A.; Allen, J.L.; Dahal, N.; Nei, Y.W.; Codreanu, S.G.; Jordan, A.T.; Palmer, L.D.; et al. An Integrated, High-Throughput Strategy for Multiomic Systems Level Analysis. J. Proteome Res. 2018, 17, 3396–3408. [Google Scholar] [CrossRef]
- Grigoryan, H.; Edmands, W.; Lu, S.S.; Yano, Y.; Regazzoni, L.; Iavarone, A.T.; Williams, E.R.; Rappaport, S.M. Adductomics Pipeline for Untargeted Analysis of Modifications to Cys34 of Human Serum Albumin. Anal. Chem. 2016, 88, 10504–10512. [Google Scholar] [CrossRef] [Green Version]
- Rappaport, S.M. Redefining environmental exposure for disease etiology. NPJ Syst. Biol. Appl. 2018, 4. [Google Scholar] [CrossRef]
- Grigoryan, H.; Edmands, W.M.B.; Lan, Q.; Carlsson, H.; Vermeulen, R.; Zhang, L.; Yin, S.N.; Li, G.L.; Smith, M.T.; Rothman, N.; et al. Adductomic signatures of benzene exposure provide insights into cancer induction. Carcinogenesis 2018, 39, 661–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.X.; Wheelock, C.E.; Skold, C.M.; Wheelock, A.M. Integration of multi-omics datasets enables molecular classification of COPD. Eur. Respir. J. 2018, 51. [Google Scholar] [CrossRef] [PubMed]
- Cambiaghi, A.; Diaz, R.; Martinez, J.B.; Odena, A.; Brunelli, L.; Caironi, P.; Masson, S.; Baselli, G.; Ristagno, G.; Gattinoni, L.; et al. An Innovative Approach for The Integration of Proteomics and Metabolomics Data In Severe Septic Shock Patients Stratified for Mortality. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Orth, J.D.; Thiele, I.; Palsson, B.O. What is flux balance analysis? Nat. Biotechnol. 2010, 28, 245–248. [Google Scholar] [CrossRef]
- Cho, I.; Blaser, M.J. The human microbiome: At the interface of health and disease. Nat. Rev. Genet. 2012, 13, 260–270. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, K.; Wu, W.; Giannoulatou, E.; Ho, J.W.K.; Li, L. Host and microbiome multi-omics integration: Applications and methodologies. Biophys. Rev. 2019, 11, 55–65. [Google Scholar] [CrossRef]
- Wikoff, W.R.; Anfora, A.T.; Liu, J.; Schultz, P.G.; Lesley, S.A.; Peters, E.C.; Siuzdak, G. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl. Acad. Sci. USA 2009, 106, 3698–3703. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, H.K.; Gudmundsdottir, V.; Nielsen, H.B.; Hyotylainen, T.; Nielsen, T.; Jensen, B.A.; Forslund, K.; Hildebrand, F.; Prifti, E.; Falony, G.; et al. Human gut microbes impact host serum metabolome and insulin sensitivity. Nature 2016, 535, 376–381. [Google Scholar] [CrossRef]
- Wandro, S.; Osborne, S.; Enriquez, C.; Bixby, C.; Arrieta, A.; Whiteson, K. The Microbiome and Metabolome of Preterm Infant Stool Are Personalized and Not Driven by Health Outcomes, Including Necrotizing Enterocolitis and Late-Onset Sepsis. mSphere 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Stewart, C.J.; Embleton, N.D.; Marrs, E.C.L.; Smith, D.P.; Fofanova, T.; Nelson, A.; Skeath, T.; Perry, J.D.; Petrosino, J.F.; Berrington, J.E.; et al. Longitudinal development of the gut microbiome and metabolome in preterm neonates with late onset sepsis and healthy controls. Microbiome 2017, 5, 75. [Google Scholar] [CrossRef]
- Ottosson, F.; Brunkwall, L.; Ericson, U.; Nilsson, P.M.; Almgren, P.; Fernandez, C.; Melander, O.; Orho-Melander, M. Connection Between BMI-Related Plasma Metabolite Profile and Gut Microbiota. J. Clin. Endocrinol. Metab. 2018, 103, 1491–1501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, H.K.; Forslund, S.K.; Gudmundsdottir, V.; Petersen, A.O.; Hildebrand, F.; Hyotylainen, T.; Nielsen, T.; Hansen, T.; Bork, P.; Ehrlich, S.D.; et al. A computational framework to integrate high-throughput ‘-omics’ datasets for the identification of potential mechanistic links. Nat. Protoc. 2018, 13, 2781–2800. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, D.; Bernstein, J.A.; Khurana Hershey, G.K.; Rothenberg, M.E.; Mersha, T.B. Leveraging Multilayered “Omics” Data for Atopic Dermatitis: A Road Map to Precision Medicine. Front. Immunol. 2018, 9, 2727. [Google Scholar] [CrossRef] [PubMed]
- Ghaemi, M.S.; DiGiulio, D.B.; Contrepois, K.; Callahan, B.; Ngo, T.T.M.; Lee-McMullen, B.; Lehallier, B.; Robaczewska, A.; McIlwain, D.; Rosenberg-Hasson, Y.; et al. Multiomics modeling of the immunome, transcriptome, microbiome, proteome and metabolome adaptations during human pregnancy. Bioinformatics 2019, 35, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Lee-Sarwar, K.; Kelly, R.S.; Lasky-Su, J.; Moody, D.B.; Mola, A.R.; Cheng, T.Y.; Comstock, L.E.; Zeiger, R.S.; O’Connor, G.T.; Sandel, M.T.; et al. Intestinal microbial-derived sphingolipids are inversely associated with childhood food allergy. J. Allergy Clin. Immunol. 2018, 142, 335–338. [Google Scholar] [CrossRef] [PubMed]
- Tseng, G.C.; Ghosh, D.; Zhou, X.J. Integrating Omics Data; Cambridge University Press: New York, NY, USA, 2015; p. 488. [Google Scholar] [CrossRef]
- Bersanelli, M.; Mosca, E.; Remondini, D.; Giampieri, E.; Sala, C.; Castellani, G.; Milanesi, L. Methods for the integration of multi-omics data: Mathematical aspects. BMC Bioinform. 2016, 17, 15. [Google Scholar] [CrossRef] [PubMed]
- Pearson, K.; Galton, F., VII. Note on regression and inheritance in the case of two parents. Proc. R. Soc. Lond. 1895, 58, 240–242. [Google Scholar] [CrossRef]
- Arkin, A.; Shen, P.; Ross, J. A Test Case of Correlation Metric Construction of a Reaction Pathway from Measurements. Science 1997, 277, 1275–1279. [Google Scholar] [CrossRef] [Green Version]
- Steuer, R.; Kurths, J.; Fiehn, O.; Weckwerth, W. Observing and interpreting correlations in metabolomic networks. Bioinformatics 2003, 19, 1019–1026. [Google Scholar] [CrossRef]
- Lee, H.K.; Hsu, A.K.; Sajdak, J.; Qin, J.; Pavlidis, P. Coexpression analysis of human genes across many microarray data sets. Genome Res. 2004, 14, 1085–1094. [Google Scholar] [CrossRef]
- Acharjee, A.; Kloosterman, B.; de Vos, R.C.; Werij, J.S.; Bachem, C.W.; Visser, R.G.; Maliepaard, C. Data integration and network reconstruction with ~omics data using Random Forest regression in potato. Anal. Chim. Acta 2011, 705, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Adourian, A.; Jennings, E.; Balasubramanian, R.; Hines, W.M.; Damian, D.; Plasterer, T.N.; Clish, C.B.; Stroobant, P.; McBurney, R.; Verheij, E.R.; et al. Correlation network analysis for data integration and biomarker selection. Mol. Biosyst. 2008, 4, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.Y.; Fauman, E.B.; Petersen, A.K.; Krumsiek, J.; Santos, R.; Huang, J.; Arnold, M.; Erte, I.; Forgetta, V.; Yang, T.P.; et al. An atlas of genetic influences on human blood metabolites. Nat. Genet. 2014, 46, 543–550. [Google Scholar] [CrossRef] [Green Version]
- Kuo, T.C.; Tian, T.F.; Tseng, Y.J. 3Omics: A web-based systems biology tool for analysis, integration and visualization of human transcriptomic, proteomic and metabolomic data. BMC Syst. Biol. 2013, 7, 64. [Google Scholar] [CrossRef] [PubMed]
- Spearman, C. The Proof and Measurement of Association between Two Things. Am. J. Psychol. 1904, 15, 72–101. [Google Scholar] [CrossRef]
- Floegel, A.; Wientzek, A.; Bachlechner, U.; Jacobs, S.; Drogan, D.; Prehn, C.; Adamski, J.; Krumsiek, J.; Schulze, M.B.; Pischon, T.; et al. Linking diet, physical activity, cardiorespiratory fitness and obesity to serum metabolite networks: Findings from a population-based study. Int. J. Obes. (Lond.) 2014, 38, 1388–1396. [Google Scholar] [CrossRef] [PubMed]
- Tulipani, S.; Palau-Rodriguez, M.; Minarro Alonso, A.; Cardona, F.; Marco-Ramell, A.; Zonja, B.; Lopez de Alda, M.; Munoz-Garach, A.; Sanchez-Pla, A.; Tinahones, F.J.; et al. Biomarkers of Morbid Obesity and Prediabetes by Metabolomic Profiling of Human Discordant Phenotypes. Clin. Chim. Acta 2016, 463, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Brunel, H.; Gallardo-Chacon, J.J.; Buil, A.; Vallverdu, M.; Soria, J.M.; Caminal, P.; Perera, A. MISS: A non-linear methodology based on mutual information for genetic association studies in both population and sib-pairs analysis. Bioinformatics 2010, 26, 1811–1818. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Langfelder, P.; Horvath, S. Comparison of co-expression measures: Mutual information, correlation, and model based indices. BMC Bioinform. 2012, 13, 328. [Google Scholar] [CrossRef]
- Zhang, X.; Zhao, X.M.; He, K.; Lu, L.; Cao, Y.; Liu, J.; Hao, J.K.; Liu, Z.P.; Chen, L. Inferring gene regulatory networks from gene expression data by path consistency algorithm based on conditional mutual information. Bioinformatics 2012, 28, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Zhang, Y.; Hu, W.; Tan, H.; Wang, X. Inferring nonlinear gene regulatory networks from gene expression data based on distance correlation. PLoS ONE 2014, 9, e87446. [Google Scholar] [CrossRef] [PubMed]
- Lauritzen, S.L. Graphical Models; Clarendon Press: Oxford, UK, 1996; Volume 17. [Google Scholar]
- Schafer, J.; Strimmer, K. A shrinkage approach to large-scale covariance matrix estimation and implications for functional genomics. Stat. Appl. Genet. Mol. Biol. 2005, 4. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.; Hastie, T.; Tibshirani, R. Sparse inverse covariance estimation with the graphical lasso. Biostatistics 2008, 9, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.K.; Rowe, H.C.; Hansen, B.G.; Kliebenstein, D.J. The complex genetic architecture of the metabolome. PLoS Genet. 2010, 6, e1001198. [Google Scholar] [CrossRef] [PubMed]
- Krumsiek, J.; Suhre, K.; Illig, T.; Adamski, J.; Theis, F.J. Gaussian graphical modeling reconstructs pathway reactions from high-throughput metabolomics data. BMC Syst. Biol. 2011, 5, 21. [Google Scholar] [CrossRef] [PubMed]
- Krumsiek, J.; Suhre, K.; Evans, A.M.; Mitchell, M.W.; Mohney, R.P.; Milburn, M.V.; Wagele, B.; Romisch-Margl, W.; Illig, T.; Adamski, J.; et al. Mining the unknown: A systems approach to metabolite identification combining genetic and metabolic information. PLoS Genet. 2012, 8, e1003005. [Google Scholar] [CrossRef] [PubMed]
- Castro, C.; Krumsiek, J.; Lehrbach, N.J.; Murfitt, S.A.; Miska, E.A.; Griffin, J.L. A study of Caenorhabditis elegans DAF-2 mutants by metabolomics and differential correlation networks. Mol. Biosyst. 2013, 9, 1632–1642. [Google Scholar] [CrossRef]
- Krumsiek, J.; Mittelstrass, K.; Do, K.T.; Stuckler, F.; Ried, J.; Adamski, J.; Peters, A.; Illig, T.; Kronenberg, F.; Friedrich, N.; et al. Gender-specific pathway differences in the human serum metabolome. Metabolomics 2015, 11, 1815–1833. [Google Scholar] [CrossRef] [Green Version]
- Benedetti, E.; Pucic-Bakovic, M.; Keser, T.; Wahl, A.; Hassinen, A.; Yang, J.Y.; Liu, L.; Trbojevic-Akmacic, I.; Razdorov, G.; Stambuk, J.; et al. Network inference from glycoproteomics data reveals new reactions in the IgG glycosylation pathway. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- de la Fuente, A.; Bing, N.; Hoeschele, I.; Mendes, P. Discovery of meaningful associations in genomic data using partial correlation coefficients. Bioinformatics 2004, 20, 3565–3574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Opgen-Rhein, R.; Strimmer, K. Inferring Gene Dependency Networks from Genomic Longitudinal Data: A Functional Data Approach. REVSTAT 2006, 4, 53–65. [Google Scholar]
- Allen, J.D.; Xie, Y.; Chen, M.; Girard, L.; Xiao, G. Comparing statistical methods for constructing large scale gene networks. PLoS ONE 2012, 7, e29348. [Google Scholar] [CrossRef] [PubMed]
- Wille, A.; Zimmermann, P.; Vranova, E.; Furholz, A.; Laule, O.; Bleuler, S.; Hennig, L.; Prelic, A.; von Rohr, P.; Thiele, L.; et al. Sparse graphical Gaussian modeling of the isoprenoid gene network in Arabidopsis thaliana. Genome Biol. 2004, 5. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Gong, Q.; Bohnert, H.J. An Arabidopsis gene network based on the graphical Gaussian model. Genome Res. 2007, 17, 1614–1625. [Google Scholar] [CrossRef] [PubMed]
- Werhli, A.V.; Grzegorczyk, M.; Husmeier, D. Comparative evaluation of reverse engineering gene regulatory networks with relevance networks, graphical gaussian models and bayesian networks. Bioinformatics 2006, 22, 2523–2531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reverter, A.; Chan, E.K. Combining partial correlation and an information theory approach to the reversed engineering of gene co-expression networks. Bioinformatics 2008, 24, 2491–2497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, J.; Li, H. A Sparse Conditional Gaussian Graphical Model for Analysis of Genetical Genomics Data. Ann. Appl. Stat. 2011, 5, 2630–2650. [Google Scholar] [CrossRef]
- Zhang, Y.; Ouyang, Z.; Zhao, H. A Statistical Framework for Data Integration through Graphical Models with Application to Cancer Genomics. Ann. Appl. Stat. 2017, 11, 161–184. [Google Scholar] [CrossRef]
- Edwards, D.; de Abreu, G.C.; Labouriau, R. Selecting high-dimensional mixed graphical models using minimal AIC or BIC forests. BMC Bioinform. 2010, 11, 18. [Google Scholar] [CrossRef]
- Kiiveri, H.T. Multivariate analysis of microarray data: Differential expression and differential connection. BMC Bioinform. 2011, 12, 42. [Google Scholar] [CrossRef] [PubMed]
- Sedgewick, A.J.; Shi, I.; Donovan, R.M.; Benos, P.V. Learning mixed graphical models with separate sparsity parameters and stability-based model selection. BMC Bioinform. 2016, 17, 175. [Google Scholar] [CrossRef] [PubMed]
- Zierer, J.; Pallister, T.; Tsai, P.C.; Krumsiek, J.; Bell, J.T.; Lauc, G.; Spector, T.D.; Menni, C.; Kastenmuller, G. Exploring the molecular basis of age-related disease comorbidities using a multi-omics graphical model. Sci. Rep. 2016, 6, 37646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravasz, E.; Somera, A.L.; Mongru, D.A.; Oltvai, Z.N.; Barabasi, A.L. Hierarchical organization of modularity in metabolic networks. Science 2002, 297, 1551–1555. [Google Scholar] [CrossRef] [PubMed]
- Hartwell, L.H.; Hopfield, J.J.; Leibler, S.; Murray, A.W. From molecular to modular cell biology. Nature 1999, 402, C47–C52. [Google Scholar] [CrossRef] [PubMed]
- Spirin, V.; Mirny, L.A. Protein complexes and functional modules in molecular networks. Proc. Natl. Acad. Sci. USA 2003, 100, 12123–12128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, M.E.; Girvan, M. Finding and evaluating community structure in networks. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2004, 69, 026113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Mitra, K.; Carvunis, A.R.; Ramesh, S.K.; Ideker, T. Integrative approaches for finding modular structure in biological networks. Nat. Rev. Genet. 2013, 14, 719–732. [Google Scholar] [CrossRef]
- Liu, B.; Pop, M. MetaPath: Identifying differentially abundant metabolic pathways in metagenomic datasets. BMC Proc. 2011, 5, 101–112. [Google Scholar] [CrossRef]
- Do, K.T.; Pietzner, M.; Rasp, D.J.; Friedrich, N.; Nauck, M.; Kocher, T.; Suhre, K.; Mook-Kanamori, D.O.; Kastenmuller, G.; Krumsiek, J.; et al. Phenotype-driven identification of modules in a hierarchical map of multifluid metabolic correlations. NPJ Syst. Biol. Appl. 2017, 3, 28. [Google Scholar] [CrossRef] [PubMed]
- Do, K.T.; Rasp, D.J.N.; Kastenmuller, G.; Suhre, K.; Krumsiek, J. MoDentify: Phenotype-driven module identification in metabolomics networks at different resolutions. Bioinformatics 2019, 35, 532–534. [Google Scholar] [CrossRef] [PubMed]
- Heckerman, D.; Chickering, M. Learning Bayesian networks: The combination of knowledge and statistical data. Mach. Learn. 1995, 20, 197–243. [Google Scholar] [CrossRef] [Green Version]
- Rodin, A.S.; Boerwinkle, E. Mining genetic epidemiology data with Bayesian networks I: Bayesian networks and example application (plasma apoE levels). Bioinformatics 2005, 21, 3273–3278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heckerman, D.; Gieger, D. Learning Bayesian Networks: A unification for discrete and Gaussian domains. In Proceedings of the Eleventh Annual Conference on Uncertainty in Artificial Intelligence, Montreal, QC, Canada, 18–20 August 1995. [Google Scholar]
- Ritchie, M.D.; Holzinger, E.R.; Li, R.; Pendergrass, S.A.; Kim, D. Methods of integrating data to uncover genotype-phenotype interactions. Nat. Rev. Genet. 2015, 16, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Kass, R.E.; Raftery, A.E. Bayes Factors. J. Am. Stat. Assoc. 1995, 90, 773–795. [Google Scholar] [CrossRef]
- McGeachie, M.J.; Chang, H.H.; Weiss, S.T. CGBayesNets: Conditional Gaussian Bayesian network learning and inference with mixed discrete and continuous data. PLoS Comput. Biol. 2014, 10, e1003676. [Google Scholar] [CrossRef]
- Illig, T.; Gieger, C.; Zhai, G.; Romisch-Margl, W.; Wang-Sattler, R.; Prehn, C.; Altmaier, E.; Kastenmuller, G.; Kato, B.S.; Mewes, H.W.; et al. A genome-wide perspective of genetic variation in human metabolism. Nat. Genet. 2010, 42, 137–141. [Google Scholar] [CrossRef]
- Davey Smith, G.; Hemani, G. Mendelian randomization: Genetic anchors for causal inference in epidemiological studies. Hum. Mol. Genet. 2014, 23, R89–R98. [Google Scholar] [CrossRef]
- Relton, C.L.; Davey-Smith, G. Two-step epigenetic Mendelian randomization: A strategy for establishing the causal role of epigenetic processes in pathways to disease. Int. J. Epidemiol. 2012, 41, 161–176. [Google Scholar] [CrossRef]
- Richmond, R.C.; Hemani, G.; Tilling, K.; Davey Smith, G.; Relton, C.L. Challenges and novel approaches for investigating molecular mediation. Hum. Mol. Genet. 2016, 25, R149–R156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.-T.; Vanderweele, T.J.; Lin, X. Joint analysis of SNP and gene expression data in genetic association studies of complex diseases. Ann. Appl. Stat. 2014, 8, 352–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- VanderWeele, T.; Vansteelandt, S. Mediation Analysis with Multiple Mediators. Epidemiol. Methods 2013, 2, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Steen, J.; Loeys, T.; Moerkerke, B.; Vansteelandt, S. Flexible Mediation Analysis With Multiple Mediators. Am. J. Epidemiol. 2017, 186, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Chu, S.H.; Loucks, E.B.; Kelsey, K.T.; Gilman, S.E.; Agha, G.; Eaton, C.B.; Buka, S.L.; Huang, Y.-T. Sex-specific epigenetic mediators between early life social disadvantage and adulthood BMI. Epigenomics 2018, 16, 321. [Google Scholar] [CrossRef]
- Loucks, E.B.; Huang, Y.-T.; Agha, G.; Chu, S.; Eaton, C.B.; Gilman, S.E.; Buka, S.L.; Kelsey, K.T. Epigenetic Mediators Between Childhood Socioeconomic Disadvantage and Mid-Life Body Mass Index: The New England Family Study. Psychosom. Med. 2016, 78, 1053–1065. [Google Scholar] [CrossRef]
- Bouhaddani, S.E.; Houwing-Duistermaat, J.; Salo, P.; Perola, M.; Jongbloed, G.; Uh, H.W. Evaluation of O2PLS in Omics data integration. BMC Bioinform. 2016, 17, 11. [Google Scholar] [CrossRef]
- Bylesjo, M.; Eriksson, D.; Kusano, M.; Moritz, T.; Trygg, J. Data integration in plant biology: The O2PLS method for combined modeling of transcript and metabolite data. Plant J. 2007, 52, 1181–1191. [Google Scholar] [CrossRef]
- Kirwan, G.M.; Johansson, E.; Kleemann, R.; Verheij, E.R.; Wheelock, A.M.; Goto, S.; Trygg, J.; Wheelock, C.E. Building multivariate systems biology models. Anal. Chem. 2012, 84, 7064–7071. [Google Scholar] [CrossRef]
- Lofstedt, T.; Hoffman, D.; Trygg, J. Global, local and unique decompositions in OnPLS for multiblock data analysis. Anal. Chim. Acta 2013, 791, 13–24. [Google Scholar] [CrossRef]
- Reinke, S.N.; Galindo-Prieto, B.; Skotare, T.; Broadhurst, D.I.; Singhania, A.; Horowitz, D.; Djukanovic, R.; Hinks, T.S.C.; Geladi, P.; Trygg, J.; et al. OnPLS-Based Multi-Block Data Integration: A Multivariate Approach to Interrogating Biological Interactions in Asthma. Anal. Chem. 2018, 90, 13400–13408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lock, E.F.; Hoadley, K.A.; Marron, J.S.; Nobel, A.B. Joint and Individual Variation Explained (Jive) for Integrated Analysis of Multiple Data Types. Ann. Appl. Stat. 2013, 7, 523–542. [Google Scholar] [CrossRef] [PubMed]
- Van Deun, K.; Van Mechelen, I.; Thorrez, L.; Schouteden, M.; De Moor, B.; van der Werf, M.J.; De Lathauwer, L.; Smilde, A.K.; Kiers, H.A. DISCO-SCA and properly applied GSVD as swinging methods to find common and distinctive processes. PLoS ONE 2012, 7, e37840. [Google Scholar] [CrossRef] [PubMed]
- Gaynanova, I.; Li, G. Structural Learning and Integrative Decomposition of Multi-View Data. arXiv 2017, arXiv:1707.06573. [Google Scholar]
- Song, Y.; Westerhuis, J.A.; Smilde, A.K. Separating common (global and local) and distinct variation in multiple mixed types data sets. arXiv 2019, arXiv:1902.06241. [Google Scholar]
- Wheelock, A.M.; Wheelock, C.E. Trials and tribulations of ‘omics data analysis: Assessing quality of SIMCA-based multivariate models using examples from pulmonary medicine. Mol. Biosyst. 2013, 9, 2589–2596. [Google Scholar] [CrossRef] [PubMed]
- Rohart, F.; Gautier, B.; Singh, A.; Le Cao, K.A. mixOmics: An R package for ’omics feature selection and multiple data integration. PLoS Comput. Biol. 2017, 13, e1005752. [Google Scholar] [CrossRef]
- Fukushima, A. DiffCorr: An R package to analyze and visualize differential correlations in biological networks. Gene 2013, 518, 209–214. [Google Scholar] [CrossRef] [Green Version]
- Siddiqui, J.K.; Baskin, E.; Liu, M.; Cantemir-Stone, C.Z.; Zhang, B.; Bonneville, R.; McElroy, J.P.; Coombes, K.R.; Mathe, E.A. IntLIM: Integration using linear models of metabolomics and gene expression data. BMC Bioinform. 2018, 19, 81. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Croft, D.; Mundo, A.F.; Haw, R.; Milacic, M.; Weiser, J.; Wu, G.; Caudy, M.; Garapati, P.; Gillespie, M.; Kamdar, M.R.; et al. The Reactome pathway knowledgebase. Nucleic Acids Res. 2014, 42, D472–D477. [Google Scholar] [CrossRef] [PubMed]
- Slenter, D.N.; Kutmon, M.; Hanspers, K.; Riutta, A.; Windsor, J.; Nunes, N.; Melius, J.; Cirillo, E.; Coort, S.L.; Digles, D.; et al. WikiPathways: A multifaceted pathway database bridging metabolomics to other omics research. Nucleic Acids Res. 2018, 46, D661–D667. [Google Scholar] [CrossRef]
- Brunk, E.; Sahoo, S.; Zielinski, D.C.; Altunkaya, A.; Drager, A.; Mih, N.; Gatto, F.; Nilsson, A.; Preciat Gonzalez, G.A.; Aurich, M.K.; et al. Recon3D enables a three-dimensional view of gene variation in human metabolism. Nat. Biotechnol. 2018, 36, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Wishart, D.S. Web-based inference of biological patterns, functions and pathways from metabolomic data using MetaboAnalyst. Nat. Protoc. 2011, 6, 743–760. [Google Scholar] [CrossRef] [PubMed]
- Kankainen, M.; Gopalacharyulu, P.; Holm, L.; Oresic, M. MPEA—Metabolite pathway enrichment analysis. Bioinformatics 2011, 27, 1878–1879. [Google Scholar] [CrossRef] [PubMed]
- Karnovsky, A.; Weymouth, T.; Hull, T.; Tarcea, V.G.; Scardoni, G.; Laudanna, C.; Sartor, M.A.; Stringer, K.A.; Jagadish, H.V.; Burant, C.; et al. Metscape 2 bioinformatics tool for the analysis and visualization of metabolomics and gene expression data. Bioinformatics 2012, 28, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Wishart, D.S. MetPA: A web-based metabolomics tool for pathway analysis and visualization. Bioinformatics 2010, 26, 2342–2344. [Google Scholar] [CrossRef]
- Kamburov, A.; Cavill, R.; Ebbels, T.M.; Herwig, R.; Keun, H.C. Integrated pathway-level analysis of transcriptomics and metabolomics data with IMPaLA. Bioinformatics 2011, 27, 2917–2918. [Google Scholar] [CrossRef]
- Sun, H.; Wang, H.; Zhu, R.; Tang, K.; Gong, Q.; Cui, J.; Cao, Z.; Liu, Q. iPEAP: Integrating multiple omics and genetic data for pathway enrichment analysis. Bioinformatics 2014, 30, 737–739. [Google Scholar] [CrossRef]
- Zhang, B.; Hu, S.; Baskin, E.; Patt, A.; Siddiqui, J.K.; Mathe, E.A. RaMP: A Comprehensive Relational Database of Metabolomics Pathways for Pathway Enrichment Analysis of Genes and Metabolites. Metabolites 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Wishart, D.S. Using MetaboAnalyst 3.0 for Comprehensive Metabolomics Data Analysis. Curr. Protoc. Bioinform. 2016, 55. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-de-Diego, R.; Tarazona, S.; Martinez-Mira, C.; Balzano-Nogueira, L.; Furio-Tari, P.; Pappas, G.J., Jr.; Conesa, A. PaintOmics 3: A web resource for the pathway analysis and visualization of multi-omics data. Nucleic Acids Res. 2018, 46, W503–W509. [Google Scholar] [CrossRef] [PubMed]
- Wanichthanarak, K.; Fan, S.; Grapov, D.; Barupal, D.K.; Fiehn, O. Metabox: A Toolbox for Metabolomic Data Analysis, Interpretation and Integrative Exploration. PLoS ONE 2017, 12, e0171046. [Google Scholar] [CrossRef]
- Xiong, Q.; Ancona, N.; Hauser, E.R.; Mukherjee, S.; Furey, T.S. Integrating genetic and gene expression evidence into genome-wide association analysis of gene sets. Genome Res. 2012, 22, 386–397. [Google Scholar] [CrossRef] [PubMed]
- Kamburov, A.; Pentchev, K.; Galicka, H.; Wierling, C.; Lehrach, H.; Herwig, R. ConsensusPathDB: Toward a more complete picture of cell biology. Nucleic Acids Res. 2011, 39, D712–D717. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Fjell, C.D.; Mayer, M.L.; Pena, O.M.; Wishart, D.S.; Hancock, R.E. INMEX—A web-based tool for integrative meta-analysis of expression data. Nucleic Acids Res. 2013, 41, W63–W70. [Google Scholar] [CrossRef] [PubMed]
- Chu, S.H.; Huang, Y.-T. Integrated genomic analysis of biological gene sets with applications in lung cancer prognosis. BMC Bioinform. 2017, 18, 336. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-T.; Pan, W.-C. Hypothesis test of mediation effect in causal mediation model with high-dimensional continuous mediators. Biometrics 2015, 72, 402–413. [Google Scholar] [CrossRef]
- Zhao, Y.; Luo, X. Pathway lasso: Estimate and select sparse mediation pathways with high dimensional mediators. arXiv 2016, arXiv:1603.07749. [Google Scholar]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vazquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef] [PubMed]
- Hao, T.; Ma, H.W.; Zhao, X.M.; Goryanin, I. Compartmentalization of the Edinburgh Human Metabolic Network. BMC Bioinform. 2010, 11, 393. [Google Scholar] [CrossRef] [PubMed]
- Mardinoglu, A.; Agren, R.; Kampf, C.; Asplund, A.; Uhlen, M.; Nielsen, J. Genome-scale metabolic modelling of hepatocytes reveals serine deficiency in patients with non-alcoholic fatty liver disease. Nat. Commun. 2014, 5, 3083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shlomi, T.; Cabili, M.N.; Herrgard, M.J.; Palsson, B.O.; Ruppin, E. Network-based prediction of human tissue-specific metabolism. Nat. Biotechnol. 2008, 26, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Blazier, A.S.; Papin, J.A. Integration of expression data in genome-scale metabolic network reconstructions. Front. Physiol. 2012, 3, 299. [Google Scholar] [CrossRef] [Green Version]
- Magnusdottir, S.; Heinken, A.; Kutt, L.; Ravcheev, D.A.; Bauer, E.; Noronha, A.; Greenhalgh, K.; Jager, C.; Baginska, J.; Wilmes, P.; et al. Generation of genome-scale metabolic reconstructions for 773 members of the human gut microbiota. Nat. Biotechnol. 2017, 35, 81–89. [Google Scholar] [CrossRef]
- Heinken, A.; Ravcheev, D.A.; Baldini, F.; Heirendt, L.; Fleming, R.M.T.; Thiele, I. Personalized modeling of the human gut microbiome reveals distinct bile acid deconjugation and biotransformation potential in healthy and IBD individuals. BioRxiv 2017. [Google Scholar] [CrossRef]
- Heirendt, L.; Arreckx, S.; Pfau, T.; Mendoza, S.N.; Richelle, A.; Heinken, A.; Haraldsdottir, H.S.; Wachowiak, J.; Keating, S.M.; Vlasov, V.; et al. Creation and analysis of biochemical constraint-based models using the COBRA Toolbox v.3.0. Nat. Protoc. 2019, 14, 639–702. [Google Scholar] [CrossRef] [Green Version]
- Yizhak, K.; Benyamini, T.; Liebermeister, W.; Ruppin, E.; Shlomi, T. Integrating quantitative proteomics and metabolomics with a genome-scale metabolic network model. Bioinformatics 2010, 26, i255–i260. [Google Scholar] [CrossRef] [Green Version]
- Jamshidi, N.; Palsson, B.O. Mass action stoichiometric simulation models: Incorporating kinetics and regulation into stoichiometric models. Biophys. J. 2010, 98, 175–185. [Google Scholar] [CrossRef]
- Chadeau-Hyam, M.; Ebbels, T.M.; Brown, I.J.; Chan, Q.; Stamler, J.; Huang, C.C.; Daviglus, M.L.; Ueshima, H.; Zhao, L.; Holmes, E.; et al. Metabolic profiling and the metabolome-wide association study: Significance level for biomarker identification. J. Proteome Res. 2010, 9, 4620–4627. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chu, S.H.; Huang, M.; Kelly, R.S.; Benedetti, E.; Siddiqui, J.K.; Zeleznik, O.A.; Pereira, A.; Herrington, D.; Wheelock, C.E.; Krumsiek, J.; et al. Integration of Metabolomic and Other Omics Data in Population-Based Study Designs: An Epidemiological Perspective. Metabolites 2019, 9, 117. https://doi.org/10.3390/metabo9060117
Chu SH, Huang M, Kelly RS, Benedetti E, Siddiqui JK, Zeleznik OA, Pereira A, Herrington D, Wheelock CE, Krumsiek J, et al. Integration of Metabolomic and Other Omics Data in Population-Based Study Designs: An Epidemiological Perspective. Metabolites. 2019; 9(6):117. https://doi.org/10.3390/metabo9060117
Chicago/Turabian StyleChu, Su H., Mengna Huang, Rachel S. Kelly, Elisa Benedetti, Jalal K. Siddiqui, Oana A. Zeleznik, Alexandre Pereira, David Herrington, Craig E. Wheelock, Jan Krumsiek, and et al. 2019. "Integration of Metabolomic and Other Omics Data in Population-Based Study Designs: An Epidemiological Perspective" Metabolites 9, no. 6: 117. https://doi.org/10.3390/metabo9060117
APA StyleChu, S. H., Huang, M., Kelly, R. S., Benedetti, E., Siddiqui, J. K., Zeleznik, O. A., Pereira, A., Herrington, D., Wheelock, C. E., Krumsiek, J., McGeachie, M., Moore, S. C., Kraft, P., Mathé, E., Lasky-Su, J., & on behalf of the Consortium of Metabolomics Studies Statistics Working Group. (2019). Integration of Metabolomic and Other Omics Data in Population-Based Study Designs: An Epidemiological Perspective. Metabolites, 9(6), 117. https://doi.org/10.3390/metabo9060117