

Exponentially Correlated Hylleraas–Configuration Interaction Studies of Atomic Systems. III. Upper and Lower Bounds to He-Sequence Oscillator Strengths for the Resonance 1S→1P Transition


Abstract
:1. Introduction
2. Variational Calculations
Method of Calculation
3. Oscillator Strengths
Transition Moment Integrals
4. Upper and Lower Bounds to Oscillator Strengths
Method of Calculation
5. Wave Functions
5.1. Comparison with Orthogonal Hy-CI
5.2. Comparison of E-Hy-CI and Hy-CI

{kind=link}
| Technique | Author | N | Energy (Hartree) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Hy-CI 1 | This work | 0 | 16 | 2.28 | 1 | 15 | 1.085 | 480 | −2.1238 2778 5160 1660 5130 | |
| Hy-CI | This work | 0 | 16 | 2.272 | 1 | 15 | 1.085 | 480 | −2.1238 2778 5160 4328 8615 | |
| Hy-CI | This work | 0 | 16 | 2.270 | 1 | 15 | 1.085 | 480 | −2.1238 2778 5160 4106 3517 | |
| Hy-CI | This work | 0 | 16 | 2.272 | 1 | 15 | 1.300 | 480 | −2.1238 2780 3592 4727 1690 | |
| Hy-CI | This work | 0 | 16 | 2.272 | 1 | 15 | 1.291 | 480 | −2.1238 2780 3657 1217 9787 | |
| Hy-CI | This work | 0 | 16 | 2.272 | 1 | 15 | 1.290 | 480 | −2.1238 2780 3655 3377 1118 | |
| Hy-CI | This work | 0 | 16 | 2.272 | 1 | 15 | 1.291 | 480 | −2.1238 2780 3657 1217 9787 | |
| E-Hy-CI | This work | 0 | 16 | 2.272 | 1 | 15 | 1.291 | 0.5 | 960 | −2.1238 4308 3927 0924 8309 |
| Hy-CI | This work | 0 | 19 | 2.272 | 1 | 19 | 1.291 | 722 | −2.1238 2782 6872 0251 1341 | |
| E-Hy-CI | This work | 0 | 19 | 2.272 | 1 | 19 | 1.291 | 0.5 | 1444 | −2.1238 4308 5062 4897 8422 |
| Hy-CI | This work | 1 | 15 | 2.000 | 2 | 14 | 0.75 | 900 | −2.1238 4303 4769 4360 5998 | |
| Hy-CI | This work | 1 | 15 | 1.955 | 2 | 14 | 0.75 | 900 | −2.1238 4303 4770 5696 3685 | |
| Hy-CI | This work | 1 | 15 | 1.900 | 2 | 14 | 0.75 | 900 | −2.1238 4303 4768 6097 2667 | |
| Hy-CI | This work | 1 | 15 | 1.955 | 2 | 14 | 2.30 | 900 | −2.1238 4307 3318 1068 7228 | |
| Hy-CI | This work | 1 | 15 | 1.955 | 2 | 14 | 2.25 | 900 | −2.1238 4307 3463 3468 4622 | |
| Hy-CI | This work | 1 | 15 | 1.955 | 2 | 14 | 2.20 | 900 | −2.1238 4307 2960 1221 3901 | |
| Hy-CI | This work | 1 | 15 | 1.955 | 2 | 14 | 2.25 | 900 | −2.1238 4307 3463 3468 4622 | |
| E-Hy-CI | This work | 1 | 15 | 1.955 | 2 | 14 | 2.25 | 0.5 | 1800 | −2.1238 4308 6441 2634 4197 |
| Hy-CI | This work | 1 | 19 | 1.955 | 2 | 19 | 2.25 | 1444 | −2.1238 4307 5774 3704 6662 | |
| O-Hy-CI | Zhang et al. [17] | 6 2 | 1.169 3 | 4 4 | 0.28419 3 | 514 | −2.1238 4308 6457 05 | |||
| E-Hy-CI | This work | 1 | 19 | 1.955 | 2 | 19 | 2.25 | 0.5 | 2888 | −2.1238 4308 6497 9644 7703 |
| Hy-CI | This work | 2 | 19 | 2.40 | 3 | 18 | 2.30 | 2128 | −2.1238 4308 6479 8938 8807 | |
| E-Hy-CI | This work | 2 | 19 | 2.40 | 3 | 18 | 2.30 | 0.5 | 4256 | −2.1238 4308 6498 0912 9534 |
| Hy-CI | This work | 3 | 18 | 2.40 | 4 | 15 | 2.30 | 2668 | −2.1238 4308 6491 8108 5041 | |
| E-Hy-CI | This work | 3 | 18 | 2.40 | 4 | 15 | 2.30 | 0.5 | 5336 | −2.1238 4308 6498 0989 3227 |
| Hy-CI | This work | 4 | 15 | 2.40 | 5 | 14 | 2.30 | 3088 | −2.1238 4308 6491 9211 6091 | |
| E-Hy-CI | This work | 4 | 15 | 2.40 | 5 | 14 | 2.30 | 0.5 | 6176 | −2.1238 4308 6498 1004 0079 |
| Hy-CI | This work | 5 | 14 | 2.40 | 6 | 14 | 2.30 | 3480 | −2.1238 4308 6491 9773 5726 | |
| E-Hy-CI | This work | 5 | 14 | 2.40 | 6 | 14 | 2.30 | 0.5 | 6960 | −2.1238 4308 6498 1008 1958 |
| Hy-CI | This work | 6 | 14 | 2.40 | 7 | 14 | 2.30 | 3872 | −2.1238 4308 6492 0145 5741 | |
| E-Hy-CI | This work | 6 | 14 | 2.40 | 7 | 14 | 2.30 | 0.5 | 7744 | −2.1238 4308 6498 1009 6317 |
| O-Hy-CI | Plute (1984) [47] | 90 | −2.1238 4260 | |||||||
| Hy | AW 5 (1974) [37] | 2.00 | 0.88 | 137 | −2.1238 4303 14 | |||||
| Hy | SLPR 6 (1965) [48] | 560 | −2.1238 4308 5800 | |||||||
| O-Hy-CI | Zhang et al. [17] | 6 2 | 1.169 3 | 4 4 | 0.28419 3 | 514 | −2.1238 4308 6457 05 | |||
| Hy | Drake (1996) [43] | 804 | −2.1238 4308 6498 091 | |||||||
| E-Hy-CI | This work | 6 | 14 | 2.40 | 7 | 14 | 2.30 | 0.5 | 7744 | −2.1238 4308 6498 1009 6317 |
| Reference | Aznabaev et al. [50] | 22,000 | −2.1238 4308 6498 1013 5924 | |||||||
| (E-Hy) 7 | (2018) | … 7333 1423 74 |
- Hy-CI -wave 4 decimal place precision becomes 8 decimal place E-Hy-CI -wave precision (> Hy-CI -wave precision),
- Hy-CI -wave 7 decimal place precision becomes 11 decimal place E-Hy-CI -wave precision (> Hy-CI l = 7 precision),
- already at the E-Hy-CI -wave expansion, the result is better than the Hy-CI l = 7 result.
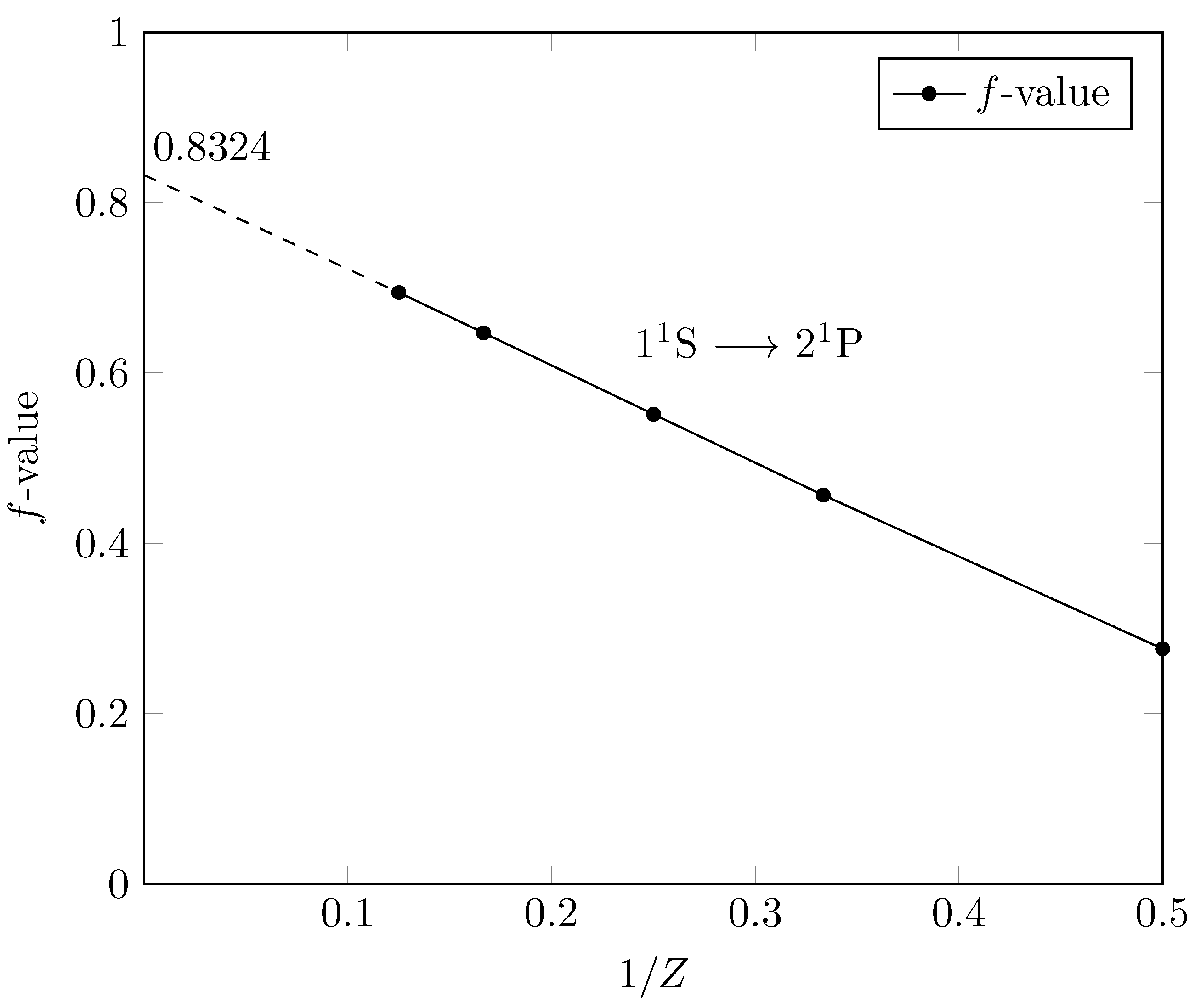
6. Isoelectronic Sequences
7. f-Values and Bounds
| State | Technique | Author | N () | E () (Hartree) | N () | E () (Hartree) | f-Value |
|---|---|---|---|---|---|---|---|
| He | E-Hy-CI | This work | 1088 | −2.9037 2435 4189 0888 5088 58 | 960 | −2.1238 4308 3927 0924 8309 | 0.2761 6466 27 ±0.0012 |
| He | E-Hy-CI | This work | 1520 | −2.9037 2435 4275 7390 6714 30 | 1444 | −2.1238 4308 5062 4897 8422 | 0.2761 6463 24 ±0.0012 |
| He | E-Hy-CI | This work | 3040 | −2.9037 2437 7034 1043 9282 03 | 2888 | −2.1238 4308 6497 9644 7703 | 0.2761 6470 03 ±0.0000 028 |
| He | E-Hy-CI | Recommended | 0.2761 647(28) | ||||
| He | Hy | SPA 1 (1971) [58] | 1078 | −2.9037 2437 48 | 364 | −2.1238 4308 26 | 0.2761 6 |
| He | Hy-CI | AW 2 (1974) [37] | 135 | −2.9037 2436 62 | 137 | −2.1238 4303 14 | 0.2761 ±0.0014 |
| He | Hy | KH 3 (1984) [60] | 138 | −2.9037 2437 51 | 140 | −2.1238 4308 | 0.2761 6 |
| He | E-Hy | CT 4 (1992) [53] | 100 | −2.9037 2437 36 | 100 | −2.1238 4308 02 | 0.2761 7 |
| He | Hy | DM 5 (2007) [61] | 0.2761 6 | ||||
| He | MCHF 6 | LLWQ 7 (2021) [28] | 0.2762 9 | ||||
| He | MCDHF 8 | LLWQ 9 (2021) [28] | 0.2763 2 | ||||
| He | Reference 9 | WF 10 (2009) [59] | 0.2762 5 | ||||
| Li II | E-Hy-CI | This work | 1088 | −7.2799 1332 7713 3474 7954 38 | 960 | −4.9933 5106 5519 3789 3549 | 0.4566 2972 51 ±0.0016 |
| Li II | E-Hy-CI | This work | 1520 | −7.2799 1332 8098 0780 5105 69 | 1444 | −4.9933 5106 5763 9277 0706 | 0.4566 2972 83 ±0.0016 |
| Li II | E-Hy-CI | This work | 3040 | −7.2799 1341 2669 2298 8163 74 | 2888 | −4.9933 5107 7779 9718 5893 | 0.4566 2984 06 ±0.0000 018 |
| Li II | E-Hy-CI | Recommended | 0.4566 298(18) | ||||
| Li II | Hy | SPA (1971) [58] | 1078 | −7.2799 1341 03 | 364 | −4.9933 5107 46 | 0.4566 |
| Li II | Hy-CI | AW (1974) [37] | 135 | −7.2799 1339 60 | 137 | −4.9933 5102 30 | 0.4566 ±0.0010 |
| Li II | E-Hy | CT (1992) [53] | 100 | −7.2799 1340 96 | 100 | −4.9933 5107 21 | 0.4566 27 |
| Li II | Reference | WF (2009) [59] | 0.4566 8 | ||||
| Be III | E-Hy-CI | This work | 1088 | −13.6555 6607 5022 1216 4675 55 | 960 | −9.1107 7158 4011 3245 7902 | 0.5515 5367 16 ±0.0019 |
| Be III | E-Hy-CI | This work | 1520 | −13.6555 6607 5806 8585 0501 71 | 1444 | −9.1107 7158 4761 1835 3319 | 0.5515 5367 29 ±0.0019 |
| Be III | E-Hy-CI | This work | 3040 | −13.6555 6623 8423 4190 7983 70 | 2888 | −9.1107 7162 2916 2965 8769 | 0.5515 5380 86 ±0.0000 021 |
| Be III | E-Hy-CI | Recommended | 0.5515 538(21) | ||||
| Be III | Hy | SPA (1971) [58] | 1078 | −13.6555 6623 60 | 364 | −9.1107 7161 94 | 0.5515 5 |
| Be III | E-Hy | CT (1992) [53] | 100 | −13.6555 6623 40 | 100 | −9.1107 7161 42 | 0.5515 55 |
| C V | E-Hy-CI | This work | 1088 | −32.4062 4629 6959 6060 3495 12 | 960 | −21.0933 3219 8896 1611 3442 | 0.6470 6730 80 ±0.0018 |
| C V | E-Hy-CI | This work | 1520 | −32.4062 4629 8488 0267 6781 26 | 1444 | −21.0933 3220 0942 1122 7462 | 0.6470 6730 91 ±0.0016 |
| C V | E-Hy-CI | This work | 3040 | −32.4062 4660 1898 1683 2715 76 | 2888 | −21.0933 3231 3387 9577 8140 | 0.6470 6746 83 ±0.0000 022 |
| C V | E-Hy-CI | Recommended | 0.6470 674(22) | ||||
| C V | Hy | SPA (1971) [58] | 1078 | −32.4062 4659 96 | 364 | −21.0933 3230 94 | 0.6470 7 |
| C V | E-Hy | CT (1992) [53] | 100 | −32.4062 4659 80 | 100 | −21.0933 3230 09 | 0.6470 67 |
| O VII | E-Hy-CI | This work | 1088 | −59.1565 9471 2148 2530 7571 52 | 960 | −38.0747 3504 8048 8747 6964 | 0.6944 4942 27 ±0.0014 |
| O VII | E-Hy-CI | This work | 1520 | −59.1565 9471 4241 7214 8946 79 | 1444 | −38.0747 3505 0653 6634 6644 | 0.6944 4942 33 ±0.0014 |
| O VII | E-Hy-CI | This work | 3040 | −59.1565 9512 2757 3982 3860 09 | 2888 | −38.0747 3523 5874 9991 6695 | 0.6944 4959 41±0.0000 022 |
| O VII | E-Hy-CI | Recommended | 0.6944 496 (22) | ||||
| O VII | Hy | SPA (1971) [58] | 1078 | −59.1565 9512 02 | 364 | −38.0747 3523 21 | 0.6944 5 |
| O VII | E-Hy | CT (1992) [53] | 100 | −59.1565 9511 90 | 100 | −38.0747 3522 16 | 0.6944 49 |
8. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
| State | N | Energy (Hartree) | ||||||
|---|---|---|---|---|---|---|---|---|
| He I 1 | 0 | 16 | 2.20 | 16 | 25.0 | 0.5 | 1088 | −2.9037 2435 4189 0888 5088 58 |
| He I 1 | 0 | 19 | 2.20 | 19 | 25.0 | 0.5 | 1520 | −2.9037 2435 4275 7390 6714 30 |
| He I 1 | 1 | 19 | 3.05 | 19 | 40.5 | 0.5 | 3040 | −2.9037 2437 7034 1043 9282 03 |
| He I 1 | 2 | 19 | 3.50 | 19 | 40.5 | 0.5 | 4560 | −2.9037 2437 7034 1195 9808 72 |
| He I 1 | 3 | 18 | 3.90 | 18 | 40.5 | 0.5 | 5928 | −2.9037 2437 7034 1195 9830 82 |
| He I 1 | 4 | 15 | 4.50 | 15 | 40.5 | 0.5 | 6888 | −2.9037 2437 7034 1195 9830 89 |
| He I 1 | 5 | 14 | 5.20 | 15 | 40.5 | 0.5 | 7728 | −2.9037 2437 7034 1195 9830 92 |
| He I 1 | 6 | 14 | 6.00 | 15 | 40.5 | 0.5 | 8568 | −2.9037 2437 7034 1195 9830 94 |
| Li II 1 | 0 | 16 | 3.30 | 16 | 37.5 | 0.5 | 1088 | −7.2799 1332 7713 3474 7954 38 |
| Li II 1 | 0 | 19 | 3.30 | 19 | 37.5 | 0.5 | 1520 | −7.2799 1332 8098 0780 5105 69 |
| Li II 1 | 1 | 19 | 4.575 | 19 | 60.75 | 0.5 | 3040 | −7.2799 1341 2669 2298 8163 74 |
| Li II 1 | 2 | 19 | 5.25 | 19 | 60.75 | 0.5 | 4560 | −7.2799 1341 2669 3059 6348 60 |
| Li II 1 | 3 | 18 | 5.85 | 18 | 60.75 | 0.5 | 5928 | −7.2799 1341 2669 3059 6491 28 |
| Li II 1 | 4 | 15 | 6.75 | 15 | 60.75 | 0.5 | 6888 | −7.2799 1341 2669 3059 6491 59 |
| Li II 1 | 5 | 14 | 7.80 | 15 | 60.75 | 0.5 | 7728 | −7.2799 1341 2669 3059 6491 62 |
| Li II 1 | 6 | 14 | 9.00 | 15 | 60.75 | 0.5 | 8568 | −7.2799 1341 2669 3059 6491 66 |
| Be III 1 | 0 | 16 | 4.40 | 16 | 50.0 | 0.5 | 1088 | −13.6555 6607 5022 1216 4675 55 |
| Be III 1 | 0 | 19 | 4.40 | 19 | 50.0 | 0.5 | 1520 | −13.6555 6607 5806 8585 0501 71 |
| Be III 1 | 1 | 19 | 6.10 | 19 | 81.0 | 0.5 | 3040 | −13.6555 6623 8423 4190 7983 71 |
| Be III 1 | 2 | 19 | 7.00 | 19 | 81.0 | 0.5 | 4560 | −13.6555 6623 8423 5866 9800 72 |
| Be III 1 | 3 | 18 | 7.80 | 18 | 81.0 | 0.5 | 5928 | −13.6555 6623 8423 5867 0206 92 |
| Be III 1 | 4 | 15 | 9.00 | 15 | 81.0 | 0.5 | 6888 | −13.6555 6623 8423 5867 0207 73 |
| Be III 1 | 5 | 14 | 10.40 | 15 | 81.0 | 0.5 | 7728 | −13.6555 6623 8423 5867 0207 76 |
| Be III 1 | 6 | 14 | 12.00 | 15 | 81.0 | 0.5 | 8568 | −13.6555 6623 8423 5867 0207 77 |
| C V 1 | 0 | 16 | 6.60 | 16 | 75.0 | 0.5 | 1088 | −32.4062 4629 6959 6060 3495 12 |
| C V 1 | 0 | 19 | 6.60 | 19 | 75.0 | 0.5 | 1520 | −32.4062 4629 8488 0267 6781 26 |
| C V 1 | 1 | 19 | 9.15 | 19 | 121.5 | 0.5 | 3040 | −32.4062 4660 1898 1683 2715 76 |
| C V 1 | 2 | 19 | 10.50 | 19 | 121.5 | 0.5 | 4560 | −32.4062 4660 1898 5302 9749 39 |
| C V 1 | 3 | 18 | 11.70 | 18 | 121.5 | 0.5 | 5928 | −32.4062 4660 1898 5303 1052 76 |
| C V 1 | 4 | 15 | 13.50 | 15 | 121.5 | 0.5 | 6888 | −32.4062 4660 1898 5303 1055 18 |
| C V 1 | 5 | 14 | 15.60 | 15 | 121.5 | 0.5 | 7728 | −32.4062 4660 1898 5303 1055 21 |
| C V 1 | 6 | 14 | 18.00 | 15 | 121.5 | 0.5 | 8568 | −32.4062 4660 1898 5303 1055 17 |
| O VII 1 | 0 | 16 | 8.80 | 16 | 100.0 | 0.5 | 1088 | −59.1565 9471 2148 2530 7571 52 |
| O VII 1 | 0 | 19 | 8.80 | 19 | 100.0 | 0.5 | 1520 | −59.1565 9471 4241 7214 8946 79 |
| O VII 1 | 1 | 19 | 12.20 | 19 | 162.0 | 0.5 | 3040 | −59.1565 9512 2757 3982 3860 09 |
| O VII 1 | 2 | 19 | 14.00 | 19 | 162.0 | 0.5 | 4560 | −59.1565 9512 2757 9255 3478 69 |
| O VII 1 | 3 | 18 | 15.60 | 18 | 162.0 | 0.5 | 5928 | −59.1565 9512 2757 9255 5849 99 |
| O VII 1 | 4 | 15 | 18.00 | 15 | 162.0 | 0.5 | 6888 | −59.1565 9512 2757 9255 5854 37 |
| O VII 1 | 5 | 14 | 20.80 | 15 | 162.0 | 0.5 | 7728 | −59.1565 9512 2757 9255 5854 39 |
| O VII 1 | 6 | 14 | 24.00 | 15 | 162.0 | 0.5 | 8568 | −59.1565 9512 2757 9255 5854 43 |
| State | N | Energy (hartree) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| He I 2 | 0 | 16 | 2.272 | 1 | 15 | 1.291 | 0.5 | 960 | −2.1238 4308 3927 0924 8309 |
| He I 2 | 0 | 19 | 2.272 | 1 | 19 | 1.291 | 0.5 | 1444 | −2.1238 4308 5062 4897 8422 |
| He I 2 | 1 | 19 | 1.955 | 2 | 19 | 2.25 | 0.5 | 2888 | −2.1238 4308 6497 9644 7703 |
| He I 2 | 2 | 19 | 2.40 | 3 | 18 | 2.30 | 0.5 | 4256 | −2.1238 4308 6498 0912 9534 |
| He I 2 | 3 | 18 | 2.40 | 4 | 15 | 2.30 | 0.5 | 5336 | −2.1238 4308 6498 0989 3227 |
| He I 2 | 4 | 15 | 2.40 | 5 | 14 | 2.30 | 0.5 | 6176 | −2.1238 4308 6498 1004 0079 |
| He I 2 | 5 | 14 | 2.40 | 6 | 14 | 2.30 | 0.5 | 6960 | −2.1238 4308 6498 1008 1958 |
| He I 2 | 6 | 14 | 2.40 | 7 | 14 | 2.30 | 0.5 | 7744 | −2.1238 4308 6498 1009 6317 |
| Li II 2 | 0 | 16 | 3.408 | 1 | 15 | 1.9365 | 0.5 | 960 | −4.9933 5106 5519 3789 3549 |
| Li II 2 | 0 | 19 | 3.408 | 1 | 19 | 1.9365 | 0.5 | 1444 | −4.9933 5106 5763 9277 0706 |
| Li II 2 | 1 | 19 | 2.9325 | 2 | 19 | 3.375 | 0.5 | 2888 | −4.9933 5107 7779 9718 5893 |
| Li II 2 | 2 | 19 | 3.60 | 3 | 18 | 3.45 | 0.5 | 4256 | −4.9933 5107 7779 9724 9272 |
| Li II 2 | 3 | 18 | 3.60 | 4 | 15 | 3.45 | 0.5 | 5336 | −4.9933 5107 7780 0140 3745 |
| Li II 2 | 4 | 15 | 3.60 | 5 | 14 | 3.45 | 0.5 | 6176 | −4.9933 5107 7780 0153 5568 |
| Li II 2 | 5 | 14 | 3.60 | 6 | 14 | 3.45 | 0.5 | 6960 | −4.9933 5107 7780 0155 3584 |
| Li II 2 | 6 | 14 | 3.60 | 7 | 14 | 3.45 | 0.5 | 7744 | −4.9933 5107 7780 0159 3316 |
| Be III 2 | 0 | 16 | 4.544 | 1 | 15 | 2.582 | 0.5 | 960 | −9.1107 7158 4011 3245 7902 |
| Be III 2 | 0 | 19 | 4.544 | 1 | 19 | 2.582 | 0.5 | 1444 | −9.1107 7158 4761 1835 3319 |
| Be III 2 | 1 | 19 | 3.910 | 2 | 19 | 4.500 | 0.5 | 2888 | −9.1107 7162 2916 2965 8769 |
| Be III 2 | 2 | 19 | 4.910 | 3 | 18 | 4.600 | 0.5 | 4256 | −9.1107 7162 2916 2983 6771 |
| Be III 2 | 3 | 18 | 4.800 | 4 | 15 | 4.600 | 0.5 | 5336 | −9.1107 7162 2916 4364 1540 |
| Be III 2 | 4 | 15 | 4.800 | 5 | 14 | 4.600 | 0.5 | 6176 | −9.1107 7162 2916 4383 9458 |
| Be III 2 | 5 | 14 | 4.800 | 6 | 14 | 4.600 | 0.5 | 6960 | −9.1107 7162 2916 4397 2592 |
| Be III 2 | 6 | 14 | 4.800 | 7 | 14 | 4.600 | 0.5 | 7744 | −9.1107 7162 2916 4406 7410 |
| C V 2 | 0 | 16 | 6.816 | 1 | 15 | 3.873 | 0.5 | 960 | −21.0933 3219 8896 1611 3442 |
| C V 2 | 0 | 19 | 6.816 | 1 | 19 | 3.873 | 0.5 | 1444 | −21.0933 3220 0942 1122 7462 |
| C V 2 | 1 | 19 | 5.865 | 2 | 19 | 6.750 | 0.5 | 2888 | −21.0933 3231 3387 9577 8140 |
| C V 2 | 2 | 19 | 7.200 | 3 | 18 | 6.900 | 0.5 | 4256 | −21.0933 3231 3387 9617 4846 |
| C V 2 | 3 | 18 | 7.200 | 4 | 15 | 6.900 | 0.5 | 5336 | −21.0933 3231 3388 3922 5630 |
| C V 2 | 4 | 15 | 7.200 | 5 | 14 | 6.900 | 0.5 | 6176 | −21.0933 3231 3388 3968 4013 |
| C V 2 | 5 | 14 | 7.200 | 6 | 14 | 6.900 | 0.5 | 6960 | −21.0933 3231 3388 3993 9969 |
| C V 2 | 6 | 14 | 7.200 | 7 | 14 | 6.900 | 0.5 | 7744 | −21.0933 3231 3388 4009 9911 |
| O VII 2 | 0 | 16 | 10.224 | 1 | 15 | 5.8095 | 0.5 | 960 | −38.0747 3504 8048 8747 6964 |
| O VII 2 | 0 | 19 | 10.224 | 1 | 19 | 5.8095 | 0.5 | 1444 | −38.0747 3505 0653 6634 6644 |
| O VII 2 | 1 | 19 | 8.7975 | 2 | 19 | 10.125 | 0.5 | 2888 | −38.0747 3523 5874 9991 6695 |
| O VII 2 | 2 | 19 | 10.800 | 3 | 18 | 9.200 | 0.5 | 4256 | −38.0747 3523 5875 0015 0005 |
| O VII 2 | 3 | 18 | 10.800 | 4 | 15 | 10.350 | 0.5 | 5336 | −38.0747 3523 5875 4462 8699 |
| O VII 2 | 4 | 15 | 10.800 | 5 | 14 | 10.350 | 0.5 | 6176 | −38.0747 3523 5875 4922 2755 |
| O VII 2 | 5 | 14 | 10.800 | 6 | 14 | 10.350 | 0.5 | 6960 | −38.0747 3523 5875 5174 8235 |
| O VII 2 | 6 | 14 | 10.800 | 7 | 14 | 10.350 | 0.5 | 7744 | −38.0747 3523 5875 5321 5035 |
| 1 | Some authors refer to this as fully correlated [5]. |
| 2 | |
| 3 | |
| 4 | |
| 5 | Actually, the configuration designation is rigorous only for Russell–Saunders () coupling in a central field. We retain the terminology here to conform to standard spectroscopic notation with the understanding that, by 1 and 1s2p , we mean the lowest terms of those respective symmetries (the He 1 ground state and the 2 lowest state of symmetry, respectively). |
| 6 | In light atoms, the relativistic correction is important for an accurate transition energy but has a small effect on the line strength [26]. For a detailed treatment of line strengths (proportional to transition rates), see Morton et al. [27], where the corrections are shown to be less than 0.1% for helium, and Liu et al. [28], where they are shown to be ≈0.01% for helium. |
| 7 | In this paper, we use f-value and oscillator strength interchangably. |
| 8 | Computed using truncations of our final non-relativistic and wave functions. |
| 9 | For an introduction to relativistic theory as implemented in GRASP, see [57]. |
References
- Sims, J.S.; Hagstrom, S. Combined Configuration-Interaction—Hylleraas-Type Wave-Function Study of the Ground State of the Beryllium Atom. Phys. Rev. A 1971, 4, 908–916. [Google Scholar] [CrossRef]
- Woźnicki, W. On the method of constructing the variational wave functions for many-electron atoms. In Theory of Electronic Shells in Atoms and Molecules; Jucys, A., Ed.; Mintis: Vilnius, Lithuania, 1971; pp. 103–106. [Google Scholar]
- Hylleraas, E.A. Neue berechnung der energie des heliums im grundzustande, sowie des tiefsten terms von ortho-helium. Z. Phys. 1929, 54, 347–366. [Google Scholar] [CrossRef]
- Bunge, C.F. Electronic Wave Functions for Atoms. II. Some Aspects of the Convergence of the Configuration Interaction Expansion for the Ground States of the He Isoelectronic Series. Theor. Chem. Acta 1970, 16, 124–144. [Google Scholar]
- Jentschura, U.D.; Adkins, G.S. Quantum Electrodynamics: Atoms, Lasers and Gravity; World Scientific Publishing Company: Singapore, 2023. [Google Scholar]
- Sims, J.S.; Padhy, B.; Ruiz, M.B. Exponentially correlated Hylleraas-configuration interaction nonrelativistic energy of the 1S ground state of the helium atom. Int. J. Quantum Chem. 2020, 121, e26470. [Google Scholar]
- Sims, J.S.; Padhy, B.; Ruiz, M.B. Exponentially correlated Hylleraas-configuration interaction studies of atomic systems. II. Non-relativistic energies of the 1 1S through 6 1S states of the Li+ ion. Int. J. Quantum Chem. 2022, 122, e26823. [Google Scholar] [CrossRef]
- Wang, C.; Mei, P.; Kurokawa, Y.; Nakashima, H.; Nakatsuji, H. Analytical evaluations of exponentially correlated unlinked one-center, three- and four-electron integrals. Phys. Rev. A 2012, 85, 042512. [Google Scholar] [CrossRef]
- Hirschfelder, J.O. Removal of electron-electron poles from many-electron Hamiltonians. J. Chem. Phys. 1963, 39, 3145–3146. [Google Scholar] [CrossRef]
- Padhy, B. Kinetic energy matrix elements for a two-electron atom with extended Hylleraas-CI wave function. Orissa J. Phys. 2018, 25, 9–21. [Google Scholar]
- Padhy, B. Analytic evaluation of two-electron atomic integrals involving extended Hylleraas-CI functions with STO basis. Orissa J. Phys. 2018, 25, 99–112. [Google Scholar]
- Harris, F.E. Exponentially correlated wave functions for four-body systems. Adv. Quantum Chem. 2016, 73, 81–102. [Google Scholar]
- Harris, F.E. Matrix elements for explicitly-correlated atomic wave functions. In Concepts, Methods and Applications of Quantum Systems in Chemistry and Physics. Progress in Theoretical Chemistry and Physics; Wang, Y., Thachuk, M., Krems, R., Maruani, J., Eds.; Springer: New York, NY, USA, 2018; Volume 31. [Google Scholar]
- Ruiz, M.B.; Sims, J.S.; Padhy, B. High-precision Hy-CI and E-Hy-CI studies of atomic and molecular properties. Adv. Quantum Chem. 2021, 83, 171–208. [Google Scholar]
- Chung, H.-K.; Braams, B.J.; Bartschat, K.; Csaszar, A.G.; Drake, G.W.F.; Kirchner, T.; Kokoouline, V.; Tennyson, J. Uncertainty Estimates for Theoretical Atomic and Molecular Data. J. Phys. D Appl. Phys. 2016, 49, 363002. [Google Scholar] [CrossRef] [Green Version]
- Löwdin, P.O. Angular momentum wavefunctions constructed by projector operators. Rev. Mod. Phys. 1964, 36, 966–976. [Google Scholar] [CrossRef]
- Zhang, Y.Z.; Gao, Y.C.; Jiao, L.G.; Liu, F.; Ho, Y.K. Linear dependence in Hylleraas-configuration interaction calculations of He atom. Int. J. Quantum Chem. 2019, 120, e26136. [Google Scholar] [CrossRef]
- Jiao, L.G.; Zan, L.R.; Zhu, L.; Zhang, Y.Z.; Ho, Y.K. High-precision calculation of the geometric quantities of two-electron atoms based on the Hylleraas-configuration-interaction basis. Phys. Rev. A 2019, 100, 022509. [Google Scholar] [CrossRef]
- Condon, E.U.; Shortley, G.H. The Theory of Atomic Spectra; Cambridge University Press: Cambridge, UK, 1963. [Google Scholar]
- Sims, J.S.; Ruiz, M.B. Parallel generalized real symmetric-definite eigenvalue problem. J. Res. Natl. Inst. Stand. Technol. 2020, 125, 125032. [Google Scholar] [CrossRef]
- Message Passing Interface Forum. MPI: A Message-Passing Interface Standard. Int. J. Supercomput. Appl. High Perform. Comput. 1994, 8, 159–416. [Google Scholar]
- Sims, J.S.; Hagstrom, S.A. High-precision Hy-CI variational calculations for the ground state of neutral helium and helium-like ions. Int. J. Quantum Chem. 2002, 90, 1600–1609. [Google Scholar] [CrossRef]
- Sims, J.S.; Hagstrom, S.A. Hylleraas-configuration interaction study of the 2 2S ground state of neutral lithium and the first five excited 2S states. Phys. Rev. A 2009, 80, 052507. [Google Scholar] [CrossRef] [Green Version]
- Sims, J.S.; Hagstrom, S.A. Hylleraas-configuration interaction study of the 1S ground state of neutral beryllium. Phys. Rev. A 2011, 83, 032518. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, L. Relative multiplet strengths in L-S coupling. Astrophys. J. 1935, 82, 1–25. [Google Scholar] [CrossRef]
- Fischer, C.F. Evaluating the accuracy of theoretical transition data. J. Phys. B At. Mol. Opt. Phys. 2009, T134, 014019. [Google Scholar] [CrossRef]
- Morton, D.C.; Moffatt, P.; Drake, G.W.F. Relativistic corrections to the He I transition rates. Can. J. Phys. 2011, 89, 129–134. [Google Scholar] [CrossRef]
- Liu, Q.; Li, J.; Wang, J.; Qu, Y. Effect of electron correlation and Breit interaction on energies, oscillator strengths, and transition rates for low-lying states of helium. Chin. Phys. Lett. 2021, 38, 113101. [Google Scholar] [CrossRef]
- Sims, J.S.; Whitten, R.C. Upper and lower bounds to atomic and molecular properties. I. Be-sequence oscillator strengths (Dipole-length formulation) for the 1s2 2s2 1S → 1s2 2s2p 1P transition. Phys. Rev. A 1973, 8, 2220–2230. [Google Scholar] [CrossRef]
- Sims, J.S.; Hagstrom, S.A. One center rij integrals over Slater-type orbitals. J. Chem. Phys. 1971, 55, 4699–4710. [Google Scholar] [CrossRef]
- Sims, J.S.; Hagstrom, S.A. Mathematical and computational science issues in high precision Hylleraas-configuration interaction variational calculations: I. Three-electron integrals. J. Phys. B At. Mol. Opt. Phys. 2004, 37, 1519–1540. [Google Scholar] [CrossRef]
- Sims, J.S.; Hagstrom, S.A. Mathematical and computational science issues in high precision Hylleraas-configuration interaction variational calculations: III. Four-electron integrals. J. Phys. B At. Mol. Opt. Phys. 2015, 48, 175003. [Google Scholar] [CrossRef]
- Chandrasekhar, S. Some remarks on the negative hydrogen ion and its absorption coefficient. Astrophys. J. 1944, 100, 176. [Google Scholar] [CrossRef]
- Chandrasekhar, S. Theoretical molecular transition probabilities. I. The V-N transition in H2. J. Chem. Phys. 1961, 34, 1224–1231. [Google Scholar]
- Weinhold, F. Calculation of upper and lower bounds to oscillator strengths. J. Chem. Phys. 1971, 54, 1874–1881. [Google Scholar] [CrossRef]
- Weinhold, F. Upper and lower bounds to quantum-mechanical properties. Adv. Quant. Chem. 1972, 6, 299–331. [Google Scholar]
- Anderson, M.T.; Weinhold, F. Dipole oscillator strengths, with rigorous limits of error, for He and Li+. Phys. Rev. A 1974, 9, 118–128. [Google Scholar] [CrossRef]
- Sims, J.S.; Hagstrom, S.A.; Rumble, J.R., Jr. Upper and lower bounds to atomic and molecular properties. III. Lithium oscillator strengths for various doublet S→doublet P transitions. Phys. Rev. A 1976, 13, 242–250. [Google Scholar] [CrossRef]
- Rumble, J.R., Jr.; Sims, J.S.; Purdy, D.R. Upper and lower bounds for the oscillator strength of the X → C transition of the hydrogen molecule. J. Phys. B: At. Mol. Opt. Phys. 1977, 10, 2553–2559. [Google Scholar] [CrossRef]
- Roginsky, D.V.I.; Laughin, C.; Cohen, M. Improved lower and upper bounds for dipole transition moments. J. Phys. B At. Mol. Phys. 1986, 19, 1115–1123. [Google Scholar] [CrossRef]
- Roginsky, D.V.I.; Weiss, A.W. Calculations of tighter error bounds for theoretical atomic-oscillator strengths. Phys. Rev. A 1988, 38, 1760–1766. [Google Scholar] [CrossRef] [PubMed]
- Drake, G.W.F. High precision theory of atomic helium. Phys. Scr. 1999, T83, 83–92. [Google Scholar] [CrossRef]
- Drake, G.W.F. High precision calculations for helium. In Atomic, Molecular, and Optical Physics Handbook; Drake, G.W.F., Ed.; API Press: New York, NY, USA, 1996; pp. 154–171. [Google Scholar]
- Szasz, L. Atomic many-body problem. I. General theory of correlated wave functions. Phys. Rev. 1962, 126, 169–181. [Google Scholar] [CrossRef]
- Szasz, L. Pair correlations in the Be atom. Phys. Lett. 1963, 126, 263–264. [Google Scholar] [CrossRef]
- Szasz, L.; Byrne, J. Atomic many-body problem. III. The calculation of Hylleraas-type correlated wave functions for the beryllium atom. Phys. Rev. 1967, 158, 34–48. [Google Scholar] [CrossRef]
- Plute, E.J., Jr. Orthogonal Combined Configuration Interaction—Hylleraas Study of Two Electron Atoms. Ph.D. Thesis, Indiana University, Bloomington, IN, USA, 1984. [Google Scholar]
- Schiff, B.; Lifson, H.; Pekeris, C.L.; Rabinowitz, P. 2 1,3P, 3 1,3P, and 4 1,3P states of He and the 2 1P State of Li+. Phys. Rev. 1965, 140, 1104–1121. [Google Scholar] [CrossRef]
- Kato, T. On the eigenfunctions of many-particle systems in quantum mechanics. Commun. Pure Appl. Math. 1957, 10, 151–177. [Google Scholar] [CrossRef]
- Aznabaev, D.T.; Bekbaev, A.K.; Korobov, V.I. Nonrelativistic energy levels of helium atoms. Phys. Rev. A 2018, 98, 012510. [Google Scholar] [CrossRef] [Green Version]
- Jiao, L.G.; Jilin University, Changchun, China. Personal communication, 2021.
- Nakashima, H.; Nakatsuji, H. Solving the Schrödinger equation for helium atom and its isoelectronic ions with the free iterative complement interaction method. J. Chem. Phys. 2008, 128, 154107. [Google Scholar] [CrossRef] [Green Version]
- Cann, N.M.; Thakkar, A.J. Oscillator strengths for S→P and P→D transitions in heliumlike ions. Phys. Rev. A 1992, 46, 5397–5404. [Google Scholar] [CrossRef]
- Jönsson, P.; He, X.; Fischer, C.F.; Grant, I.P. The GRASP2K atomic structure package. Comput. Phys. Commun. 2007, 177, 597–622. [Google Scholar] [CrossRef]
- Fischer, C.F.; Gaigalas, G.; Jönsson, P.; Bierón, J. Grasp2018—A Fortran 95 version of the general relativistic atomic structure package. Comput. Phys. Commun. 2019, 237, 184–187. [Google Scholar] [CrossRef]
- Jönsson, P.; Gaigalas, G.; Bierón, J.; Fischer, C.F.; Grant, I.P. New version: GRASP2K relativistic atomic structure package. Comput. Phys. Commun. 2013, 184, 2197–2203. [Google Scholar] [CrossRef] [Green Version]
- Jönsson, P.; Godefroid, M.; Gaigalas, G.; Ekman, J.; Grumer, J.; Li, W.; Li, J.; Brage, T.; Grant, I.P.; Bierón, J.; et al. An Introduction to Relativistic Theory as Implemented in GRASP. Atoms 2023, 11, 7. [Google Scholar] [CrossRef]
- Schiff, B.; Pekeris, C.L.; Accad, Y. f values for transitions between the low-lying S and P states of the helium isoelectronic sequence up to Z = 10. Phys. Rev. A 1971, 4, 885–893. [Google Scholar] [CrossRef]
- Wiese, W.L.; Fuhr, J.R. Accurate atomic transition probabilities for hydrogen through lithium. J. Phys. Chem. Ref. Data 2009, 38, 565–719. [Google Scholar] [CrossRef] [Green Version]
- Kono, A.; Hattori, S. Accurate oscillator strengths for neutral helium. Phys. Rev. A 1984, 29, 2981–2988. [Google Scholar] [CrossRef]
- Drake, G.W.F.; Morton, D.C. A Multiplet Table for Neutral Helium (4He I) with Transition Rates. Astrophys. J. Supp. Ser. 2007, 170, 251–260. [Google Scholar] [CrossRef] [Green Version]
- Fischer, C.F. A multi-configuration Hartree-Fock program. Comput. Phys. Commun. 1970, 1, 151–166. [Google Scholar] [CrossRef]
- Fischer, C.F.; Brage, T.; Jönsson, P. Computational atomic structure: An MCHF approach. In Concepts, Methods and Applications of Quantum Systems in Chemistry and Physics. Progress in Theoretical Chemistry and Physics; Routledge: New York, NY, USA, 1997. [Google Scholar]
- Fischer, C.F. The MCHF atomic-structure package. Comput. Phys. Commun. 2000, 128, 635–636. [Google Scholar] [CrossRef]
- Fischer, C.F. The MCHF atomic-structure package. Comput. Phys. Commun. 2013, 184, 2197–2203. [Google Scholar]
- Wiese, W.L.; Weiss, A.W. Regularities in atomic oscillator strengths. Phys. Rev. 1968, 175, 50–65. [Google Scholar] [CrossRef]
- Weiss, A.W. Oscillator Strengths for the Helium Isoelectronic Sequence. J. Res. Natl. Inst. Stand. Technol. 1967, 71A, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Lüchow, A.; Kleindienst, H. Accurate upper and lower bounds to the 2S states of the lithium atom. Int. J. Quantum Chem. 1994, 51, 211–224. [Google Scholar] [CrossRef]
- King, F.W. Lower bound for the nonrelativistic ground state energy of the lithium atom. J. Chem. Phys. 1995, 102, 8053–8058. [Google Scholar] [CrossRef]
- Ireland, R.T.; Jeszenszki, P.; Mâtyus, E.; Martinazzo, R.; Ronto, M.; Pollak, E. Lower Bounds for Nonrelativistic Atomic Energies. ACS Phys. Chem. Au 2022, 2, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Pollak, E.; Martinazzo, R. Lower Bounds for Coulombic Systems. ACS Phys. Chem. Au 2021, 17, 1535–1547. [Google Scholar] [CrossRef]
- Ronto, M.; Jeszenszki, P.; Mâtyus, E.; Pollak, E. Lower bounds on par with upper bounds for few-electron atomic energies. Phys. Rev. A 2023, 107, 012204. [Google Scholar] [CrossRef]

| State | Technique | Author | N | Energy (Hartree) |
|---|---|---|---|---|
| He 1 | E-Hy-CI | Sims et al. (2020) [6] | 8568 | −2.9037 2437 7034 1195 9830 94 |
| He 1 | ICI | Nakashima and Nakatsuji (2008) [52] | 22,709 | −2.9037 2437 7034 1195 9831 12 |
| Li II 1 | E-Hy-CI | Sims et al. (2021) [7] | 8568 | −7.2799 1341 2669 3059 6491 66 |
| Li II 1 | ICI | Nakashima and Nakatsuji (2008) [52] | 22,709 | −7.2799 1341 2669 3059 6491 95 |
| Be III 1 | E-Hy-CI | This work | 8568 | −13.6555 6623 8423 5867 0207 77 |
| Be III 1 | ICI | Nakashima and Nakatsuji (2008) [52] | 9682 | −13.6555 6623 8423 5867 0208 17 |
| C V 1 | E-Hy-CI | This work | 8568 | −32.4062 4660 1898 5303 1055 17 |
| C V 1 | ICI | Nakashima and Nakatsuji (2008) [52] | 9682 | −32.4062 4660 1898 5303 1055 74 |
| O VII 1 | E-Hy-CI | This work | 8568 | −59.1565 9512 2757 9255 5854 43 |
| O VII 1 | ICI | Nakashima and Nakatsuji (2008) [52] | 9682 | −59.1565 9512 2757 9255 5854 99 |
| He 2 | E-Hy-CI | This work | 6176 | −2.1238 4308 6498 1004 0079 |
| He 2 | E-Hy-CI | This work | 6960 | −2.1238 4308 6498 1008 1958 |
| He 2 | E-Hy-CI | This work | 7744 | −2.1238 4308 6498 1009 6317 |
| He 2 | Extrapolated | This work | −2.1238 4308 6498 1010(3) | |
| He 2 | E-Hy | Aznabaev et al. (2018) [50] | 22,000 | −2.1238 4308 6498 1013 5925 |
| Li II 2 | E-Hy-CI | This work | 6176 | −4.9933 5107 7780 0153 5568 |
| Li II 2 | E-Hy-CI | This work | 6960 | −4.9933 5107 7780 0155 3584 |
| Li II 2 | E-Hy-CI | This work | 7744 | −4.9933 5107 7780 0159 3316 |
| Li II 2 | Extrapolated | This work | −4.9933 5107 7780 0165(3) | |
| Li II 2 | ECS 1 | Cann and Thakkar (1992) [53] | 100 | −4.9933 5107 21 |
| Be III 2 | E-Hy-CI | This work | 6176 | −9.1107 7162 2916 4383 9458 |
| Be III 2 | E-Hy-CI | This work | 6960 | −9.1107 7162 2916 4397 2592 |
| Be III 2 | E-Hy-CI | This work | 7744 | −9.1107 7162 2916 4406 7410 |
| Be III 2 | Extrapolated | This work | −9.1107 7162 2916 442(2) | |
| Be III 2 | ECS | Cann and Thakkar (1992) [53] | 100 | −9.1107 7161 42 |
| C V 2 | E-Hy-CI | This work | 6176 | −21.0933 3231 3388 3968 4013 |
| C V 2 | E-Hy-CI | This work | 6960 | −21.0933 3231 3388 3993 9969 |
| C V 2 | E-Hy-CI | This work | 7744 | −21.0933 3231 3388 4009 9911 |
| C V 2 | Extrapolated | This work | −21.0933 3231 3388 403(2) | |
| C V 2 | ECS | Cann and Thakkar (1992) [53] | 100 | −21.0933 3230 09 |
| O VII 2 | E-Hy-CI | This work (QP) | 7744 | −38.0747 3523 5875 4074 |
| O VII 2 | E-Hy-CI | This work | 6176 | −38.0747 3523 5875 4922 2755 |
| O VII 2 | E-Hy-CI | This work | 6960 | −38.0747 3523 5875 5174 8235 |
| O VII 2 | E-Hy-CI | This work | 7744 | −38.0747 3523 5875 5321 5034 |
| O VII 2 | Extrapolated | This work | −38.0747 3523 5875 55(2) | |
| O VII 2 | ECS | Cann and Thakkar (1992) [53] | 100 | −38.0747 3522 16 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sims, J.S.; Padhy, B.; Ruiz, M.B.R. Exponentially Correlated Hylleraas–Configuration Interaction Studies of Atomic Systems. III. Upper and Lower Bounds to He-Sequence Oscillator Strengths for the Resonance 1S→1P Transition. Atoms 2023, 11, 107. https://doi.org/10.3390/atoms11070107
Sims JS, Padhy B, Ruiz MBR. Exponentially Correlated Hylleraas–Configuration Interaction Studies of Atomic Systems. III. Upper and Lower Bounds to He-Sequence Oscillator Strengths for the Resonance 1S→1P Transition. Atoms. 2023; 11(7):107. https://doi.org/10.3390/atoms11070107
Chicago/Turabian StyleSims, James S., Bholanath Padhy, and María Belén Ruiz Ruiz. 2023. "Exponentially Correlated Hylleraas–Configuration Interaction Studies of Atomic Systems. III. Upper and Lower Bounds to He-Sequence Oscillator Strengths for the Resonance 1S→1P Transition" Atoms 11, no. 7: 107. https://doi.org/10.3390/atoms11070107
APA StyleSims, J. S., Padhy, B., & Ruiz, M. B. R. (2023). Exponentially Correlated Hylleraas–Configuration Interaction Studies of Atomic Systems. III. Upper and Lower Bounds to He-Sequence Oscillator Strengths for the Resonance 1S→1P Transition. Atoms, 11(7), 107. https://doi.org/10.3390/atoms11070107